
Abstract
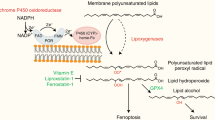
The accumulation of lipid peroxides is recognized as a determinant of the occurrence of ferroptosis. However, the sensors and amplifying process of lipid peroxidation linked to ferroptosis remain obscure. Here we identify PKCβII as a critical contributor of ferroptosis through independent genome-wide CRISPR–Cas9 and kinase inhibitor library screening. Our results show that PKCβII senses the initial lipid peroxides and amplifies lipid peroxidation linked to ferroptosis through phosphorylation and activation of ACSL4. Lipidomics analysis shows that activated ACSL4 catalyses polyunsaturated fatty acid-containing lipid biosynthesis and promotes the accumulation of lipid peroxidation products, leading to ferroptosis. Attenuation of the PKCβII–ACSL4 pathway effectively blocks ferroptosis in vitro and impairs ferroptosis-associated cancer immunotherapy in vivo. Our results identify PKCβII as a sensor of lipid peroxidation, and the lipid peroxidation–PKCβII–ACSL4 positive-feedback axis may provide potential targets for ferroptosis-associated disease treatment.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others

Data availability
Previously published data from 49 patients with melanoma who underwent tumour biopsies before starting immunotherapy that were re-analysed here are available at the Gene Expression Omnibus under the accession code GSE91061 (ref. 28). Data of the CRISPR screening are available as Supplementary Table 1. Data of the kinase screening are available as Supplementary Table 2. Lipidomics data are available as Supplementary Tables 3 and 4. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
References
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Stockwell, B. R. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Tang, D., Chen, X., Kang, R. & Kroemer, G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125 (2021).
Jiang, X., Stockwell, B.R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Zhang, Y. et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 20, 1181–1192 (2018).
Gao, M. et al. Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363 (2019).
Wang, W. et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019).
Lei, G. et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 30, 146–162 (2020).
Yuan, H., Li, X., Zhang, X., Kang, R. & Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 478, 1338–1343 (2016).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Yang, W. S. & Stockwell, B. R. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 26, 165–176 (2016).
Stockwell, B. R. & Jiang, X. The chemistry and biology of ferroptosis. Cell Chem. Biol. 27, 365–375 (2020).
Stockwell, B. R., Jiang, X. & Gu, W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 30, 478–490 (2020).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
Lee, H. et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22, 225–234 (2020).
Yan, B. et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol. Cell 81, 355–369 (2021).
Wenzel, S. E. et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641 (2017).
Chu, B. et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 21, 579–591 (2019).
Mochly-Rosen, D., Das, K. & Grimes, K. V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 11, 937–957 (2012).
Nakajima, T. Positive and negative regulation of radiation-induced apoptosis by protein kinase C. J. Radiat. Res. 49, 1–8 (2008).
Maher, P. How protein kinase C activation protects nerve cells from oxidative stress-induced cell death. J. Neurosci. 21, 2929–2938 (2001).
Steinberg, S. F. Mechanisms for redox-regulation of protein kinase C. Front. Pharmacol. 6, 128 (2015).
Gopalakrishna, R. & Jaken, S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 28, 1349–1361 (2000).
Hassannia, B., Vandenabeele, P. & Vanden Berghe, T. Targeting ferroptosis to iron out cancer. Cancer Cell 35, 830–849 (2019).
Hisanaga, Y. et al. Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J. Biol. Chem. 279, 31717–31726 (2004).
Smith, M. E., Saraceno, G. E., Capani, F. & Castilla, R. Long-chain acyl-CoA synthetase 4 is regulated by phosphorylation. Biochem. Biophys. Res. Commun. 430, 272–277 (2013).
Riaz, N. et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 171, 934–949 (2017).
Hart, T. et al. Evaluation and design of genome-wide CRISPR/SpCas9 Knockout Screens. G3 7, 2719-2727 (2017).
Mair, B. et al. Essential gene profiles for human pluripotent stem cells identify uncharacterized genes and substrate dependencies. Cell Rep. 27, 599–615 (2019).
Parker, P. J. et al. Equivocal, explicit and emergent actions of PKC isoforms in cancer. Nat. Rev. Cancer 21, 51–63 (2021).
Alkon, D. L., Sun, M. K. & Nelson, T. J. PKC signaling deficits: a mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci. 28, 51–60 (2007).
Yan, S. F., Harja, E., Andrassy, M., Fujita, T. & Schmidt, A. M. Protein kinase C β/early growth response-1 pathway: a key player in ischemia, atherosclerosis, and restenosis. J. Am. Coll. Cardiol. 48, A47–A55 (2006).
Altman, A. & Kong, K. F. Protein kinase C enzymes in the hematopoietic and immune systems. Annu. Rev. Immunol. 34, 511–538 (2016).
Acknowledgements
This study was supported by the Natural Science Foundation of China (grant nos 81630079 (to X.-F.Z.), 81803006 (to H.-L.Z.), 81772624 (to R.D.), 81972855 (to X.-F.Z.) and 82173092 (to R.D.)), Natural Science Foundation of Guangdong Province (grant no. 2021A1515010092 to R.D.), National Postdoctoral Program for Innovative Talents (grant no. BX2021391 to Z.-L.L.), China Postdoctoral Science Foundation (grant no. 2020M683107 to T.D.). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
X.-F.Z. and H.-L.Z. conceived the idea. H.-L.Z. and B.-X.H. performed most experiments. Z.-L.L., Z.-P.Y., X.-D.P. and G.-K.F. performed the animal experiments. X.L., X.-Y.Z. and Y.-H.C. performed the lipidomic analyses. Y.H. and T.D. analysed the clinical data. H.-L.Z., R.D. and X.-F.Z. wrote the manuscript. All co-authors have seen and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks Marcus Conrad and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 PKC kinase inhibitors inhibit ferroptosis.
(a) An immunoblot showing the levels of PKCα phosphorylation in MDA-MB-231 or HT1080 cells subjected to the indicated treatments for 16 h. p-PKCα, phospho-PKCα. The experiment was repeated three times, independently, with similar results. (b,c) Cell death (left) and lipid peroxidation (right) measurements in HT1080 cells subjected to the indicated treatments for 20 h. Erastin, 4 μM; RSL3, 50 nM; Go6983, 5 μM; Enza, 5 μM enzastaurin. (d,e) Cell death (left) and lipid peroxidation (right) measurements in MEF cells subjected to the indicated treatments for 16 h. Erastin, 2 μM; RSL3, 25 nM; Go6983, 5 μM; Enza, 5 μM enzastaurin. (f) Analysis of radical trapping/antioxidant potential of Go6983 and enzastaurin (Enza) compared to α-tocopherol. Antioxidant activity was assessed using the DPPH assay. Absorption was measured at 517 nm. (g) Representative co-autoxidations of STY-BODIPY (1 μM)-embedded liposomes of egg PC (5 mM) initiated by di-tert-undecyl hyponitrite (0.2 mM, black) and inhibited by either Go6983 (20 μM, red), enzastaurin (20 μM, blue), or α-tocopherol (20 μM, green). (h) Cell death (left) and lipid peroxidation (right) measurements in the indicated cells treated with 5 μM erastin or 2.5 μM RLS3 for 20 h. The mean ± s.d. of n = 3 independent experiments are shown. Statistical analysis was performed using an unpaired two-tailed t test.
Extended Data Fig. 2 PKCβII promote ferroptosis.
(a-l) Cell viability measurements in MDA-MB-231 and HT1080 cells subjected to the indicated treatments. Error bars are mean ± s.d., n = 3 independent repeats. Statistical analysis was performed using a two-way ANOVA analysis.
Extended Data Fig. 3 PKCβII has a direct impact on ferroptosis sensitivity.
An immunoblot showing the expression of PKCβII in HS578T (a), MDA-MB-468 (g), MCF7 (m) and BT474 (s) cells subjected to the indicated treatments. Data in a, g, m, s are representative of n = 3 biologically independent experiments. (b-e, h-k, n-q, t-w) Cell viability measurements in cells subjected to the indicated treatments. (f, l, r, x) Lipid peroxidation measurements in the indicated cells subjected to the indicated treatments for 20 h. f and l, Erastin, 10 μM; RSL3, 6 μM, Time, 24 h. r and x, Erastin, 15 μM; RSL3, 6 μM, Time, 24 h. b-e, h-k, n-q, t-w, error bars are mean ± s.d., n = 3 independent repeats. Statistical analysis was performed using a two-way ANOVA analysis. f, l, r, x, the mean ± s.d. of n = 3 independent experiments are shown. Statistical analysis was performed using an unpaired two-tailed t test.
Extended Data Fig. 4 PKCβII activation is essential for ferroptosis.
(a-b) Cell death measurements in MDA-MB-231 (a) and HT1080 (b) cells subjected to the indicated treatments. (c) An immunoblot showing the expression of membrane-PKCα, βI, γ, δ, ζ in MDA-MB-231 cells treated with erastin at the indicated concentration for 16 h. (d) Lipid peroxidation measurement in the indicated MDA-MB-231 cells or HT1080 cells treated with erastin or RLS3 for 20 h. For MDA-MB-231 cells, erastin, 5 μM; RSL3, 2.5 μM. For HT1080 cells, erastin, 4 μM; RSL3, 50 nM. The mean ± s.d. of n = 3 independent experiments are shown. Statistical analysis was performed using an unpaired two-tailed t test. (e,f) An immunoblot showing the expression of membrane-PKCβII in MDA-MB-231 (e) or HT1080 cells (f) subjected to the indicated treatments for 16 h. For MDA-MB-231 cells, erastin, 5 μM; RSL3, 2.5 μM. For HT1080 cells, erastin, 4 μM; RSL3, 50 nM. (g,h) Endogenous PKCβII was immunoprecipitated from MDA-MB-231 (g) and HT1080 (h) cells subjected to the indicated treatments for 16 h, followed by immunoblotting using a phospho-Ser/Thr antibody. For MDA-MB-231 cells, erastin, 5 μM; RSL3, 2.5 μM. For HT1080 cells, erastin, 4 μM; RSL3, 50 nM. a, b, d, the mean ± s.d. of n = 3 independent experiments are shown. Statistical analysis was performed using an unpaired two-tailed t test. Data in c, e-h, are representative of n = 3 biologically independent experiments.
Extended Data Fig. 5 PKCβII did not affect the protein level of GPX4 or cellular divalent iron levels.
(a) An immunoblot showing the expression of GPX4 in MDA-MB-231 cells subjected to the indicated treatments for 18 h. (b) Cellular divalent iron levels were assayed in the indicated MDA-MB-231 cells. (c) Cellular divalent iron levels were assayed in MDA-MB-231 cells subjected to the indicated treatments. Go6983, 5 μM; Enza, 5 μM. (d) The potential phosphorylation sites of ACSL4 predicted by GPS and the SCANSITE program. (e) An immunoblot showing the levels of ACSL4 in the indicated cells. (f-i) Endogenous ACSL4 was immunoprecipitated from cells subjected to the indicated treatments for 16 h, followed by immunoblotting using an ACSL4 phospho-T328 antibody. For MDA-MB-231 cells, erastin, 5 μM; RSL3, 2.5 μM. For HT1080 cells, erastin, 4 μM; RSL3, 50 nM. b, c, the mean±s.d. of n = 3 independent experiments are shown. Statistical analysis was performed using an unpaired two-tailed t test. Data in a, e-i, are representative of n = 3 biologically independent experiments.
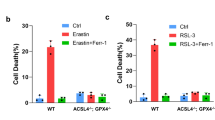
Extended Data Fig. 6 Fer-1 or Lipro-1 blocks PKCβII–ACSL4 pathway through eliminating lipid peroxides.
(a) MDA-MB-231 and HT1080 cells were cotransfected with the FLAG-ACSL4 plasmid and HA-ACSL4 plasmid. Then, these cells were subjected to the indicated treatments for 16 h, and FLAG-ACSL4 was immunoprecipitated and immunoblotted using an HA antibody. For MDA-MB-231 cells, erastin, 5 μM; RSL3, 2.5 μM; Fer-1, 5 μM; Lipro-1, 5 μM. For HT1080 cells, erastin, 4 μM; RSL3, 50 nM; Fer-1, 5 μM; Lipro-1, 5 μM. (b) MDA-MB-231 and HT1080 cells subjected to the indicated treatments for 16 h. Cell lysates were immunoblotted under non-reducing conditions. For MDA-MB-231 cells, erastin, 5 μM; RSL3, 2.5 μM; Fer-1, 5 μM; Lipro-1, 5 μM. For HT1080 cells, erastin, 4 μM; RSL3, 50 nM; Fer-1, 5 μM; Lipro-1, 5 μM. Data in a,b are representative of n = 3 biologically independent experiments. (c) The relative signal intensity of PE species containing mono-unsaturated and saturated fatty acids in cells were subjected to the indicated treatments for 16 h. Erastin, 5 μM; Enza, 5 μM. Data show the mean±s.d. of n = 6 independent experiments. Statistical analysis was performed using unpaired two-tailed t tests.
Extended Data Fig. 7 T328 phosphorylation is critical for the pro-ferroptotic functions of ACSL4.
Vector control, ACSL4-WT, ACSL4-T328A or ACSL4-T328D plasmids were transfected into ACSL4-knockout MDA-MB-231 and HT1080 cells. An immunoblot showing the levels of ACSL4 in the indicated cells. Data are representative of n = 3 biologically independent experiments. (b,c) Cell death (b) or lipid peroxidation (c) measurements in the indicated cells treated with 4 μM erastin or 50 nM RSL3 for 20 h. The P values correspond to the comparison between the ACSL4-T328A and matched ACSL4-WT cells under erastin or RSL3 treatment. The mean±s.d. n = 3 of independent experiments are shown. Statistical analysis was performed using an unpaired two-tailed t test.
Extended Data Fig. 8 The ACSL4-T328D variant stably expressed in PKCβII knockout cells suffice to re-sensitize to ferroptosis.
(a, f) An immunoblot showing the expression of ACSL4 in MDA-MB-231 (a) and HT1080 (f) cells subjected to the indicated treatments. (b-e, g-j) Cell viability measurements in cells subjected to the indicated treatments. (k,l) Lipid peroxidation (k) and cell death (l) measurements in cardiomyocytes cells subjected to the indicated treatments. (m) Endogenous ACSL4 was immunoprecipitated from cardiomyocytes cells subjected to the indicated treatments, followed by immunoblotting using an ACSL4 phospho-T328 antibody. Data in a, f, m, are representative of n = 3 biologically independent experiments. b-e, g-j, error bars are mean±s.d., n = 3 independent repeats. Statistical analysis was performed using a two-way ANOVA analysis. K,l, the mean±s.d. of n = 3 independent experiments are shown. Statistical analysis was performed using an unpaired two-tailed t test.
Extended Data Fig. 9 PKCβII knockout reduces the efficacy of immunotherapy.
(a) Vector control, ACSL4-WT or ACSL4-T328A plasmids were transfected into ACSL4-knockout B16 cells. An immunoblot showing the levels of ACSL4 in the indicated cells. (b) PKCβ was knockout in 4T1 cells using CRISPER-Cas9 technology. An immunoblot showing the levels of PKCβII in the indicated cells. Data in a,b are representative of n = 3 biologically independent experiments. (c) Representative TIL dot plots from a representative mouse for each group are shown (left). The percentage of TILs for each group was calculated (right). The mean±s.d. are shown; n = 6 biologically independent mice. Statistical analysis was performed using an unpaired two-tailed t test.
Extended Data Fig. 10 PKCβII enhances the efficacy of immunotherapy by facilitating ferroptosis.
(a,b) Tumour growth of mouse B16-PKCβII-KO-Vector or B16-PKCβII-KO- PKCβII tumour xenografts in C57 mice following treatment with mouse anti-PD1 antibody or lipoxstatin alone or in combination. (n = 6 mice per group). Tumour volumes (a) and tumour weights (b) upon autopsy on day 15 were calculated. (c) Relative lipid peroxidation in tumour cells isolated from B16-PKCβII-KO-Vector or B16-PKCβII-KO- PKCβII tumours. (d-g) The percentage of TILs for each group was calculated. The mean±s.d. are shown; n = 6 biologically independent mice. Statistical analysis was performed using an unpaired two-tailed t test.
Supplementary information
Supplementary Table 1
Supplementary Table 1. Data of the CRISPR screening. Supplementary Table 2. Data of the kinase screening. Supplementary Table 3. Lipidomic data 1. Supplementary Table 4. Lipidomic data 2. Supplementary Table 5. Correlation between ACSL4 and the immunotherapeutic response.
Source data
Source Data Fig. 1
Unprocessed western blots and/or gels.
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Unprocessed western blots and/or gels.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Unprocessed western blots and/or gels.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Unprocessed western blots and/or gels.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Unprocessed western blots and/or gels.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 1
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
About this article

Cite this article
Zhang, HL., Hu, BX., Li, ZL. et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol 24, 88–98 (2022). https://doi.org/10.1038/s41556-021-00818-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41556-021-00818-3
This article is cited by
-
Identifying CTH and MAP1LC3B as ferroptosis biomarkers for prognostic indication in gastric cancer decoding
Scientific Reports (2024)
-
Polyamine-mediated ferroptosis amplification acts as a targetable vulnerability in cancer
Nature Communications (2024)
-
The cell biology of ferroptosis
Nature Reviews Molecular Cell Biology (2024)
-
Metabolism-regulated ferroptosis in cancer progression and therapy
Cell Death & Disease (2024)
-
Ferroptosis: potential targets and emerging roles in pancreatic diseases
Archives of Toxicology (2024)
