
Abstract
The integral membrane protein ATG9A plays a key role in autophagy. It displays a broad intracellular distribution and is present in numerous compartments, including the plasma membrane (PM). The reasons for the distribution of ATG9A to the PM and its role at the PM are not understood. Here, we show that ATG9A organizes, in concert with IQGAP1, components of the ESCRT system and uncover cooperation between ATG9A, IQGAP1 and ESCRTs in protection from PM damage. ESCRTs and ATG9A phenocopied each other in protection against PM injury. ATG9A knockouts sensitized the PM to permeabilization by a broad spectrum of microbial and endogenous agents, including gasdermin, MLKL and the MLKL-like action of coronavirus ORF3a. Thus, ATG9A engages IQGAP1 and the ESCRT system to maintain PM integrity.
Similar content being viewed by others


Main
The autophagy1,2 and ESCRT systems3,4,5 both act in the remodelling of cellular membranes and contribute to a range of intracellular homeostatic functions and biological processes. The classical panel of ESCRT activities is diverse3,4,5 and affects many organelles and functions, including plasma membrane (PM) repair6,7,8,9. Autophagy in turn, along with its variations10, contributes to a range of intracellular homeostatic activities11 and is impaired in medical conditions2 that often have strong inflammatory components12.
The canonical autophagy pathway turns over defective and surplus cytoplasmic components and contributes to protein and organellar quality control1. It also has a parallel, purely metabolic function13. The mammalian autophagy pathway depends on ATG factors organized in a network of protein modules1, extensive lipid transactions14,15,16,17 and protein–lipid and protein–protein interactions within the modules and between the modules1. When autophagy is set in motion, the modules interlock1. After initiation and subsequent stages, degradative autophagy terminates in the formation of autolysosomes18.
Until very recently16,17,19, the core mammalian autophagy factor ATG9A has been less understood. High-resolution cryogenic electron microscopy (cryo-EM) structure and functional studies have revealed that both yeast Atg9 and mammalian ATG9A are lipid scramblases that play a role in autophagosome expansion16,17,19. This is consistent with yeast Atg9 localizing at the tips of a growing phagophore20, where it also organizes several components of the Atg machinery, including Atg2 (ref. 20). However, mammalian ATG9A is present in numerous intracellular compartments, including the trans-Golgi network and early and recycling endosomes21,22, and traffics through the secretory pathway to the PM and to the endocytic pathway from the PM23,24. The complex intracellular localization and trafficking of ATG9A in mammalian cells suggests the existence of additional functions of ATG9A that are yet to be defined.
Here, we report a previously unappreciated function of ATG9A associated with its trafficking through the PM. ATG9A protects cells against PM damage caused by a spectrum of exogenous and endogenous agents, including permeabilization by gasdermin and mixed lineage kinase domain like (MLKL), which generate pores at the PM25 or perturb PM integrity26,27, respectively, during programmed cell death processes of pyroptosis28,29 and necroptosis30,31. We furthermore define the ATG9A–IQGAP1 apparatus that integrates with the ESCRT system3,4,5 to cooperatively heal areas of PM damage.
Results
ATG9A protects cells against PM damage
We hypothesized that ATG9A, which traffics through numerous membranous compartments21,23,32,33,34, functions in membrane damage homeostasis. Owing to its presence on the PM23,24,35, we tested its role in protection against PM injury. A propidium iodide (PI) uptake assay6 was adapted to enable quantification of PM damage by high-content microscopy (HCM) of adherent cells (Fig. 1a). Knocking out ATG9A in Huh7 cells (ATG9AHuh7-KO) (Fig. 1b) rendered them more susceptible to injury by digitonin, saponin or streptolysin O (SLO) (Fig. 1c,d and Extended Data Fig. 1a). Transfection with GFP–ATG9A or FLAG–ATG9A rescued ATG9AHuh7-KO cells (Extended Data Fig. 1b,c).

a, Schematic of PM permeabilization/damage quantification by HCM and PI+ (nuclei) staining. b, Immunoblot of ATG9AHuh7-KO cells. Image is representative of three independent experiments. c,d, Image examples (white masks, algorithm-defined cell boundaries; yellow masks, computer-identified PI+ nuclei) (c) and HCM quantification (d) of PM permeabilization (by saponin (Sap), digitonin (Dig) or SLO) in ATG9AHuh7-WT and ATG9AHuh7-KO cells. Data show the percentage of cells positive for PI (mean ± s.e.m.; n = 5 biologically independent samples, two-way ANOVA Sidak’s test). Ctrl, control. Scale bars, 10 µm. e, Schematic of the PMHAL assay, which uses a HT probe for quantification of PM permeabilization/damage by HCM. MIL staining is scored, while MPL staining is used as a control for the HT probe. f, PMHAL images (confocal) of ATG9AHuh7-WT and ATG9AHuh7-KO cells showing HT MPL and MIL staining with or without PM damage (Dig). Scale bars, 10 µm. g, PMHAL assay and HCM quantification (GFP+MIL+ puncta intensity) of PM permeabilization in ATG9AHuh7-WT and ATG9AHuh7-KO cells subjected to PM damage. Data shown as the mean ± s.e.m.; n = 5 biologically independent samples, two-way ANOVA Sidak’s test. h, HCM quantification of PM permeabilization (PI, Dig) of ATG9AHuh7-WT and ATG9AHuh7-KO cells washed with 5 mM EGTA and incubated in a Ca2+-free HBSS medium with (+) or without (–) added 3.6 mM Ca2+. Data show the percentage of cells positive for PI (mean ± s.e.m.; n = 5 biologically independent samples, two-way ANOVA Sidak’s test). i, Schematic of how ATG9A protects cells against PM damage.
We employed additional methods and developed an assay (PMHAL) to quantify PM damage based on a HaloTag probe (GFP–HT) and membrane permeant and impermeant fluorescent chloroalkane ligands (MPL and MIL, respectively; Fig. 1e). In damaged cells, GFP–HT labels with both ligands, whereas in undamaged cells, it labels only with MPL. Untreated wild-type Huh7 cells (ATG9AHuh7-WT) and ATG9AHuh7-KO cells expressing GFP–HT were stained with MPL, whereas when they were treated with digitonin, ATG9AHuh7-WT cells stained weakly and ATG9AHuh7-KO cells stained strongly with MIL (Fig. 1f,g and Extended Data Fig. 1d–f). Using dextran-10k (Dx-10) as another probe for PM permeability, we observed increased staining in ATG9AHuh7-KO cells relative to ATG9AHuh7-WT cells (Extended Data Fig. 1g). Changes in endocytosis did not cause these differences, as ATG9AHuh7-KO and ATG9AHuh7-WT cells internalized equivalent amounts of the endocytic probe DQ-Red BSA (Extended Data Fig. 1h). Endosomal multivesicular body biogenesis was not altered in ATG9AHuh7-KO cells, as quantified by lysobisphosphatidic acid (LBPA) staining visualized in enlarged vesicles induced by Rab5Q79L (Extended Data Fig. 1i,j). PM tension, a measure of PM lipid ordering36, was not altered in ATG9AHuh7-KO cells relative to ATG9AHuh7-WT cells (Extended Data Fig. 1k,l) based on equal fluorescence lifetimes of Flipper-TR, a membrane tension probe36. In the absence of extracellular Ca2+, both ATG9AHuh7-KO and ATG9AHuh7-WT cells showed equal levels of PM damage, whereas with the added free Ca2+ during digitonin exposure, ATG9AHuh7-KO cells experienced relatively more damage compared with the ATG9AHuh7-WT cells (Fig. 1h and Extended Data Fig. 1m). Thus, ATG9A protects cells against PM damage through an active, Ca2+-dependent process elicited after damage rather than by passively affecting PM sensitivity to damaging agents.
Additional cell types were tested. Increased sensitivity to digitonin-mediated PM damage was observed in ATG9AMCF7-KO cells versus parental ATG9AMCF7-WT cells and in HeLa cells knocked down for ATG9A (Extended Data Fig. 1n–u). We tested primary cells using murine bone-marrow-derived macrophages (BMMs). BMMs from Atg9afl/fl LysM-Cre+ mice were also more sensitive to PM damage caused by digitonin or SLO relative to BMMs from Atg9afl/fl LysM-Cre− mice (Extended Data Fig. 2a–d). In summary, ATG9A protects cells against PM damage (Fig. 1i).
ATG9A protects the PM against gasdermin pores
We tested the role of ATG9A in protection against PM permeabilization in the context of a known physiological process. During pyroptosis, gasdermin pores form on the PM after proteolytic processing of gasdermin-D (GSDMD), which entails the liberation of the amino-terminal fragment (GSDMD-NT) and its subsequent oligomerization into a pore-like structure at the PM37. Overexpressing GSDMD-NT increased PI uptake more in ATG9AHuh7-KO cells than in parental ATG9AHuh7-WT cells (Fig. 2a), which was paralleled by reduced staining with calcein, a cell viability reporter (Fig. 2a, inset, and Extended Data Fig. 2e). We next tested the effects of ATG9A on PM permeabilization following activation of endogenous GSDMD by electroporated or transfected lipopolysaccharide (LPS) into U2OS cells and BMMs. Processing of endogenous GSDMD monitored by GSDMD-NT release was equal in LPS-electroporated ATG9AU2OS-KO versus ATG9AU2OS-WT cells and in LPS-primed and then LPS-transfected Atg9afl/fl LysM-Cre+ BMMs versus Atg9afl/fl LysM-Cre− BMMs (Fig. 2b,c). In each case, ATG9AKO cells were more susceptible than ATG9AWT cells to activated endogenous gasdermin in a time- and dose-dependent manner (Fig. 2d–g). Thus, ATG9A protects cells against PM permeabilization caused by gasdermin pores (Fig. 2h).

a, HCM quantification of PM permeabilization (PI staining) and cell viability (Live/Dead, calcein+ cells) of ATG9AHuh7-WT and ATG9AHuh7-KO cells transfected with FLAG–GSDMD-NT. Data show the percentage of FLAG+ cells that were PI+ or calcein+ (inset). Data shown as the mean ± s.e.m.; n = 6 biologically independent samples, unpaired t-test. b, Immunoblot analysis of ATG9AU2OS-KO cells and endogenous GSDMD cleavage (GSDMDNT). c, Immunoblot analysis of endogenous GSDMD cleavage (GSDMDNT) in BMMs from Atg9afl/fl LysM-Cre− and Atg9afl/fl LysM-Cre+ mice. d, HCM quantification of PM permeabilization (PI staining) of ATG9AU2OS-WT and ATG9AU2OS-KO cells electroporated with LPS to induce endogenous GSDMD cleavage. Data shown as the mean ± s.e.m.; n = 6 biologically independent samples, one-way ANOVA Tukey’s test. UT, untreated. e, Cell death analysis of supernatants of ATG9AU2OS-WT and ATG9AU2OS-KO cells electroporated with LPS. Data show the percentage of LDH release (mean ± s.e.m.; n = 6 biologically independent samples, one-way ANOVA Tukey’s test). f, HCM quantification of PM permeabilization (PI staining) of Atg9afl/fl LysMCre– and Cre+ BMMs transfected with LPS to induce endogenous GSDMD cleavage. Data shown as the mean ± s.e.m.; n = 6 biologically independent samples, one-way ANOVA Tukey’s test. g, Cell death analysis of supernatants of Atg9aKO (LysMCre+) and Atg9aWT (LysMCre−) BMMs after LPS priming and transfection. Data shown the percentage of LDH release (mean ± s.e.m.; n = 6 biologically independent samples, one-way ANOVA Tukey’s test). h, Schematic of how ATG9A protects cells against PM damage.
ATG9A translocation to the PM protects cells from damage
At least a fraction of ATG9A undergoes vesicular trafficking to and from the PM23,24,35. Using MyrPalm–EGFP as a PM marker6, an increased presence of ATG9A was detected at the PM following damage with digitonin, SLO, saponin or by glass-bead-inflicted injury (GBI) (Fig. 3a,b and Extended Data Fig. 3a). Super-resolution total internal reflection fluorescence (TIRF) microscopy confirmed the appearance of ATG9A at the PM after injury (Fig. 3c and Extended Data Fig. 3b), and this was further biochemically ascertained by surface biotinylation assays38 (Extended Data Fig. 3c). ATG9A translocation to the PM and PM protection against damage were sensitive to N-ethylmaleimide (NEM), an antagonist of SNARE-based membrane fusion (Extended Data Fig. 3d–g).

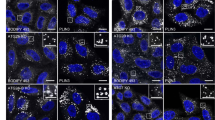
a, ATG9A localization in HeLa cells expressing FLAG–ATG9A and MyrPalm–EGFP untreated control (Ctrl) or treated with Dig. Scale bars, 10 µm. b, HCM analysis of FLAG–ATG9A and MyrPalm–EGFP colocalization in HeLa cells, after starvation (EBSS) or PM damage with Dig, SLO or by GBI. Data show the mean ± s.e.m.; n = 5 biologically independent samples, one-way ANOVA Dunnett’s test. c, Super-resolution TIRF microscopy analysis of FLAG–ATG9A and MyrPalm–EGFP (HeLa). Scale bars, 1 µm. Arrows indicate ATG9A fluorescence at the plasma membrane. d, Stable HEK293TAPEX2–ATG9A cells were exposed (Dig) or not (Ctrl) to PM damage. White arrowheads indicate PM areas showing deposits of DAB (APEX2 activity product) with or without adjacent DAB-positive vesicles. Scale bars, 1 µm. e, Quantification of the percentage of PM with DAB deposits (APEX2–ATG9A) in untreated (Ctrl) and Dig-treated HEK293TAPEX2–ATG9A cells. Data shown as the mean ± s.e.m.; n = 45 random PM profiles, unpaired t-test. f, Images of PM permeabilization (PI; red) in HeLa cells expressing MyrPalm–EGFP (green) and FLAG–ATG9AY8F (blue). White arrows and arrowheads indicate FLAG–ATG9AY8F transfected and untransfected cells, respectively. Scale bars, 10 µm. g, HCM quantification of PM permeabilization (PI) in HeLa cells expressing GFP–ATG9AWT or GFP–ATG9AY8F. Data shown as the mean ± s.e.m.; n = 5 biologically independent samples, two-way ANOVA Sidak’s test.
We carried out ultrastructural analysis by electron microscopy (EM) using APEX2 as an EM tag. A Flp-In-FLAG–APEX2–ATG9ATetON cell line (HEK293TAPEX2–ATG9A; Extended Data Fig. 3h) showed increased 3,3′-diaminobenzidine (DAB) precipitates at the PM when cells were subjected to PM damage (Fig. 3d,e and Extended Data Fig. 3i–n). Thus, ATG9A translocates to the PM following its damage (Extended Data Fig. 3o).
The Y8F mutation retards ATG9A removal from the PM during its trafficking24. Expression of FLAG–ATG9AY8F partially protected cells against PM damage (Fig. 3f,g and Extended Data Fig. 4a). Physiological enhancement of the presence of ATG9A at the PM in cells pulsed with human epidermal growth factor (hEGF)24 increased protection against PM damage, an effect abrogated in ATG9AHuh7-KO cells (Extended Data Fig. 4b–d). Thus, the presence of ATG9A at the PM confers protection against PM injury (Extended Data Fig. 4e,f).
The partner of ATG9A, IQGAP1, confers protection against PM damage
Proteomics analysis of ATG9A-containing membranes have been reported32,39. Here, we identified specific interactors of ATG9A during PM damage using APEX2–ATG9A as a tool for proximity biotinylation of closely apposed partners40. The APEX2–ATG9A construct was compatible with known functions of ATG9A; that is, APEX2–ATG9A rescued the LC3 lipidation defect in ATG9AHuh7-KO cells under autophagy-inducing conditions (Extended Data Fig. 5a). The HEK293TAPEX2–ATG9A cells (Extended Data Figs. 3h and 5b) were treated with digitonin, SLO or GBI followed by APEX2 biotinylation reaction40,41, and ATG9A partners were identified by liquid chromatography with tandem mass spectrometry (LC–MS/MS) analysis of the biotinylated proteins (Supplementary Tables 1–9, Fig. 4a and Extended Data Fig. 5c–f).

a, Volcano plot of ATG9A partners and changes in their proximity during PM damage (Dig, HEK293TAPEX2–ATG9A). The x axis shows the log2(fold change) (Dig/Ctrl ratio; spectral counts), the y axis shows −log10(P values); t-test (n = 3 biological replicates per group). Green and red dots indicate increase and decrease in proximity to ATG9A after Dig treatment, respectively. Orange dots indicate values below the statistical significance cut-off (P ≥ 0.05). IQGAP1 and adaptor proteins are highlighted as purple circles. Bubble size represents a normalized value for the total amount of spectral counts for the protein indicated. b, Co-IP (anti-FLAG) analysis of FLAG–ATG9A and GFP–IQGAP1 (HEK293T) with or without PM damage (Dig, SLO or GBI). One of three independent experiments shown. c, GST pulldown analysis using radiolabelled [35S]Myc–IQGAP1 and GST–ATG9A1–584. Images are representative of three biologically independent experiments. Data in the graph shown the mean ± s.e.m.; n = 3 independent experiments, unpaired t-test. CBB, Coomassie Brilliant Blue. AR, autoradiograph. d, HCM quantification of PM permeabilization (PI, HeLa IQGAP1 knockdown). si Scr, scrambled siRNA. Data shown as the mean ± s.e.m.; n = 5 biologically independent samples, two-way ANOVA Sidak’s test. e, Schematic summary of the findings in this figure.
Among the ATG9A interactors identified herein were IQGAPs42, with IQGAP1 showing the highest number of peptides and dynamic changes during PM damage (Fig. 4a, Extended Data Fig. 5c,d and Supplementary Tables 1, 3 and 5). The increased IQGAP1–ATG9A association during PM damage was confirmed by co-immunoprecipitation (co-IP) experiments (Fig. 4b and Extended Data Fig. 6a). FLAG–ATG9A and IQGAP1 colocalized at the PM following treatment with digitonin (Extended Data Fig. 6b,c). ATG9A and IQGAP1 directly interact, as established in glutathione S-transferase (GST) pulldown assays (Fig. 4c). In these experiments, GST fusion with full-size ATG9A could not be efficiently expressed, but a GST–ATG9A lacking the last 255 residues of its long carboxy-terminal domain was stable and bound IQGAP1. Recent cryo-EM structures of human ATG9A have revealed the organization of complex cytosolic domains16,17,19. When we tested cytosolic domains individually, which are adjacent in the three-dimensional cryo-EM structure of ATG9A, they showed a capacity to associate with IQGAP1 in GST pulldown assays (Extended Data Fig. 6d–f).
IQGAP1 knockdown (Extended Data Fig. 6g) increased PI uptake (Fig. 4d) and prevented ATG9A translocation to the PM following injury (Extended Data Fig. 6h). Thus, IQGAP1 controls ATG9A recruitment to the PM during damage. As with ATG9A, IQGAP1 knockdown did not reduce multivesicular body levels, as quantified by LBPA staining (Extended Data Fig. 6i,j). IQGAP1 responds to Ca2+ fluxes42, and the role of ATG9A protection against PM damage depended on extracellular Ca2+ (Fig. 1h). In the absence of Ca2+, ATG9A translocation to injured PM and co-IP between ATG9A and IQGAP1 were lost (Extended Data Fig. 6k–n). Thus, Ca2+, IQGAP1 and ATG9A together confer protection against PM damage (Fig. 4e).
ATG9A interacts with ESCRTs
Proximity biotinylation proteomics analyses uncovered ESCRTs as partners of ATG9A (Supplementary Tables 1, 3, 5, 8 and 10). Proteins from most ESCRT subcomplexes3,4,5 were detected in proximity to ATG9A, including TSG101 and ALIX (also known as PDCD6IP) (Supplementary Tables 8–10). These data suggest a previously unappreciated property of ATG9A to associate with multiple ESCRT components. We confirmed that ATG9A interacts with TSG101 and the ALIX V domain in co-IP assays (Fig. 5a and Extended Data Figs. 6a and 7a,b). Interactions between APEX2–ATG9A and endogenous TSG101 and ALIX were additionally assessed in a modified proximity biotinylation assay, which consisted of affinity purification on avidin beads of APEX2 proximity-biotinylated proteins and their detection by western blotting (BioWeB assay; Fig. 5b). Using BioWeB, we observed that endogenous TSG101 (Fig. 5c,d), but not ALIX (Extended Data Fig. 7c), was enriched in the vicinity of ATG9A in HEK293TAPEX2–ATG9A cells subjected to PM damage. The TSG101 enrichment in co-IP and BioWeB assays with ATG9A reflects, at least in part, an increased association of IQGAP1 with ATG9A during PM damage (Extended Data Fig. 6a), and this is in keeping with the reported binding of IQGAP1 to TSG101 (ref. 43). Consistent with this result, IQGAP1 knockdown indicated a decrease in TSG101 in immunoprecipitates of endogenous ATG9A from cells subjected to PM damage injury (Extended Data Fig. 8a).

a, Co-IP analysis (anti-FLAG) of GFP–ATG9A and FLAG–TSG101 interactions during PM damage (Dig). One of three independent experiments shown. b, Schematic of the BioWeB assay (HEK293TAPEX2–ATG9A tetracycline (Tet)-inducible cells) for capture, elution and detection by immunoblotting of endogenous proteins that are proximal to APEX2–ATG9A in different conditions. c, BioWeB analysis of changes in TSG101 proximity to APEX2–ATG9A during PM damage (Dig, SLO, GBI, HEK293TAPEX2–ATG9A). d, Quantification of the ratios of eluted TSG101 band intensities versus TSG101 in the input relative to c (mean ± s.e.m.; n = 4 biologically independent experiments, unpaired t-test). e, Co-IP analysis (anti-GFP) of FLAG–CHMP4A and GFP–ATG9A interactions during PM damage (Dig, HEK293T). One of three independent experiments shown. f, Co-IP analysis (anti-GFP) of interactions between Myc–CHMP4B and GFP–ATG9A during PM damage (Dig, HEK293T). Asterisk indicates a nonspecific band. One of three independent experiments shown. g,h, Quantification of TIRF microscopy images of mCherry–CHMP4B recruitment to PM (MyrPalm–EGFP) during PM damage (Dig) in ATG9AHuh7-WT cells pretreated with NEM or NEM + DTT. CHMP4B total fluorescence intensity (g) and CHMP4B puncta number in the TIRF field (h). Data shown as the mean ± s.e.m.; n = 5 independent images, unpaired t-test. i–k, TIRF microscopy images (i) and quantification (j,k) of mCherry–CHMP4B (red) recruitment to PM (MyrPalm–EGFP, green) following damage (Dig) in ATG9AHuh7-WT and ATG9AHuh7-KO cells. Scale bars, 5 µm. Data of CHMP4B total fluorescence intensity (j) and CHMP4B puncta number in the TIRF field (k) shown as the mean ± s.e.m.; n = 5 biologically independent samples, unpaired t-test. l, HCM quantification of mCherry–CHMP4B overlap with PM (MyrPalm–EGFP) in cells washed with 5 mM EGTA and incubated in a Ca2+-free HBSS medium with (+) or without (−) added 3.6 mM Ca2+ during PM damage (Dig). Data show the overlap area between CHMP4B and MyrPalm (mean ± s.e.m.; n = 4 biologically independent samples, one-way ANOVA Tukey’s test).
We tested whether ESCRT-III effector components acting downstream of TSG101 and ALIX3 can be detected in protein complexes with ATG9A. FLAG–CHMP4A and Myc–CHMP4B were detected in co-IP assays with GFP–ATG9A after PM damage (Fig. 5e,f). Using TIRF microscopy and quantifying CHMP4B profiles at the PM, their increase after injury was sensitive to NEM (Fig. 5g,h and Extended Data Fig. 8b), which parallels the sensitivity of ATG9A profiles (Extended Data Fig. 3d–f). Increases in both CHMP4A and CHMP4B profiles, detectable at the PM during damage, depended on Ca2+ and ATG9A, as visualized by confocal microscopy (Extended Data Fig. 8c,d) and quantified by TIRF microscopy and HCM (Fig. 5i-l). GFP–ATG9A and mCherry–CHMP4B appeared juxtaposed following GSDMD-NT-induced PM damage (Extended Data Fig. 8e). Thus, components of the ESCRT machinery associate with ATG9A and appear together on the PM, and these relationships increase in response to PM damage.
ESCRTs and ATG9A cooperate to protect against PM damage
The ESCRT components play a role in protection against PM damage6,7,8,9. We tested whether ESCRTs participate in protection against PM injury conferred by ATG9A. TSG101 knockdown increased the sensitivity to PM damage by digitonin, whereas ALIX knockdown did not, and combined knockdown of ALIX and TSG101 showed no increase in damage relative to single knockdown of TSG101 (Fig. 6a–c). CRISPR-based knockout of ALIX did not increase the susceptibility to digitonin injury (Fig. 6a–c). Prior publications have indicated that ALIX does not contribute to protection against certain types of PM permeabilization, such as those caused by effectors of necroptosis and pyroptosis8,9. We could not test overlapping or additive effects of ATG9A and TSG101 because we observed that TSG101 was destabilized in ATG9AKO cells and in cells subjected to ATG9A knockdown. We nevertheless tested contributions of CHMP2A, which is a key downstream ESCRT-III factor that is often used to establish the role of ESCRT-dependent membrane remodelling in different cellular processes6,9,44. Knocking down CHMP2A increased the sensitivity of ATG9AHuh7-WT cells but not of ATG9AHuh7-KO cells to digitonin and GSDMD (Fig. 6d–g). Moreover, CHMP2A directly bound IQGAP1 (GST pulldowns; Fig. 6h,i). Thus, ATG9A, IQGAP1 and ESCRTs are components of the same pathway protecting cells from PM damage (Fig. 6j).

a, Confirmation by immunoblotting of ALIX and TSG101 knockdown as well as CRISPR–Cas9 KO of ALIX in HeLa cells. One of three independent experiments shown. b, Example of HCM images of PM permeabilization (PI, Dig) in HeLa cells after knockdown of ALIX, TSG101, ALIX + TSG101 or CRISPR–Cas9 KO of ALIX (white masks, algorithm-defined cell boundaries; red masks, computer-identified PI+ nuclei). Scale bars, 10 µm. c, HCM quantification of PM permeabilization (PI, Dig, HeLa) after knockdown of ALIX, TSG101, ALIX + TSG101 or in ALIX CRISPR knockout (ALIXKO) and ALIXWT HeLa cells. Data shown as the mean ± s.e.m.; n = 6 biologically independent samples, one-way ANOVA Tukey’s test. d, Confirmation by immunoblotting of CHMP2A knockdown in ATG9AHuh7-WT and ATG9AHuh7-KO cells (one of three independent experiments shown). e, Example HCM images of PM permeabilization (PI, Dig) in ATG9AHuh7-WT and ATG9AHuh7-KO cells after knockdown of CHMP2A (white masks, algorithm-defined cell boundaries; red masks, computer-identified PI+ nuclei). Scale bars, 10 µm. f, HCM quantification of PM permeabilization (PI, Dig) in ATG9AHuh7-WT and ATG9AHuh7-KO cells after CHMP2A knockdown. Data shown as the mean ± s.e.m.; n = 6 biologically independent samples, unpaired t-test. g, HCM quantification of PM permeabilization (PI, FLAG–GSDMD-Full length (FL) or -NT fragment transfection) in ATG9AHuh7-WT and ATG9AHuh7-KO cells after CHMP2A knockdown. Data show the percentage of FLAG+ cells that were PI+ (mean ± s.e.m.; n = 6 biologically independent samples, unpaired t-test). h, GST pulldown analysis of in vitro translated and radiolabelled [35S]Myc–IQGAP1 with GST, GST–CHMP2A and GST–CHMP4B fusions. i, Quantification of the binding percentage of IQGAP1 relative to the GST constructs in h. Data shown as the mean ± s.e.m.; n = 3 biologically independent experiments, unpaired t-test. j, Schematic summary of findings presented in Fig. 5 and this figure.
Contributions of other ATG9A partners and ATG9A functions
In addition to ESCRTs and IQGAPs, our proximity biotinylation proteomics analyses revealed additional ATG9A interactors, such as Rab GTPases and adaptor proteins (Supplementary Tables 7 and 8), as well as several ATG proteins (Supplementary Table 10).
ATG2A and ATG2B have been perceived as ATG9A interactors, as in yeast20, and act in lipid transport14. We detected ATG2 peptides with APEX2–ATG9A (Supplementary Table 10). When ATG2A or ATG2B were knocked out by CRISPR, this did not affect sensitivity to digitonin or endogenous gasdermin activation (Extended Data Fig. 9a–e). Downregulating another lipid-modifying factor found on ATG9A membranes, PI4KB39, did not affect PM damage (Extended Data Fig. 9f,g). A lipid scramblase activity has been recently reported for ATG9A16,17. We tested whether the ATG9A mutant M33 (autophagy-defective and scramblase-defective; K321L, R322L, E323L, T419W)17 lost the ability to protect the PM from damage. However, ATG9A-M33 complemented ATG9AHuh7-KO cells in the digitonin PM-damage test (Extended Data Fig. 9h). Thus, the activities of ATG9A that are defective in the M33 mutant17 do not appear to be essential for protection against PM damage.
Additional ATG9A partners observed in the proximity biotinylation proteomics analyses were confirmed for interactions with ATG9A, and some were tested for effects on PM sensitivity to damage (Extended Data Fig. 9i–q).
ATG9A affects membrane dynamics and shedding
PM repair is often accompanied by membrane rearrangements and emission of membranous particles6,7,45. Localized laser-induced PM injury, albeit different from other non-confined types of PM damage in this work, was employed for live microscopy observations. GFP–ATG9A coalesced at the interface between the cytosol and protruding membranes that became diffusionally separated (Supplementary Video 1 and Extended Data Fig. 10a). Following PM damage with digitonin, particles were released that showed a relatively homogenous peak at 95 nm in ATG9AHuh7-WT cells, which shifted to smaller sizes in ATG9AHuh7-KO cells (75 nm; Fig. 7a,b and Extended Data Fig. 10b,c). The particles released by ATG9AHuh7-KO cells were enriched for the membrane marker CD63 relative to the particles released from ATG9AHuh7-WT cells (Extended Data Fig. 10d). Thus, ATG9A participates in dynamic events at the PM and affects the size and type of membranous particles emitted during PM damage.

a, Nanoparticle tracking analysis. Concentration (y axis) and size distribution (x axis) of enriched EVs in supernatants after PM (Dig) from ATG9AHuh7-WT (Ctrl: blue; Dig: green) and ATG9AHuh7-KO (Ctrl: pink; Dig: red) cells. One of three independent experiments shown. b, Percentage of particles in the 10–88-nm bin (G1 in a) and the 89–350-nm bin (G2 in a) after PM damage (Dig). Data show nanoparticle sizing (>800 frames per sample), n = 3 biologically independent experiments; mean ± s.e.m.; two-way ANOVA Sidak’s test. c, HCM quantification of PM permeabilization (PI) in ATG9AHuh7-WT and ATG9AHuh7-KO cells expressing Hras25 PM-targeted MLKL–Venus (PM) or non-targeted MLKL–Venus (NT). Data show the percentage of Venus+ cells positive for PI (mean ± s.e.m., n = 5 biologically independent samples, two-way ANOVA Sidak’s test). d, HCM quantification of PM permeabilization (PI) in ATG9AHuh7-WT and ATG9AHuh7-KO cells expressing SARS-CoV-2 FLAG–ORF3a (FLAG–ORF3aCov-2). Data show the percentage of cells positive for PI (mean ± s.e.m., n = 6 biologically independent samples, two-way ANOVA Tukey’s test). e, HCM complementation analysis of PM sensitivity to permeabilization by SARS-CoV-2 ORF3a in ATG9AHuh7-KO cells co-transfected with SARS-CoV-2 GFP–ORF3a and ATG9A–FLAG (WT or M33 scramblase mutant). PI+ cells quantified after gating on GFP+ cells (HCM, mean ± s.e.m., n = 6 biologically independent samples, one-way ANOVA Tukey’s test). f, HCM quantification of PM permeabilization (PI) in ATG9AHuh7-WT and ATG9AHuh7-KO cells exposed to virulent Mtb Erdman at m.o.i. of 0 (Ctrl), 5 or 10 for 1 h. Data show the percentage of cells positive for PI (mean ± s.e.m., n = 6 biologically independent samples, two-way ANOVA Sidak’s test). g, Overall schematic summary. Following PM damage, ATG9A (recruited by Ca2+ influx and IQGAP1) organizes the ESCRT machinery at the site of injury where ESCRT-III effectors (CHMP4A/B and CHMP2A) remodel membranes to bud EVs carrying away the PM pore/damaged area.
ATG9A protects the PM from damage by diverse biological agents
Our findings that ATG9A protects against endogenous PM-permeabilizing agents such as GSDMD-NT (Fig. 2) correlate with the reports that the ESCRT system protects cells during gasdermin-mediated pyroptotic cell death8. Another cell death process, necroptosis, is associated with MLKL-dependent loss of PM integrity and it too is countered by ESCRT-III9. We expressed the characterized PM-targeting MLKL system (full-length MLKL–Venus tagged with the HRas25 PM-targeting motif), which is sufficient to cause a loss of PM integrity27, and detected increased PM permeability in ATG9AHuh7-KO cells compared with ATG9AHuh7-WT cells (Fig. 7c). Moreover, ATG9A protected cells against PM leakage caused by SARS-CoV-2 ORF3a, an activity previously reported for SARS-CoV-1 (ref. 46) (Fig. 7d). In complementation experiments, ATG9AWT rescued ATG9AHuh7-KO cells subjected to PM damage induced by SARS-CoV-2 ORF3a (Fig. 7e). Mirroring the results from the complementation experiments with digitonin treatment, ATG9A-M33 rescued ATG9AHuh7-KO cells damaged by SARS-CoV-2 ORF3a (Fig. 7e). In conclusion, ATG9A counters physiological PM perturbations associated with pyroptotic and necroptotic programmed cell death pathways.
External biological agents, such as infectious agents including bacteria, viruses and protozoan parasites, can cause host cell membrane damage. The intracellular pathogen Mycobacterium tuberculosis (Mtb) causes partial phagosomal permeabilization, whereas a subset of autophagy factors play a role in controlling Mtb infection47,48. However, Mtb also causes PM damage while interacting with host cells49,50. We therefore tested whether ATG9A can protect cells from PM damage caused by Mtb. To avoid complications from intracellular Mtb effects, we used nonphagocytic Huh7 cells, exposed them to virulent Mtb and quantified PI staining, which showed increased Mtb-inflicted PM damage in the absence of ATG9A (Fig. 7f and Extended Data Fig. 10e). As a control, we used the nonvirulent derivative of Mtb subspecies, Mycobacterium bovis (BCG), and detected neither PM damage nor increase dependent on ATG9A (Extended Data Fig. 10f,g). Thus, ATG9A plays a protective role against PM damage caused by Mtb.
Discussion
In this study, we uncovered a hitherto unknown role of ATG9A in PM repair. In response to Ca2+ influx due to PM damage, IQGAP1, ATG9A and ESCRTs are recruited to the sites of injury for repair (Fig. 7g). IQGAP1, a known Ca2+ responder42, recruits ATG9A to the damaged PM, and together they organize the ESCRT machinery for PM repair. ATG9A has been traditionally viewed as an important autophagy factor, whereas our results show that it should also be considered as a peripheral factor cooperating with the ESCRT system. ATG9A and IQGAP1 combine with their ESCRT effectors in protection against GSDMD and MLKL pores8,9 to prevent excessive necrotic death. This may be of significance in controlling inflammation and tissue damage under various pathophysiological conditions.
Our MS analyses uncovered the hitherto unknown association of ATG9A with IQGAPs, a class of proteins that, along with their binding partner calmodulin, act as Ca2+-sensing factors51,52,53,54,55, with IQGAP1 better known for its ability to govern cytoskeletal rearrangements at the leading edge of the PM in migrating cells56,57. The ATG9A–IQGAP1 interactions defined here and the previously known functions of IQGAP1 are furthermore compatible with the reported role of ATG9A in cell migration58. Others have described a process whereby Ca2+ recruits ALG-2 and ESCRTs to repair the PM after laser-induced PM damage7. However, ALG-2 does not play a role in protection against GSDMD or MLKL-induced PM damage8,9.
In addition to ESCRT components, our proteomics findings include a variety of previously reported ATG9A partners identified by conventional biochemical methods, thereby validating the use of APEX2–ATG9A. The repertoire of ATG9A interactors includes a suite of Rab GTPases, which probably reflect the multiple trafficking routes taken by ATG9A. This may include the small vesicles observed subcortically during PM damage that we propose ferry ATG9A en route to or from PM. The Rab GTPases identified in proximity to ATG9A include RAB7A, which is known to colocalize with ATG9A21, and a RAB7-specific GTPase activating protein (GAP), TBC1D15 (ref. 59), thereby underscoring the role of RAB7 in ATG9A trafficking that is also reflected in the observed sensitivity of RAB7-depleted cells to PM damage. By MS, we also detected dynamic changes in associations and confirmed several by co-IP assays with the adaptor proteins AP-1, AP-2, AP-3 (with the AP-3 interaction being identified here) and AP-4 (refs. 24,35,60). We interpret these relationships as indicative of increased ATG9A trafficking during PM damage.
Our findings suggest that ATG9A confers protection against diverse PM-damaging products and activities, including those of microbial pathogens. ATG9A protects cells from PM injury caused by Mtb49,50,61. Likewise, SARS-CoV-1 ORF3a46 and, as shown here, ORF3a of SARS-CoV-2 cause PM damage, and ATG9A protects against this activity. ORF3a, apart from its membrane-permeabilization activities, plays a role in lysosomal damage and deacidification, with a proposed contribution to nonlytic viral egress via endosomal and lysosomal exocytosis62. Thus, ATG9A activities at the PM intersect with a spectrum of microbial pathogenesis processes. This is in keeping with a growing recognition of the expanding effects of the components of the autophagy apparatus in various cellular processes2,10. The physical and functional cooperation of ATG9A with IQGAP1 and the ESCRT machinery represents another example of non-canonical functions of the ATG factors and broadens the fundamental scope and translational potential of both the ATG and the ESCRT systems.
Methods
Antibodies and reagents
The following antibodies were from Abcam: GFP (ab290) (1:1,000 for western blotting (WB)); GFP (ab38689) (1:100 for immunoprecipitation (IP)); GM130 (ab1299) (1:1,000 for WB); GSDMD (ab209845, for BMMs) (1:1,000 for WB); PDCD6/ALG-2 (ab133326) (1:1,000 for WB); RAB7 (ab137029) (1:1,000 for WB); and TSG101(ab83) (1:1,000 for WB). The ALIX antibody was from BioLegend (634502) (1:1,000 for WB; 1:500 for immunofluorescence (IF)) and the CD63 antibody was from BD (556019) (1:500 for WB). Antibodies from Cell Signaling Technology were ATG9A (13509) (1:1,000 for WB) and IQGAP1 (20648) (1:1,000 for WB; 1:500 for IF). Other antibodies used in this study were from the following sources: FLAG M2 (F1804) (1:1,000 for WB, 1:100 for IP), GSDMD (G7422, for U2OS) (1:1,000 for WB), LC3B (L7543) (1:2,000 for WB) and PI4KB (06-578) (1:1,000 for WB) from Sigma Aldrich; β-actin (C4) (1:1,000 for WB), c-Myc (sc-40) (1:500 for WB) and HA probe (12CA5) (1:1,000 for WB) from Santa Cruz Biotechnology; and ATG2A (23226-1-AP) (1:1,000 for WB), ATG2B (25155-1-AP) (1:1,000 for WB) and CHMP2A (10477-1-AP) (1:500 for WB) from Proteintech. HRP-labelled anti-rabbit (sc-2004) (1:2,000 for WB) and anti-mouse (sc-2005) (1:2,000 for WB) secondary antibodies, anti-rabbit Alexa Fluor-488 (A-11034), -568 (A-11036) and -647 (A-21245), and anti-mouse Alexa Fluor -488 (A-11029), -568 (A-11004) and -647 (A-21235) (1:500 for IF) were from ThermoFisher Scientific. The tags MIL (Alexa Fluor 660-conjugated, G8471) (1:1,000 for IF) and MPL (tetramethylrhodamine-conjugated, G8251) (1:1,000 for IF) were from Promega. Saponin (S4521), digitonin (D5628), SLO (SAE0089) and acid-washed glass beads (diameter of ~0.5 mm, G8772) were purchased from Sigma Aldrich. PI solution (10008351) was purchased from Cayman Chemical. Dx-10 fluorescein (D1820), DQ-Red BSA (D12051) and CellMask Deep Red PM stain (C10046) (1:1,000 for IF) were purchased from ThermoFisher Scientific. DMEM, RPMI, OptiMEM and EBSS media were from Life Technologies.
Cells and cell lines
HEK293T and HeLa cells were from the American Type Culture Collection. The Huh7 cell line was purchased from Rocky Mountain Laboratory. BMMs were isolated from femurs of Atg9afl/fl LysM-Cre+ mice and its Cre-negative littermates63 and were cultured in DMEM supplemented with mouse macrophage colony stimulating factor (5228, CST). Mice were cared following protocols approved by Institutional Animal Care and Use Committee. The study was compliant with all relevant ethical guidelines for animal research.
HEK293T Flp-In-FLAG–APEX2–ATG9ATetON (HEK293TAPEX2–ATG9A) and Hela Flp-In-mCherry–CHMP4A were generated using a modified gateway construct from T.J. MCF-7 ATG9A CRISPR KO (ATG9AMCF-7-KO) cells and their parental MCF-7 WT (ATG9AMCF-7-WT) cells were a kind gift from M. Jäättelä (University of Copenhagen). MCF-7 cells were from M. Jäättelä. Huh7 ATG9A CRISPR KO (ATG9AHuh7-KO) cells and their parental Huh7 ATG9A WT (ATG9AHuh7-WT) cells were generated by transduction of two ATG9A CRISPR–Cas9 guide RNAs (gRNAs). An identical approach was used to generate U2OS cells lacking ATG9A (ATG9AU2OS-KO), ATG2A (ATG2AU2OS-KO) or ATG2B (ATG2BU2OS-KO). These cells were a kind gift from F.R. U2OS cells were a kind gift from G. Strous (University Medical Center Utrecht, The Netherlands). HeLa ALIX CRISPR KO (ALIXHeLa-KO) cells and their parental Hela ALIX WT (ALIXHeLa-WT) cells were generated by transduction of one CRISPR–Cas9 gRNA.
Plasmids, siRNAs and transfection
Plasmids used in this study for immunoprecipitation or IF assays, such as those expressing ATG9A, ALIX (including their mutants’ deletions) and CHMP4A were cloned into pDONR221 (Gateway Technology cloning vector, Thermo Scientific) using a BP cloning reaction. Expression vectors were then made by a LR cloning reaction (Gateway, ThermoFisher) in appropriate destination (pDEST) vectors.
ATG9A mutants were generated utilizing a QuikChange site-directed mutagenesis kit (Agilent) and confirmed by sequencing (Genewiz). Small interfering RNAs (siRNAs) were from GE Dharmacon (siGENOME SMART pool). Plasmid transfections were performed using the calcium phosphate transfection method in HEK293T cells or using Lipofectamine 2000 in the other cell lines (ThermoFisher Scientific). siRNAs were delivered into cells using Lipofectamine RNAiMAX (ThermoFisher Scientific).
Generating cell lines by CRISPR–Cas9
ATG9A (ATG9AHuh7-KO and ATG9AU2OS-KO), ATG2A (ATG2AU2OS-KO), ATG2B (ATG2BU2OS-KO) and ALIX (ALIXHeLa-KO) cell lines by CRISPR–Cas9
Sequences of the two single‐guide RNA (sgRNA) used to generate ATG9AHuh7-KO cells were GACCCCCAGGAGTGTGACGG and TCTGGAAACGGAGGATGCGG. Those to generate ATG9AU2OS-KO, ATG2AU2OS-KO and ATG2BU2OS-KO were CTGTTGGTGCACGTCGCCGAGGG and CCCTGGGGGTGAATCACTATAGG, CACTGCACAGTGCGCGTGTCCGG and CCAGGGCACGGCCACCTCGATGG, and GAGGATTAGAAATGGTCTTCCGG and AAGAGCCCCATGGAACTGACAGG, respectively. The sequence of the sgRNA used to knockout ALIX (PDCD6IP) was CTTAAGTCGAGAGCCGACCG.
ATG9AHuh7-KO and ALIXHeLa-KO cells were generated by infecting target cells with the appropriate sgRNA, lentiviral vectors and lentiCRISPRv2 as previously described64. Briefly, HEK293T cells were transfected with lentiCRISPRv2 sgRNA vectors together with psPAX2 and pMD2.G at the ratio of 4 μg, 2.5 μg and 1.5 μg per 6‐cm dish. After 60 h, the supernatants containing lentiviruses were collected and spun down at 300 × g for 5 min to clear cell debris. Lentiviruses were diluted with DMEM full medium at 1:2 ratio and used to infect target cells overnight with in presence of 8 μg ml−1 of polybrene (hexadimethrine bromide) in 12‐well plates. Then, the medium with lentivirus was removed and changed to fresh medium to continue incubation for 24 h. Cells were selected on 2 μg ml−1 puromycin for 5 days before validation of the knockout. Single clones were isolated by seeding single cells in 96‐well plates after serial dilutions.
ATG9AU2OS-KO, ATG2AU2OS-KO and ATG2BU2OS-KO cells were created by first cloning the sgRNA into the pX458 plasmid (Addgene), which also allows the simultaneous expression of Cas9 and GFP. U2OS cells were transfected with the generated plasmids using Xfect (Takara Bio), and 48 h later clonally sorted by FACS based on GFP expression. Single-cell clones were then expanded and sequenced, and protein expression was assessed by immunoblotting.
Generating Flp-In cell lines
HEK293T Flp-In-FLAG–APEX2–ATG9ATetON (HEK293TAPEX2-ATG9A) and HeLa Flp-In-mCherry–CHMP4A cell lines
HEK293T or HeLa Flp-In T-REX host cells were transfected with ATG9A and CHMP4A reconstructed plasmid, respectively, and the pOG44 expression plasmid at ratio of 9:1. After 24 h of transfection, cells were washed and fresh medium added. One day later, cells were split into fresh medium containing 100 μg ml−1 hygromycin, at a confluency around 25%. Selective medium was renewed every 3–4 days until single-cell clones could be identified. Hygromycin-resistant clones were picked and expanded. Clones were tested by immunoblotting after overnight incubation in medium containing 1 μg ml−1 tetracycline to analyse the expression of FLAG–APEX2–ATG9A or mCherry–CHMP4A.
Co-IP and immunoblotting
Cells transfected with 8–10 μg of plasmids were lysed in NP-40 buffer (ThermoFisher Scientific) supplemented with protease inhibitor cocktail (Roche) and 1 mM phenylmethylsulfonyl fluoride (Sigma) for 30 min on ice. Supernatants were incubated with 2–3 μg of antibodies at 4 °C overnight. The immune complexes were captured with Dynabeads (ThermoFisher Scientific). Dynabeads were washed three times with PBS and bound proteins eluted with 2× Laemmli sample buffer (Bio-Rad) before being subjected to immunoblot analysis. Immunoblotting images were visualized and analysed using ImageLab v.6.0.0.
IF confocal microscopy
For IF confocal microscopy, cells were plated onto coverslips in 12-well plates. Cells were transfected with plasmids and treated as indicated in the figures before fixing in 4% paraformaldehyde (PFA) for 10 min followed by permeabilization with PBS 0.1% saponin for 10 min. Cells were then blocked in PBS 5% BSA, 0.05% saponin for 30 min before labelling with primary antibodies overnight at 4 °C. After washing with PBS and incubation with appropriate secondary antibodies for 1 h at room temperature, coverslips were mounted using ProLong Gold Antifade Mountant (Invitrogen) and analysed by confocal microscopy using a Zeiss LSM510 laser scanning microscope driven by Zeiss LSM 510 v.4.2 SPI software.
GST pulldown assay
Recombinant GST and GST-fusion proteins were produced in Escherichia coli SoluBL21 (Genlantis, C700200) by inducing expression in overnight cultures with 50–75 μg ml−1 isopropyl β-d-1-thiogalactopyranoside. Expressed proteins were purified by immobilization on glutathione sepharose 4 fast flow beads (GE Healthcare, 17-5132-01). For GST pulldown assays, Myc-tagged proteins were in vitro translated and radiolabelled with [35S]-methionine using a TNT T7 Reticulocyte Lysate system (Promega, l4610). Ten microlitres of in vitro translated proteins were precleared to reduce nonspecific binding with 10 μl of empty glutathione sepharose beads in 100 μl of NETN buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA and 0.5% NP-40) supplemented with complete EDTA-free protease inhibitor cocktail (Roche, 1183617001) for 30 min at 4 °C. This was followed by incubation with purified GST or GST-fusion proteins for 1–2 h at 4 °C. The mixture was washed five times with NETN buffer by centrifugation at 2,500 × g for 2 min followed by addition of 2× SDS gel loading buffer (100 mM Tris pH 7.4, 4% SDS, 20% glycerol, 0.2% bromophenol blue and 200 mM dithiothreitol (DTT) (Sigma, D0632)) and heating for 10 min. The proteins were then resolved by SDS–PAGE and the gel stained with Coomassie Brilliant Blue R-250 dye (ThermoFisher Scientific, 20278) to visualize the GST and GST-fusion proteins. The gel was vacuum-dried and the radioactive signal detected using a Bioimaging analyzer BAS-5000 (Fujifilm).
PM damage
PM damage using the detergents digitonin and saponin was performed as previously described6. Briefly, 0.05% saponin or 50–200 µg ml−1 digitonin (as indicated) diluted in DMEM–10% FBS (complete medium) was applied to cells at 37 °C for 1 min. Cells were then washed with complete medium and fixed for 3 min after the addition of detergents with 4% PFA in PBS or lysed.
PM damage using SLO was performed with a modified protocol65. Cells were washed with Ca2+-free HBSS containing 5 mM EGTA at 37 °C before washing twice in Ca2+-free HBSS. Reduced SLO (10 mM DTT at room temperature for 5 min) was diluted in Ca2+-free HBSS (200 U ml−1) and added on target cells for 10 min at 37 °C. Cells were washed in complete medium before being fixed or lysed.
PM damage induced by GBI was performed as previously described66. Acid-washed glass beads were gently poured over the cells (~35 mg of beads per well for a 24-well plate). The beads were agitated over the cells for 1 min on a rotator platform at 160 r.p.m. Thirty seconds after GBI, the cells were washed with warm PBS and fixed or lysed.
PM damage induced by the pore-forming FLAG–GSDMD-NT fragment was performed as previously described67. Huh7 cells were transfected with 100 ng per 105 cells of FLAG–GSDMD-NT plasmid (Addgene, 80951), FLAG–GSDMD (Addgene, 80950) or FLAG–GSDMD-NT-4A (Addgene, 80952) using Lipofectine 2000 in OptiMEM for 2 h. Then, cells were washed in complete medium before assessing PI uptake and cell viability (Live/Dead, Thermo Scientific) at the indicated time points.
PM damage triggered by cleavage of endogenous GSDMD was performed as previously described for BMMs and human cell lines8,29. BMMs were seeded at a density of 3 × 104 cells per well in a 96-well plate 1 day before stimulation and grown overnight at 37 °C in 5% CO2. The next day, the supernatant was removed and BMMs were primed for 4 h with 50 ng per well of LPS O55:B5. Then, 3 × 104 cells were transfected with LPS complexes prepared by mixing 100 µl of OptiMEM with ultrapure LPS O111:B4 (Invivogen) and 0.5 µl of Lipofectamine RNAiMAX (Thermo Scientific). The transfection mixture was incubated for 10 min at room temperature and subsequently added to the cells. Plates were centrifuged at 1,000 r.p.m. for 5 min at 37 °C. BMMs were incubated for the indicated times at 37 °C in 5% CO2 before measuring PM damage using the PI assay and lactate dehydrogenase (LDH) release in the supernatant (Promega, G1780). For U2OS cells, 2 µg of LPS O111:B4 (Invivogen) was mixed with 2 × 106 U2OS cells in 100 µl of Amaxa Nucleofector Kit V buffer (Lonza, VVCA-1003) and electroporated according to the manufacturer’s instructions. Cells were then plated in a 96-well plate for 6 h or 17 h before measuring LDH release in the supernatant and performing the PI assay.
PM leakage induced by SARS-CoV-2 ORF3a was performed by transfection of ORF3a (FLAG or GFP tagged in Nter) using Lipofectine 2000 in OptiMEM for 2 h. Then, cells were washed in complete medium before assessing PI uptake the next day. This plasmid was obtained by LR cloning reaction (Gateway, ThermoFisher) in a pDEST-FLAG or a pDEST-GFP vector using pDONR207 SARS-Cov-2 ORF3a (Addgene, 141271).
PM permeabilization assays
For the PI assay, cells were incubated with 100 µg ml−1 PI diluted in complete medium for 1 min at 37 °C, 2 min after the addition of saponin or digitonin, 10 min after SLO or 1 min after GBI treatment. After PI incubation, cells were fixed with 4% PFA.
For the Dx-10 staining, cells were incubated for 1 min at 37 °C with 500 µg ml−1 of Dx-10 diluted in complete medium, 2 min after the addition of digitonin and before 4% PFA fixation.
PMHAL assay
Huh7 cells were transiently transfected with the HT probe (PEX3–GFP–HT) before digitonin treatment for 1 min. Cells were then washed for 1 min in complete medium before incubation with complete medium containing the HT ligands MIL and/or MPL (1:1,000) for 1 min before 4% PFA fixation and Hoechst staining. For HCM quantification, cells transfected with the PEX3–GFP–HT probe were gated using GFP total cell fluorescence intensity. A mask was then assigned to GFP-positive puncta according to the fluorescence intensity and puncta area. The same threshold was applied to ATG9AWT and ATG9AKO cells. The fluorescence intensity of MIL or MPL colocalizing with GFP–HT puncta masks (GFP+MIL+ or GFP+MPL+, respectively) was then assessed.
Endocytosis assay
To monitor endocytosis, we used DQ-Red BSA as a fluid phase tracer. Cells were incubated with 10 µg ml−1 of DQ-Red BSA diluted in complete medium for 2 min at 37 °C after digitonin treatment. Endocytosis was stopped by 4% PFA fixation.
Bimolecular fluorescence complementation assay
HeLa cells were transiently transfected with pDEST-VNALIX and pDEST-VCATG9A followed by analysis of Venus fluorescence by HCM and confocal microscopy.
Cell surface biotinylation
ATG9A cell surface biotinylation was performed as previously described23, with an adapted protocol suitable for the analysis of proteins during PM damage66. HEK293T cells transiently expressing GFP–ATG9A were treated with PM-damaging agents (digitonin, SLO or GBI) or EBSS for 1 h. Cells were rinsed twice with ice-cold PBS2+ (PBS containing 1 mM MgCl2 and 0.1 mM CaCl2) and were subsequently incubated with freshly prepared ice-cold 0.4 mM maleimide-PEG2-biotin (ThermoFisher Scientifc) in PBS (1 mM MgCl2, 2 mM CaCl2, 150 mM NaCl) for 60 min at 4 °C with gentle agitation. Unreacted maleimide-PEG2-biotin was then quenched by washing the cells twice with ice-cold quenching buffer (PBS, 1 mM MgCl2, 0.1 mM CaCl2 and 100 mM glycine) for 20 min at 4 °C. Cells were subsequently rinsed twice with ice-cold PBS2+ and scraped into NP-40 buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% NP-40 and supplemented with protease inhibitor cocktails). After 30 min on ice, lysates were cleared by centrifugation at 16,000 × g for 10 min at 4 °C, and the protein concentration was determined using a BCA protein assay (Pierce). A total of 4 mg of protein was incubated with 3 μg of GFP antibody overnight at 4 °C. The immune complexes were captured with Dynabeads (ThermoFisher Scientific). The Dynabeads were wash with PBS three times and bound proteins were eluted with 2× Laemmli sample buffer (Bio-Rad) before analysis by immunoblotting using a HRP-conjugated streptavidin antibody.
hEGF treatment
Cells were washed three times with pre-warmed PBS and serum-starved with DMEM containing 0.1% FBS for HeLa cells or serum-free DMEM for U2OS cells. After 24 h of starvation, cells were stimulated for 30 min with hEGF in DMEM (50 ng ml−1 for HeLa cells and 100 ng ml−1 for U2OS cells).
NEM treatment
NEM treatment was performed as previously described68. Cells were washed twice in PBS supplemented with 0.1 mM CaCl2 and 0.1 mM MgCl2 (PBS+) and then treated in PBS+ on ice either with 1 mM NEM for 15 min followed by quenching with 2 mM DTT for 15 min, or with 1 mM NEM plus 2 mM DTT for 30 min. Finally, cells were washed in PBS+ and incubated in complete medium for 30 min at 37 °C before inducing PM damage.
Extracellular vesicle enrichment and analysis
ATG9AHuh7-KO and their parental ATG9AHuh7-WT cells were treated with 100 µg ml−1 digitonin diluted in OptiMEM for 1 min. Cells were washed twice with OptiMEM and the supernatant containing the released extracellular vesicle (EVs) was collected 5 min after digitonin addition. EVs were isolated using Total exosome isolation reagent (ThermoFisher Scientific) according to the manufacturer’s instructions. The obtained cell pellets were suspended in 100 µl of PBS and analysed by immunoblotting or in a nanoparticle tracking instrument.
Nanoparticle tracking analysis
Isolated EVs (as described above) from the ATG9AHuh7-KO and their parental ATG9AHuh7-WT cell culture supernatants were diluted in PBS and used for nanoparticle tracking analysis using a Nanosight NS300 instrument (NanoSight), followed by evaluation using the Nanoparticle Tracking Analysis (NTA) software. Conditions were as follows: camera type, sCMOS; detection threshold, 2; recording for 800 frames at 25 frames per s.
PM damage by mycobacteria
Mtb Erdman (Erdman) culture was prepared by thawing a frozen stock aliquot and grown in 7H9 Middlebrook liquid medium supplemented with oleic acid, albumin, dextrose and catalase (OADC; Becton Dickinson), 0.5% glycerol and 0.05% Tween-80. Cultures were grown at 37 °C. ATG9AHuh7-KO and their parental ATG9AHuh7-WT cells were co-incubated with Mtb Erdman at a multiplicity of infection (m.o.i.) of 5 or 10 (or BCG at m.o.i. of 10) for 1 h followed by PI staining (100 µg ml−1 in complete medium) and fixation in 4% PFA for 1 h to analyse PM permeability by HCM.
HCM
Cells in 96-well plates were treated as indicated in each experiment before fixing in 4% PFA. Cells were then permeabilized with 0.1% saponin in 3% BSA for 30 min followed by incubation with primary antibodies overnight at 4 °C and secondary antibodies for 1 h at room temperature. HCM with automated image acquisition and quantification was carried out using a Cellomics HCS scanner and iDEV software (ThermoFisher Scientific). Automated epifluorescence image collection was performed using a minimum of 500 cells per well and ≥5 wells per sample. Epifluorescence images were machine analysed using preset scanning parameters and object mask definitions. Hoechst 33342 staining was used for autofocus and to automatically define cellular outlines based on background staining of the cytoplasm. Primary objects (cells, regions of interests (ROIs) or targets) were algorithm-defined for shape/segmentation, maximum/minimum average intensity, total area and total intensity minimum and maximum limits to automatically identify puncta or other profiles within valid primary objects. Nuclei were defined as a ROI for PI staining. All data collection, processing (object, ROI and target mask assignments) and analyses were computer-driven independently of human operators.
TIRF dSTORM super-resolution imaging and analysis
Super-resolution imaging by dSTORM TIRF microscopy and data analyses were done as previously described69. HeLa cells transiently transfected with FLAG–ATG9A and MyrPalm–EGFP were plated on 25-mm round no. 1.5 coverslips (Warner Instruments) coated with poly-l-lysine solution (Sigma-Aldrich) and allowed to adhere overnight. After two-step fixation (first step (0.6% PFA, 0.1% glutaraldehyde (GA), 0.1% saponin) for 60 s; second step (4% PFA, 0.2% GA) for 3 h), cells were washed with 1× PBS twice and incubated with 0.1% NaBH4 for 5 min. After two washes with PBS, cells were incubated with 10 mM Tris-HCl, pH 7.5 for 5 min and blocked with 5% BSA containing 0.05% saponin for 15 min. After a wash with PBS, cells were incubated with anti-FLAG antibody overnight at 4 °C and washed with PBS three times followed by labelling with Alexa Fluor 647 (A21245, Invitrogen). The coverslip was mounted on an Attofluor cell chamber (A-7816, ThermoFisher Scientific) with 1.1 ml of imaging buffer (50 mM Tris-HCl, pH 8, 10 mM NaCl, 10% glucose, 168.8 U ml−1 glucose oxidase (Sigma), 1,404 U ml−1 catalase (Sigma) and 20 mM 2-aminoethanethiol). The chamber was sealed by placing an additional coverslip over the chamber, and the oxygen-scavenging reaction in the Imaging buffer was allowed to proceed for 20 min at room temperature before starting imaging.
Imaging was performed using a custom-built TIRF illuminating fluorescence microscope controlled by custom-written software (https://github.com/LidkeLab/matlab-instrument-control) in Matlab (MathWorks). The samples were loaded on a x-y-z piezo stage (Mad City Labs, Nano-LPS100) mounted on a manual x-y translator. Images were recorded on an iXon 897 electron-multiplying charge coupled (EMCCD) camera (Andor Technologies). The EMCCD gain was set to 100, and 256 × 256 pixel frames were collected with a pixel resolution of 0.1078 µm. A 642-nm laser (collimated from a laser diode, HL6366DG, Thorlabs) was used for sample excitation. The laser was coupled to a multi-mode fibre (P1-488PM-FC-2, Thorlabs) and focused onto the back focal plane of the objective lens (UAPON ×150 OTIRF, Olympus America). Optimal laser penetration depth was achieved by adjusting the TIRF angle, translating the laser beam away from the optical axis along the back focal plane of the objective. Sample excitation was done through a quad-band dichroic and filter set (LF405/488/561/635-A; Semrock). The fluorescence emission path included a band-pass filter (685/45, Brightline) and a quadband optical filter (Photometrics, QV2-SQ) with four filter sets (600/37, 525/45, 685/40 and 445/45, Brightline).
When imaging the first signal, for each target cell, a brightfield reference image was saved in addition to the x-y stage position coordinates. The 642-nm laser was used at ∼1 kW cm−2 to take 20 sets of 2,000 frames (a total of 40,000) at 60 Hz. After imaging all target cells, the imaging buffer was replaced with PBS, the residual fluorescence was photobleached and quenched with NaBH4 and the preparation washed twice with PBS. Before the second round of imaging, cells were blocked for 30 min, labelled with anti-GFP antibody coupled with Alexa Fluor 647 (A-31852, ThermoFisher Scientifc) at 1:500 for 1 h and washed with PBS three times. Before the second round of imaging, each target cell was located and realigned using the saved brightfield reference image as described in ref. 70.
Data were analysed via a two-dimensional (2D) localization algorithm based on maximum likelihood estimation71. The localized emitters were filtered through thresholds of maximum background photon counts of 200, minimum photon counts per frame per emitter of 250 and a data model hypothesis test72 with a minimum P value of 0.01. The accepted emitters were used to reconstruct the super-resolution image. Each emitter was represented by a 2D Gaussian function with σx and σy equal to the localization precisions, which were calculated from the Cramér–Rao Lower Bound (CRLB). Clustering analysis was performed with a Matlab code using clustering tools (http://stmc.health.unm.edu/tools-and-data/). ROIs were selected from the image. Clustering was then performed separately for each label in each ROI, using the density-based DBSCAN algorithm choosing a maximal nearest neighbour distance of 40 nm and requiring clusters to contain at least 5 observations. In all cases, most observations for each label in each ROI formed a single cluster. Cluster boundaries were produced via the Matlab “boundary” function, from which inter-label cluster distances were computed.
Photodamage and time-lapse imaging
For laser-induced PM damage, Huh7 cells transiently transfected with GFP–ATG9A using Lipofectamine 2000 were plated in a chambered coverslip (μ-Slide 8 well, ibiTreat, 80826) and allowed to adhere overnight. Cells were maintained at 37 °C with a supply of 5% CO2. Cells were incubated with medium containing 160 μg ml−1 PI immediately before photodamage. Photodamage and associated time-lapse acquisitions were performed with a Leica TCS SP8 confocal microscope equipped with a 405 laser (100 mW, set at 100%, 50 iterations) to induce PM damage. The acquisition was performed with a ×63/1.4 NA oil-immersion apochromat objective and the resulting images (1 image per 0.8 s) were processed with Leica software LAS AF, Adobe After Effects and Adobe Premiere Pro.
TIRF microscopy
TIRF microscopy images were obtained using a ×100/1.49 NA oil-immersion objective and a TIRF module built-in Eclipse TI-E inverted microscope (Nikon Instruments). A 488-nm laser and a 543-nm laser were used for excitation, and emission signals were filtered with 528 ± 19-nm and 617 ± 36-nm band-pass filters for Alexa Fluor 488 and Alexa Fluor 555 imaging, respectively. Images were collected by a QuantEM 512SC imaging camera (Photometrics) operated with NIS-Elements software (Nikon Instruments). For puncta counting, images were converted to binary images by thresholding using ImageJ (National Institutes of Health).
PM tension measurement
ATG9AHuh7-WT or ATG9AHuh7-KO cells were spread at around 70% confluence on a 8-well glass bottom microslides (Ibidi, 80826) and grown in DMEM medium supplemented with 10% FBS for 24 h. The medium was replaced with the same medium containing 2 μM of Flipper-TR probe (SC020, Spirochrome) and kept for at least 30 min before imaging. FLIM imaging was performed using a Leica TCS SP8 SMD microscope equipped with a time-correlated single-photon counting module from PicoQuant58. Excitation was performed using a pulsed 470-nm laser (PicoQuant, LDH-D-C-470) operating at 40 MHz, and the emission signal was collected through a 600/50-nm bandpass filter using a MPD-SPAD detector (Micro Photon Devices—Single Photon Avalanche Diode) and a TimeHarp 260 PICO board (PicoQuant). SymPhoTime software (PicoQuant) was then used to fit fluorescence decay data (from full images) to a dual exponential model.
Cell fixation, DAB reaction, embedding and EM analyses
HEK293TAPEX2–ATG9A cells were treated with DAB (oxidation reaction) as previously described73. Alternatively, HEK293TAPEX2–ATG9A cells were exposed to 50 µg ml−1 of digitonin for 1 min before fixing and DAB treatment. Briefly, an equal volume of double strength fixative (4% GA in 0.1 M sodium cacodylate buffer, pH 7.4) was added to the cells for 20 min at room temperature before fixing the cells with one volume of single strength fixative (2% GA in 0.1 M sodium cacodylate buffer, pH 7.4) at 4 °C for 1 h. After 5 washes with cacodylate buffer (pH 7.4) followed by 5 washes in PBS, a filtered solution of 0.5 mg ml−1 DAB (Sigma) in PBS was mixed (10,0000:3) with 30% H2O2 (Sigma) and added to the cells for 7 min. The DAB oxidation reaction was monitored using a brightfield microscope and stopped by rinsing the cells 3 times with PBS for 5 min. Cells were then processed for EM by embedding them in EPON resin as previously described74. Ultra-thin 70-nm sections were cut using a Leica EM UC7 ultramicrotome (Leica Microsystems) and collected on 150 formvar-mesh copper grids. Cell sections were examined using a CM100bio TEM (FEI). The quantification of ATG9A at the PM was performed on APEX2–ATG9A-positive cell profiles per condition in the following way: the entire length of the PM as well as the sections of the PM stained with DAB in each cell profile were measured with the ImageJ software. The average percentage of the PM positive for APEX2–ATG9A in 45 randomly selected cell profiles were analysed.
APEX2 labelling and streptavidin enrichment for LC–MS/MS analysis
HEK293TAPEX2-ATG9A cells were incubated in 500 µM biotin–phenol (AdipoGen) in complete medium before inducing PM damage. For digitonin treatment, 100 µg ml−1 digitonin diluted in complete medium was added on the cells for 1 min. Cells were washed once in complete medium before adding back biotin–phenol medium. For SLO treatment, cells were washed at 37 °C with Ca2+-free HBSS containing 5 mM EGTA followed by two more washes in Ca2+-free HBSS. SLO was reduced by 10 mM DTT 5 min at room temperature before dilution in Ca2+-free HBSS (200 U ml−1) and added on target cells for 10 min at 37 °C. Cells were then washed once in complete medium before adding biotin–phenol medium. For GBI treatment, ~1.6 g of beads were gently poured on the 10-cm petri dish containing the cells. The beads were agitated over the cells for 1 min on a rotator platform at 160 r.p.m. A 1-min pulse with 1 mM H2O2 at room temperature was stopped with quenching buffer (10 mM sodium ascorbate, 10 mM sodium azide and 5 mM Trolox in PBS). All samples were washed twice with quenching buffer, and twice with PBS.
For LC–MS/MS analysis, cell pellets were lysed in 500 µl ice-cold lysis buffer (6 M urea, 0.3 M NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM sodium ascorbate, 10 mM sodium azide, 5 mM Trolox, 1% glycerol and 25 mm Tris-HCl, pH 7.5) for 30 min by gentle pipetting. Lysates were clarified by centrifugation and protein concentrations were determined using Pierce 660 nm protein assay reagent. Streptavidin-coated magnetic beads (Pierce) were washed with lysis buffer. A total of 1 mg of each sample was mixed with 100 µl of streptavidin beads. The suspensions were gently rotated at 4 °C overnight to bind biotinylated proteins. The flow-through after enrichment was removed and the beads were washed in sequence with 1 ml IP buffer (150 mM NaCl, 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100) twice; 1 ml 1 M KCl; 1 ml of 50 mM Na2CO3; 1 ml 2 M urea in 20 mM Tris HCl, pH 8.0; and 1 ml IP buffer. Biotinylated proteins were eluted, and 10% of the sample processed for immunoblotting and 90% of the sample processed for MS.
LC–MS/MS
Digested peptides were analysed by LC–MS/MS on a Thermo Scientific Q Exactive Plus Orbitrap mass spectrometer in conjunction Proxeon Easy-nLC II HPLC (Thermo Scientific) and Proxeon nanospray source. The digested peptides were loaded onto a 100 µm × 25 mm Magic C18 100 Å 5U reverse-phase trap where they were desalted online before being separated using a 75 µm × 150 mm Magic C18 200 Å 3U reverse-phase column. Peptides were eluted using a 140-min gradient with a flow rate of 300 nl min−1. A MS survey scan was obtained for the m/z range 350–1,600, and MS/MS spectra were acquired using a top 15 method, where the top 15 ions in the MS spectra were subjected to high energy collisional dissociation. An isolation mass window of 1.6 m/z was for the precursor ion selection, and a normalized collision energy of 27% was used for fragmentation. A 15-s duration was used for the dynamic exclusion.
MS data processing and analysis
Tandem mass spectra were extracted by Proteome Discoverer v.2.2. Charge state deconvolution and deisotoping were not performed. All MS/MS samples were analysed using X! Tandem (The GPM, thegpm.org; version X! Tandem Alanine (2017.2.1.4)). X! Tandem was set up to search the UniProt Human proteome database plus 110 common laboratory contaminants and an equal number of decoy sequences (147,936 entries total) assuming the digestion enzyme trypsin. X! Tandem was searched with a fragment ion mass tolerance of 20 ppm and a parent ion tolerance of 20 ppm. Glu->pyro-Glu of the N terminus, ammonia-loss of the N terminus, gln->pyro-Glu of the N terminus, deamidated of asparagine and glutamine, oxidation of methionine and tryptophan and dioxidation of methionine and tryptophan were specified in X! Tandem as variable modifications.
Scaffold (v.Scaffold_4.9.0, Proteome Software) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 98.0% probability by the Scaffold Local false discovery rate (FDR) algorithm. Peptide identifications were also required to exceed specific database search engine thresholds. X! Tandem identifications required at least −log(E-value) of 2. Protein identifications were accepted if they could be established at greater than 5.0% probability to achieve an FDR less than 5.0% and contained at least 1 identified peptide. This resulted in a peptide decoy FDR of 0.7% and a protein decoy FDR of 0.66%. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. Raw data and Scaffold results are available from the MassIVE proteomics repository (MSV000084519) and Proteome Exchange PXD016084.
BioWeB assay: APEX2 labelling and streptavidin enrichment for immunoblotting analyses
HEK293TAPEX2–ATG9A cells were treated as described above for LC–MS/MS analysis. Cells were lysed in 500 µl ice-cold NP-40 buffer for 30 min on ice. Lysates were clarified by centrifugation and protein concentrations were determined using Pierce 660-nm protein assay reagent. One milligram of each sample was mixed with 100 µl of streptavidin magnetic beads (Pierce). The suspensions were gently rotated at 4 °C overnight to bind biotinylated proteins. The flow-through after enrichment was removed and the beads were washed in sequence with 1 ml IP buffer twice; 1 ml 1 M KCl; 1 ml of 50 mM Na2CO3; 1 ml 2 M urea in 20 mM Tris-HCl, pH8; and 1 ml IP buffer. Biotinylated proteins were eluted with 2× Laemmli sample buffer (Bio-Rad) and subjected to immunoblot analysis.
Statistics and reproducibility
Data in this paper are presented as the mean ± s.e.m. (n ≥ 3). Data were analysed with either analysis of variance (ANOVA) with Dunnett’s, Tukey’s or Sidak’s HSD post hoc test, or unpaired Student’s t-test using GraphPad Prism v.7 to determine statistical significance. No statistical methods were used to predetermine the sample sizes. The number of independent samples and any statistical tests used are indicated in the figure legends, and all the replicates reproduced the shown findings. The experiments were repeated at least three times wherever representative results are shown.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
MS raw data and Scaffold results have been deposited in the MassIVE proteomics repository (MSV000084519) (https://massive.ucsd.edu) and Proteome Xchange with the primary accession code PXD016084 (http://www.proteomexchange.org). Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Code availability
Super-resolution TIRF imaging was performed using a custom-built TIRF illuminating fluorescence microscope controlled by custom-written software (https://github.com/LidkeLab/matlab-instrument-control) in Matlab (MathWorks).
References
Morishita, H. & Mizushima, N. Diverse cellular roles of autophagy. Annu. Rev. Cell Dev. Biol. 35, 453–475 (2019).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42 (2019).
Christ, L., Raiborg, C., Wenzel, E. M., Campsteijn, C. & Stenmark, H. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci. 42, 42–56 (2017).
Hurley, J. H. ESCRTs are everywhere. EMBO J. 34, 2398–2407 (2015).
Gatta, A. T. & Carlton, J. G. The ESCRT-machinery: closing holes and expanding roles. Curr. Opin. Cell Biol. 59, 121–132 (2019).
Jimenez, A. J. et al. ESCRT machinery is required for plasma membrane repair. Science 343, 1247136 (2014).
Scheffer, L. L. et al. Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 5, 5646 (2014).
Ruhl, S. et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018).
Gong, Y. N. et al. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169, 286–300.e16 (2017).
Galluzzi, L. & Green, D. R. Autophagy-independent functions of the autophagy machinery. Cell 177, 1682–1699 (2019).
Mizushima, N. & Levine, B. Autophagy in human diseases. N. Engl. J. Med. 383, 1564–1576 (2020).
Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 54, 437–453 (2021).
Lahiri, V., Hawkins, W. D. & Klionsky, D. J. Watch what you (self-) eat: autophagic mechanisms that modulate metabolism. Cell Metab. 29, 803–826 (2019).
Valverde, D. P. et al. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 218, 1787–1798 (2019).
Maeda, S., Otomo, C. & Otomo, T. The autophagic membrane tether ATG2A transfers lipids between membranes. eLife 8, e45777 (2019).
Matoba, K. et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 27, 1185–1193 (2020).
Maeda, S. et al. Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat. Struct. Mol. Biol. 27, 1194–1201 (2020).
Zhao, Y. G. & Zhang, H. Autophagosome maturation: an epic journey from the ER to lysosomes. J. Cell Biol. 218, 757–770 (2019).
Guardia, C. M. et al. Structure of human ATG9A, the only transmembrane protein of the core autophagy machinery. Cell Rep. 31, 107837 (2020).
Gomez-Sanchez, R. et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J. Cell Biol. 217, 2743–2763 (2018).
Young, A. R. et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 119, 3888–3900 (2006).
Longatti, A. et al. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J. Cell Biol. 197, 659–675 (2012).
Puri, C., Renna, M., Bento, C. F., Moreau, K. & Rubinsztein, D. C. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 154, 1285–1299 (2013).
Zhou, C. et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 27, 184–201 (2017).
Broz, P., Pelegrin, P. & Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20, 143–157 (2020).
Wang, H. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014).
Quarato, G. et al. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol. Cell 61, 589–601 (2016).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Weinlich, R., Oberst, A., Beere, H. M. & Green, D. R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136 (2017).
Kakuta, S. et al. Small GTPase Rab1B is associated with ATG9A vesicles and regulates autophagosome formation. FASEB J. 31, 3757–3773 (2017).
Karanasios, E. et al. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat. Commun. 7, 12420 (2016).
Orsi, A. et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 23, 1860–1873 (2012).
Popovic, D. & Dikic, I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep. 15, 392–401 (2014).
Colom, A. et al. A fluorescent membrane tension probe. Nat. Chem. 10, 1118–1125 (2018).
Ding, J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016).
Mellgren, R. L. A new twist on plasma membrane repair. Commun. Integr. Biol. 4, 198–200 (2011).
Judith, D. et al. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIβ. J. Cell Biol. 218, 1634–1652 (2019).
Lam, S. S. et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015).
Jia, J. et al. Galectins control mTOR in response to endomembrane damage. Mol. Cell 70, 120–135.e8 (2018).
Hedman, A. C., Smith, J. M. & Sacks, D. B. The biology of IQGAP proteins: beyond the cytoskeleton. EMBO Rep. 16, 427–446 (2015).
Morita, E. et al. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 26, 4215–4227 (2007).
Denais, C. M. et al. Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358 (2016).
Keyel, P. A. et al. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 124, 2414–2423 (2011).
Yue, Y. et al. SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. 9, 904 (2018).
Gutierrez, M. G. et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766 (2004).
Kimmey, J. M. et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528, 565–569 (2015).
Divangahi, M. et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 10, 899–906 (2009).
Mishra, M., Adhyapak, P., Dadhich, R. & Kapoor, S. Dynamic remodeling of the host cell membrane by virulent mycobacterial sulfoglycolipid-1. Sci. Rep. 9, 12844 (2019).
Brill, S. et al. The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and Rho family GTPases. Mol. Cell. Biol. 16, 4869–4878 (1996).
Ho, Y. D., Joyal, J. L., Li, Z. & Sacks, D. B. IQGAP1 integrates Ca2+/calmodulin and Cdc42 signaling. J. Biol. Chem. 274, 464–470 (1999).
Atcheson, E. et al. IQ-motif selectivity in human IQGAP2 and IQGAP3: binding of calmodulin and myosin essential light chain. Biosci. Rep. 31, 371–379 (2011).
Hart, M. J., Callow, M. G., Souza, B. & Polakis, P. IQGAP1, a calmodulin-binding protein with a rasGAP-related domain, is a potential effector for cdc42Hs. EMBO J. 15, 2997–3005 (1996).
Joyal, J. L. et al. Calmodulin modulates the interaction between IQGAP1 and Cdc42. Identification of IQGAP1 by nanoelectrospray tandem mass spectrometry. J. Biol. Chem. 272, 15419–15425 (1997).
Mataraza, J. M. et al. IQGAP1 promotes cell motility and invasion. J. Biol. Chem. 278, 41237–41245 (2003).
Noritake, J., Watanabe, T., Sato, K., Wang, S. & Kaibuchi, K. IQGAP1: a key regulator of adhesion and migration. J. Cell Sci. 118, 2085–2092 (2005).
Claude-Taupin, A. et al. ATG9A is overexpressed in triple negative breast cancer and its in vitro extinction leads to the inhibition of pro-cancer phenotypes. Cells 7, 248 (2018).
Yamano, K., Fogel, A. I., Wang, C., van der Bliek, A. M. & Youle, R. J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 3, e01612 (2014).
Mattera, R., Park, S. Y., De Pace, R., Guardia, C. M. & Bonifacino, J. S. AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc. Natl Acad. Sci. USA 114, E10697–E10706 (2017).
Beckwith, K. S. et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat. Commun. 11, 2270 (2020).
Ghosh, S. et al. β-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 183, 1520–1535.e14 (2020).
Yamaguchi, J. et al. Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy 14, 764–777 (2018).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
Idone, V. et al. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 180, 905–914 (2008).
Mellgren, R. L. A plasma membrane wound proteome: reversible externalization of intracellular proteins following reparable mechanical damage. J. Biol. Chem. 285, 36597–36607 (2010).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Galli, T. et al. A novel tetanus neurotoxin-insensitive vesicle-associated membrane protein in SNARE complexes of the apical plasma membrane of epithelial cells. Mol. Biol. Cell 9, 1437–1448 (1998).
Pallikkuth, S. et al. Sequential super-resolution imaging using DNA strand displacement. PLoS ONE 13, e0203291 (2018).
Valley, C. C., Liu, S., Lidke, D. S. & Lidke, K. A. Sequential superresolution imaging of multiple targets using a single fluorophore. PLoS ONE 10, e0123941 (2015).
Smith, C. S., Joseph, N., Rieger, B. & Lidke, K. A. Fast, single-molecule localization that achieves theoretically minimum uncertainty. Nat. Methods 7, 373–375 (2010).
Huang, F., Schwartz, S. L., Byars, J. M. & Lidke, K. A. Simultaneous multiple-emitter fitting for single molecule super-resolution imaging. Biomed. Opt. Express 2, 1377–1393 (2011).
Martell, J. D., Deerinck, T. J., Lam, S. S., Ellisman, M. H. & Ting, A. Y. Electron microscopy using the genetically encoded APEX2 tag in cultured mammalian cells. Nat. Protoc. 12, 1792–1816 (2017).
Zhou, X. et al. Ultrastructural characterization of membrane rearrangements induced by porcine epidemic diarrhea virus infection. Viruses 9, 251 (2017).
Acknowledgements
We thank M. Jäätelä and E. Corcelle-Termeau for ATG9AMCF-7-KO cells, M. J. Wester for mathematical analysis, M. Campen and T. Young for nanoparticle tracking analysis and UTechS Photonic Bioimaging (supported by ANR-10-INSB-04-01). S.A.T. was supported by the Francis Crick Institute (Cancer Research UK, FC001187), the UK Medical Research Council (FC001187) and the Wellcome Trust (FC001187). F.R. was supported by a ZonMW TOP (91217002) and ALW Open Programme (ALWOP.310) grants. M.C.M. was supported by an ALW Open Program (ALWOP.355) grant. This work was supported by NIH grants R37AI042999 and R01AI042999, and center grant P20GM121176 to V.D.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.C.-T. and V.D. Formal analysis: A.C.-T., J.J., Z.B., M.G.-T., S.K., G.P.D.d.S., R.J., Y.G., L.A., R.P., S.P., F.W., M.S., S.A.T. and V.D. Investigation and validation: A.C.-T., J.J., Z.B., M.G.-T., S.K., G.P.D.d.S., Y.G., L.A., R.P., S.P., F.W., M.S., B.P., M.M., P.V., M.C.M., T.J. and V.D. Resources: V.D., K.A.L., Y.U., S.A.T., P.V., T.J. and F.R. Data curation: L.A., B.P., A.C.-T. and V.D. Writing (original draft): A.C.-T. and V.D. Writing (comments): S.A.T. and F.R. Visualization: A.C.-T., S.P., M.C.M. and V.D. Supervision: V.D., L.J.d.C., B.P., T.J. and F.R. Project administration and funding acquisition: V.D.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 ATG9A protects different cell lines against plasma membrane damage.
a, HCM image examples (white masks, algorithm-defined cell boundaries; yellow masks, computer-identified PI+ nuclei) of PM permeabilization (saponin (Sap) and streptolysin O (SLO)) in ATG9AHuh7-WT and ATG9AHuh7-KO cells. Scale bars, 10 µm. b,c, HCM complementation analysis of PM permeabilization sensitivity (Dig) in ATG9AHuh7-KO transfected with b, GFP or GFP-ATG9A or c, FLAG-ATG9A increasing concentrations. PI+ cells quantified after gating on GFP+ or FLAG+ cells (HCM, mean± SEM, n=5 (b), n=6 (c) biologically independent samples, two-way ANOVA Sidak’s test (b) or unpaired t test (c). d, Example of HCM images of ATG9AHuh7-WT and ATG9AHuh7-KO cells. PMHAL assay (Dig). Yellow masks, computer-identified GFP-MIL+ puncta. Scale bars, 10 µm. e,f, HCM PMHAL analysis of ATG9AHuh7-WT and ATG9AHuh7-KO cells (Dig, MPL staining). e, Yellow masks, computer-identified GFP-MPL+ puncta. Scale bars, 10 µm. f, Graph, HCM PMHAL quantification of GFP-MPL+ puncta/cell (mean±SEM, n=5 biologically independent samples, two-way ANOVA Sidak’s test). g, HCM analysis of PM permeabilization (FITC-Dextran-10k - Dx-10) in ATG9AHuh7-WT and ATG9AHuh7-KO cells upon PM damage (Dig). Data, % of cells positive for Dx-10 (mean±SEM, n=5 biologically independent samples, two-way ANOVA Sidak’s test). h, HCM analysis of the endocytic pathway (DQ-Red BSA) in ATG9AHuh7-WT and ATG9AHuh7-KO cells upon PM damage (Dig). Data, number of DQ-Red BSA profiles/cell (mean±SEM, n=5 biologically independent samples, two-way ANOVA Sidak’s test). i, HCM quantification of the overlap area between GFP-Rab5 (WT or Q79L mutant) and LBPA in ATG9AHuh7-WT and ATG9AHuh7-KO cells. Data, mean±SEM; n=6 biologically independent samples, two-way ANOVA Sidak’s test. j, Confocal images of LBPA (red) in ATG9AHuh7-WT and ATG9AHuh7-KO cells transiently expressing GFP-Rab5 (WT or Q79L, green). Scale bars, 10 µm. k,l, Analysis of PM tension in ATG9AHuh7-WT and ATG9AHuh7-KO cells using the Flipper-TR® probe (Fluorescence lifetime microscopy, FLIM). k, Representative images of the average fluorescence lifetime of Flipper-TR®. Color scale from 2 to 6 ns. Scale bars, 10 µm. l, quantification of the average lifetime of Flipper-TR from full images (mean±SEM, n=5 independent images, unpaired t test). m, HCM analysis of calcein+ cells (Live/DeadTM 30 min staining prior to Dig treatment) in ATG9AHuh7-WT and ATG9AHuh7-KO cells (washed with 5 mM EGTA and incubated in a Ca2+-free HBSS medium with (+) or without (-) added 3.6 mM Ca2+). Data, % of cells positive for calcein (mean±SEM; n=5 biologically independent samples, two-way ANOVA Sidak’s test). n, Immunoblotting of the ATG9AMCF-7-KO cells (one of 3 independent experiments). o,p, HCM analysis of PM permeabilization (PI, Dig) of ATG9AMCF-7-WT and ATG9AMCF-7-KO cells. o, Example of HCM images: white masks, algorithm-defined cell boundaries; red masks, computer-identified PI+ nuclei. Scale bars, 10 µm. p, % of cells positive for PI (mean±SEM, n=5 biologically independent samples, two-way ANOVA Sidak’s test). q, HCM analysis of PM permeabilization (Dx-10, Dig) in ATG9AMCF-7-WT and ATG9AMCF-7-KO cells. Data, % of cells positive for Dx-10 (mean±SEM, n=5 biologically independent samples, two-way ANOVA Sidak’s test). r, HCM analysis of the endocytic pathway (DQ-Red BSA, Dig) in ATG9AMCF-7-WT and ATG9AMCF-7-KO cells. Data, quantification of DQ-Red BSA profiles/cell (mean±SEM, n=5 biologically independent samples, two-way ANOVA Sidak’s test). s, Confirmation by immunoblotting of ATG9A KD in HeLa cells (one of 3 independent experiments). t,u, HCM analysis of PM permeabilization (PI, Dig). t, example of HCM images: white masks, algorithm-defined cell boundaries; red masks, computer-identified PI+ nuclei. Scale bars, 10 µm. u, % of cells positive for PI (mean±SEM, n=5 biologically independent samples, two-way ANOVA Sidak’s test).
Extended Data Fig. 2 ATG9A protects primary cells against plasma membrane damage.
a, Confirmation by immunoblotting of the Atg9aKO (LysMCre+) in BMM. One of 3 independent experiement. b, HCM analysis of PM permeabilization (Dx-10, Dig) in BMM from Atg9afl/flLysM-Cre- and Atg9afl/flLysM-Cre+ mice. Data, % of cells positive for Dx-10 (mean±SEM, n=5 biologically independent samples, two-way ANOVA Sidak’s test). c, HCM analysis of the endocytic pathway (DQ-Red BSA, Dig) in BMM from Atg9afl/flLysM-Cre- and Atg9afl/flLysM-Cre+ mice. Data, Quantification of DQ-Red BSA profiles/cell (mean±SEM; n=5 biologically independent samples, two-way ANOVA Sidak’s test). d, HCM analysis of PM permeabilization (Alexa Fluor 647-Dextran-10k -AF647 Dx-10, SLO) in BMM from Atg9afl/flLysM-Cre- and Atg9afl/flLysM-Cre+ mice. Data, % of cells positive for Dx-10 (mean±SEM, n=6 biologically independent samples, two-way ANOVA Sidak’s test). e, HCM quantification of PM permeabilization (PI staining) and cell viability (Live/DeadTM, Calcein+ cells) of ATG9AHuh7-WT and ATG9AHuh7-KO cells transfected with FLAG-GSDMD-N-terminal fragment (NT). Data, % of FLAG-positive cells that were double positive for PI and Calcein. Data, mean±SEM; n=6 biologically independent samples, unpaired t test.
Extended Data Fig. 3 Plasma membrane damage induces ATG9A translocation to PM.
a, Confocal images of FLAG-ATG9A (red) localization to PM (MyrPalm-EGFP, green) during its damage (SLO, GBI or Sap) in HeLa cells. Scale bars, 10 µm. b, Quantification (super resolution TIRF) of the distance between FLAG-ATG9A and MyrPalm-EGFP clusters before and after PM damage (Dig). c, Cell surface biotinylation analysis of ATG9A in HEK293T cells transiently expressing GFP-ATG9A (Starvation, EBSS or PM damage: Dig, SLO or GBI). GFP-ATG9A (IP anti GFP) analyzed by Streptavidin-HRP immunoblotting. One of 3 biologically independent experiments. d, TIRF microscopy images of FLAG-ATG9A (red) recruitment to PM (MyrPalm-EGFP, green) upon Dig in cells pretreated with NEM or DTT, as indicated. Scale bars, 5 µm. e,f, Quantification of ATG9A recruitment to PM in TIRF field upon Dig and/or NEM treatment. e, FLAG-ATG9A fluorescence intensity and f, ratio of FLAG-ATG9A puncta intensity with fluorescence intensity in TIRF field. Data, mean±SEM; n=10 biologically independent cells, unpaired t test. g, HCM analysis of PM permeabilization (PI, Dig) in ATG9AHuh7-WT and ATG9AHuh7-KO cells pre-treated with NEM or DTT as indicated. Data, % of cells positive for PI (mean±SEM, n=6 biologically independent samples, unpaired t test). h, Schematic of the stable cell line HEK293TAPEX2-ATG9A constructed in HEK293T Flp-In cells, expressing integrated FLAG-APEX2-ATG9A inducible by tetracycline. i-n, HEK293TAPEX2-ATG9A cells were exposed (j-n) or not (i) to Dig. i and j, overviews. k-n, magnification of areas showing deposits of diaminobenzidine (APEX2 activity product of APEX2-ATG9A) without or with adjacent diaminobenzidine-positive vesicles/tubulo-vesicular structures. E, endosome; N, Nucleus; PM, plasma membrane; white arrowheads highlight regions of the PM where APEX2-ATG9A is concentrated. Scale bars, 1 µm (i, j, n), 250 nm (Panels k-m). o, Schematic summary of the findings in Extended Data Figure 3.
Extended Data Fig. 4 ATG9A translocation to plasma membrane protects cells from PM damage.
a, Examples of HCM images of HeLa cells expressing GFP-ATG9AWT or GFP-ATG9AY8F (green) subjected to PI staining (red) after Dig treatment. Red masks, computer-identified PI+ nuclei on GFP+ cells. Scale bars, 10 µm. b, Confocal images of ATG9A localization in HeLa cells expressing FLAG-ATG9A (red) and MyrPalm-EGFP (green), serum-starved for 24 h, and then stimulated with hEGF for 30 min. Non-starved cells: full media (FM) used as control. Scale bars, 10 µm. c, HCM quantification of PM permeabilization (PI, Dig) in HeLa cells expressing GFP-ATG9A, serum-starved and stimulated with hEGF. Data, % of GFP+ cells that are PI+ (mean±SEM; n=5 biologically independent samples, two-way ANOVA Sidak’s test). d, HCM analysis of PM permeabilization (PI, Dig) in ATG9AU2OS-WT and ATG9AU2OS-KO cells serum-starved for 24 h, and then stimulated with hEGF for 30 min. Non-starved cells: full media (FM) used as control. Data, % of cells positive for PI (mean±SEM, n=5 biologically independent samples, two-way ANOVA Tukey’s test). e, Schematic summary of the findings in Fig. 3f,g. f, Schematic summary of findings in Extended Data Figure 4b-d.
Extended Data Fig. 5 ATG9A partners identified by dynamic proximity biotinylation proteomics.
a, Complementation analysis of LC3 lipidation in ATG9AHuh7-KO cells transfected with FLAG-APEX2-ATG9A. Autophagy was induced by starvation in EBSS (90 min) with or without Bafilomycin A1 (BAF). *, unspecific band (one of 3 independent experiments). b, Proximity biotinylation proteomics (process stages) for LC-MS/MS identification of APEX2-ATG9A partners and analysis of dynamic changes in their proximity during PM damage (Dig, SLO or GBI). c,d, Volcano plots, HEK293TAPEX2-ATG9A cells were incubated in full medium under control (Ctrl) or PM damage-inducing conditions SLO (c) or GBI (d). X-axis, log2 fold change (PM damage/Ctrl ratio; spectral counts); y-axis, -log10 of p-values, t-test (n=3 biologically independent samples per group). Green and red dots, increase and decrease in proximity to ATG9A after PM damage, respectively. Orange dots, values below statistical significance (for increase/decrease changes) cut-off (p ≥ 0.05). IQGAP1, RAB7A, and AP3D1 are highlighted in purple. Bubble size represents a normalized value for the total spectral counts (average of all samples) for the protein indicated. e,f, Protein species showing infinite positive (e) or negative (f) fold change in proteomic studies here, in all three conditions (Dig, SLO and GBI) tested with HEK293TAPEX2-ATG9A cells. Data, mean total peptide spectral counts (green, infinite positive-fold change group had 0 spectral counts in all 3 untreated samples; red, infinite negative-fold change group had 0 spectral counts in all 3 treated samples). IQGAP1 (e) and AP3D1 (f) are shown as non-infinite fold change comparators.
Extended Data Fig. 6 IQGAP1 and calcium contribute to ATG9A’s role in protection against plasma membrane damage.
a, CoIP analysis of endogenous ATG9A (CST, HEK293T) with TSG101 and IQGAP1 after PM damage (Dig, SLO, GBI, one of 3 independent experiments). b, Confocal images of endogenous IQGAP1 (blue) in HeLa cells expressing MyrPalm-EGFP (green) and FLAG-ATG9A (red) during PM damage (Dig). Scale bars, 10 µm. c, HCM quantification of FLAG-ATG9A and GFP-IQGAP1 overlap area in HeLa cells after PM damage by digitonin (Dig). Data, Quantification of the overlap between FLAG-ATG9A and GFP-IQGAP1 profiles (mean±SEM; n=5 biologically independent samples, unpaired t-test). d, Aspect (top view) of ATG9A CryoEM structure (PDB:7JLP; Maeda et al., 2020), revealing adjacency of extramembranous domains at the cytosolic side of ATG9A. e, Modified schematic from Guardia et al., 2020, based on CryoEM ATG9A structure. Colored circles indicate beginnings and ends of regions used in GST-pulldowns in panel f and correspond in color to domains in d. f, GST-pulldown analysis of in vitro translated and radiolabeled [35S]Myc-IQGAP1 with GST and GST fused with ATG9A cytosolic domains (residues 1-66, 495-839, 522-839, 153-289 and 723-839). CBB: Coomassie brilliant blue. Graph, quantification of the binding percentage of IQGAP1 relative to GST constructs. Data, mean±SEM; n=3 biologically independent experiments, unpaired t-test g, Confirmation by immunoblotting of IQGAP1 KD in HeLa cells. One of 3 independent experiments. h, Confocal images of ATG9A localization after IQGAP1 KD during PM damage (Dig), in HeLa cells expressing MyrPalm-EGFP (green) and FLAG-ATG9A (red). Scale bars, 10 µm. i, HCM quantification of the overlap area between GFP-Rab5 (WT or Q79L mutant) and LBPA in Huh7 cells after IQGAP1 KD. Data, mean±SEM; n=4 biologically independent samples, two-way ANOVA Tukey’s test. j, Confocal images of LBPA (red) in Huh7 cells (IQGAP1 KD) transiently expressing GFP-Rab5 (WT or Q79L, green). Scale bars, 10 µm. k, Confocal images of ATG9A localization after PM damage (Dig), in the presence or absence of Ca2+. HeLa cells expressing FLAG-ATG9A (red) and MyrPalm-EGFP (green) were washed with 5 mM EGTA and incubated in a Ca2+-free HBSS medium with (+) or without (-) added 3.6 mM Ca2+ during PM damage. Scale bars, 10 µm. l, Examples of HCM images in HeLa cells expressing GFP-IQGAP1 (green) and FLAG-ATG9A (red) after PM damage (Dig, Ca2+ treatment/conditions as in k). Yellow masks, computer-identified FLAG-ATG9A+GFP-IQGAP1+ double positive profiles. Scale bars, 10 µm. m, HCM quantification of FLAG-ATG9A and GFP-IQGAP1 colocalization in HeLa cells (Dig, Ca2+ treatment/conditions as in k). Data, overlap area between FLAG-ATG9A and GFP-IQGAP1 (mean±SEM; n=3 biologically independent samples, unpaired t-test). n, CoIP (anti FLAG) analysis of FLAG-ATG9A and GFP-IQGAP1 interaction after PM damage (Dig) in the presence or absence of Ca2+ (HEK293T, Ca2+ treatment/conditions as in k, one of 3 independent experiments).
Extended Data Fig. 7 ATG9A interacts with ALIX.
a, ALIX domains and constructs used in this study. FL, full length; Bro1, Bro1 domain; VD, V domain, PRD, proline-rich domain. Numbers, residue positions. b, CoIP analysis (anti-FLAG) of FLAG-ALIX deletion mutants with HA-ATG9A (HEK293T). One of 3 independent experiments. c, BioWeB analysis of ALIX proximity to APEX2-ATG9A (HEK293TAPEX2-ATG9A), with or without PM damage (Dig, SLO, GBI). One of 3 independent experiments.
Extended Data Fig. 8 ESCRTs cooperate with ATG9A in protection against plasma membrane damage.
a, CoIP analysis of endogenous ATG9A (CST, HEK293T) with TSG101 after IQGAP1 KD and PM damage (Dig); one of 3 independent experiments. b, TIRF microscopy images of mCherry-CHMP4B (red) recruitment to PM (MyrPalm-EGFP, green) during PM damage (Dig), in ATG9AHuh7-WT cells pretreated with NEM or NEM+DTT. Scale bars, 5 µm. c, Confocal images of HeLa Flp-In mCherry-CHMP4A (red) and MyrPalm-EGFP (green) during PM damage (Dig) (washed with 5mM EGTA and incubated in a Ca2+-free HBSS medium with (+) or without (-) added 3.6 mM Ca2+). Arrows, cell periphery. Scale bars, 10 µm. d, Confocal images of mCherry-CHMP4B (red) and MyrPalm-EGFP (green) during PM damage (Dig) in ATG9AHuh7-WT and ATG9AHuh7-KO cells. Ca2+ treatment/conditions as in panel c. Scale bars, 10 µm. White arrows indicate CHMP4B puncta localizing to the PM (MyrPalm-EGFP). e, Confocal images of GFP-ATG9A (green) and mCherry-CHMP4B (red) in cells transiently transfected with FLAG-GSDMD-NT (PM damage) or -NT-4A as control. Scale bars, 10 µm. White arrows indicate GSDMD-NT localization at PM in proximity to ATG9A and CHMP4B.
Extended Data Fig. 9 Analysis of additional ATG9A interactors and ATG9A M33 mutant in defense against PM damage.
a, Confirmation by immunoblotting of CRISPR-Cas9 in ATG2AU2OS-KO cells. One of 3 independent experiments. b, Confirmation by immunoblotting of CRISPR-Cas9 in ATG2BU2OS-KO cells. One of 3 independent experiments. c, Graph, HCM quantification of PM permeabilization (PI, Dig) in ATG2AU2OS-KO, ATG2BU2OS-KO and ATG9AU2OS-KO cells. Data, % of total cells that are PI+ cells (mean±SEM; n=3 biologically independent samples, two-way ANOVA Sidak’s test). d,e, HCM quantification of PM permeabilization (PI, d) and cell viability (LDH release in the supernatant, e) in ATG2AU2OS-KO, ATG2BU2OS-KO and ATG9AU2OS-KO cells after LPS electroporation. Data, % of total cells that are PI+ cells (d, HCM; e, % LDH release quantified, mean±SEM; n=6 biologically independent samples, unpaired t test). f, Confirmation by immunoblotting of PI4KB KD in HeLa cells (one of 3 independent experiments). g, Graph, HCM quantification of PM permeabilization (PI, Dig) in PI4KB KD cells. Data, % of total cells that are PI+ cells (mean±SEM; n=5 biologically independent samples, two-way ANOVA Sidak’s test). h, HCM complementation analysis of PM permeabilization sensitivity (Dig) in ATG9AHuh7-KO transfected with ATG9A-FLAG (WT or M33 scramblase mutant). PI+ cells quantified after gating on FLAG+ cells (HCM, mean± SEM, n=6 biologically independent samples, unpaired t test). i, Immunoblot confirmation of Rab7 KD in HeLa cells. One of 3 biologically independent experiments. j, HCM quantification of PM permeabilization (PI, Dig) in HeLa cells after Rab7 KD. Data, % of cells positive for PI (mean±SEM; n=6 biologically independent samples, two-way ANOVA Sidak’s test). k, CoIP (anti-GFP) analysis of FLAG-ATG9A and GFP-AP2M1 interaction during PM damage (Dig, HEK293T, one of 3 independent experiments). l, CoIP (anti-GFP) analysis of FLAG-ATG9A and GFP-AP4M1 interaction during PM damage (Dig, HEK293T, one of 3 independent experiments). m, Bimolecular fluorescence complementation assay schematic for analysis of ALIX and ATG9A association with split Venus fluorescent protein. ALIX and ATG9A were respectively fused with the N-terminal (VNALIX) and C-terminal (VCATG9A) fragments of Venus and expressed in HeLa cells. Venus fluorescence corresponding to ALIX and ATG9A association was then assessed by HCM and confocal microscopy. n, Confocal images of ATG9A and ALIX association using the bimolecular fluorescence complementation assay (VNALIX+VCATG9A=green) during PM damage (Dig). GM130 (Golgi staining, red). Scale bars, 10 µm. o, p, HCM quantification of (o) ATG9A and ALIX association and (p) their overlap with PM using the bimolecular fluorescence complementation assay, during PM damage (Dig). HeLa cells transiently expressing VNALIX and VCATG9A were labelled with cell outline/plasma membrane stain CellMask reagent. Data, HCM quantification of (o) Venus puncta and (p) the overlap area between Venus puncta and CellMask (mean±SEM; n=5 biologically independent samples, one-way ANOVA Dunnett’s test). q, HCM quantification of cytosolic PI staining in ATG9AHuh7-WT and ATG9AHuh7-KO cells without damage (mean±SEM; n=6 biologically independent samples, unpaired t test).
Extended Data Fig. 10 Analyses of ATG9A-dependent processes during PM damage.
a, Time-lapse confocal images of GFP-ATG9A (magenta hot) and propidium iodide (blue) upon laser-induced PM damage (ROI, yellow rectangle). Insets, ATG9A distribution at site of PM damage. Green ROI (2’10”) indicate organization of GFP-ATG9A bordering with damaged areas. Scale bar, 10 µm. b, Example images (Dig) obtained from videos during nanoparticle tracking analysis. c, Particle distribution (“D”-values) in the supernatant of ATG9AHuh7-WT and ATG9AHuh7-KO cells (Dig). D10, D50 and D90 values reflect the diameter of the particles, whereby 10%, 50% or 90% of all particles is below the size indicated on the y-axis. Data, mean±SEM; n=5 biologically independent samples, two-way ANOVA Sidak’s test. d, Immunoblotting analysis of enriched extracellular vesicles (EVs) present in the supernatants of ATG9AHuh7-WT and ATG9AHuh7-KO cells (Dig) expressing GFP or MyrPalm-EGFP and immunoblotted for GFP and CD63 (one of 3 independent experiments). e, Example of HCM images showing PM permeabilization (PI) of ATG9AHuh7-WT and ATG9AHuh7-KO cells exposed to virulent Mycobacterium tuberculosis Erdman; mutliplicity of infection (MOI; bacteria to cells ratio) 0 (Ctrl), 5 or 10. red masks, computer-identified PI+ nuclei. Scale bars, 10 µm. f, HCM quantification of PM permeabilization (PI) in ATG9AHuh7-WT and ATG9AHuh7-KO cells exposed to a nonvirulent derivative of M. tuberculosis subspecies bovis (BCG), mutliplicity of infection (MOI) 0 (Ctrl) or 10. Data, % of cells positive for PI (mean±SEM; n=6 biologically independent samples, two-way ANOVA Sidak’s test). g, Example of HCM images relative to f. Scale bars, 10 µm.
Supplementary information
Supplementary Tables
Supplementary Table 1: ATG9A partners and quantitative changes identified by proximity biotinylation and LC-MS/MS (Data Dependent Acquisition, DDA) in 293TAPEX2-ATG9A cells subjected to PM damage by digitonin (Dig). Data, spectral counts and fold change relative to control (full medium); n=3 independent biological samples per group (digitonin or full medium control)
Supplementary Table 2: List of ATG9A partners with an infinite fold change upon PM damage with digitonin. Values derived from data in Table 1.
Supplementary Table 3: ATG9A partners and quantitative changes identified by proximity biotinylation and LC-MS/MS (Data Dependent Acquisition, DDA) in 293TAPEX2-ATG9A cells subjected to PM damage by Streptolysin O (SLO). Data, spectral counts and fold change relative to control (no SLO); n=3 independent biological samples per group (SLO or no SLO control.
Supplementary Table 4: List of ATG9A partners with an infinite fold change upon PM damage by SLO. Values derived from data in Table 3.
Supplementary Table 5: ATG9A partners and quantitative changes identified by proximity biotinylation and LC-MS/MS (Data Dependent Acquisition, DDA) in 293TAPEX2-ATG9A cells subjected to PM damage by glass bead injury (GBI). Data, spectral counts and fold change relative to control (no GBI); n=3 independent biological samples per group (GBI or no GBI control.
Supplementary Table 6: List of ATG9A partners with an infinite fold change upon PM damage by GBI. Values derived from data in Table 5.
Supplementary Table 7: Summary of notable ATG9A partners and associated spectral counts. Categories include ATG proteins, known ATG9A interactors, Rab and RabGAPs, and adaptor proteins. All values based on data in Table 1.
Supplementary Table 8: Summary of the ESCRT proteins detected by LC-MS/MS (DDA) in the proximity of ATG9A and associated spectral counts. All values based on data in Table 1.
Supplementary Table 9: Percent coverage of ALIX (PDCD6I) and TSG101 proteins identified as ATG9A partners in Table 1. The peptides highlighted in yellow in ALIX and TSG101 protein sequences are represented in LC-MS/MS spectra.
Supplementary Table 10: Notable ATG9A interactors from ESCRT, ATG/autophagy factors and annexin categories. Data Independent Acquisition (DIA). Cells were treated with EBSS, LLOMe or CCCP (3 biologically independent samples each) and normalized intensity fold change (compared to control conditions) represented. Also shown are numbers of peptides per prottein entity.
Supplementary Video 1
Live microscopy of ATG9A trafficking in cells subjected to laser damage of the PM. Huh7 cells expressing GFP–ATG9A (pink) were incubated in culture medium containing 160 µg ml–1 of PI (blue). After low-intensity laser wounding (slanted rectangle at the beginning of the sequence shows the ROI used for damage), the GFP–ATG9A profiles were monitored by live microscopy. Note PI entry into the cytoplasm at the site of laser-localized damage, its redistribution over time to and entrapment in the nucleus, and diffusion separation of the damaged areas from the rest of the cell coinciding with the line of GFP–ATG9A profiles at the base of the PI+-damaged area.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 1
Unprocessed western blots and/or gels.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 2
Unprocessed western blots and/or gels.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots and/or gels.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 5
Unprocessed western blots and/or gels.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 6
Unprocessed western blots and/or gels.
Source Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 1
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 2
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 3
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 7
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 8
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 9
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 10
Statistical source data.
Source Data Extended Data Fig. 10
Unprocessed western blots and/or gels.
Rights and permissions
About this article

Cite this article
Claude-Taupin, A., Jia, J., Bhujabal, Z. et al. ATG9A protects the plasma membrane from programmed and incidental permeabilization. Nat Cell Biol 23, 846–858 (2021). https://doi.org/10.1038/s41556-021-00706-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41556-021-00706-w
This article is cited by
-
Autophagy genes in biology and disease
Nature Reviews Genetics (2023)
-
Synaptic vesicle proteins and ATG9A self-organize in distinct vesicle phases within synapsin condensates
Nature Communications (2023)
-
AP-4-mediated axonal transport controls endocannabinoid production in neurons
Nature Communications (2022)
-
Letter to Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy
Translational Stroke Research (2022)
-
Response to Letter to Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy
Translational Stroke Research (2022)
