
Abstract
Lipid nanoparticles (LNPs) for the efficient delivery of drugs need to be designed for the particular administration route and type of drug. Here we report the design of LNPs for the efficient delivery of therapeutic RNAs to the lung via nebulization. We optimized the composition, molar ratios and structure of LNPs made of lipids, neutral or cationic helper lipids and poly(ethylene glycol) (PEG) by evaluating the performance of LNPs belonging to six clusters occupying extremes in chemical space, and then pooling the lead clusters and expanding their diversity. We found that a low (high) molar ratio of PEG improves the performance of LNPs with neutral (cationic) helper lipids, an identified and optimal LNP for low-dose messenger RNA delivery. Nebulized delivery of an mRNA encoding a broadly neutralizing antibody targeting haemagglutinin via the optimized LNP protected mice from a lethal challenge of the H1N1 subtype of influenza A virus, and delivered mRNA more efficiently than LNPs previously optimized for systemic delivery. A cluster approach to LNP design may facilitate the optimization of LNPs for other administration routes and therapeutics.
Similar content being viewed by others

Main
Once delivered into the cytoplasm, exogenously administered messenger RNA can express therapeutic nucleases, antigens, antibodies or other proteins1. However, these drugs are limited by the need to deliver mRNA into the cytosol without eliciting an immune response2. Laboratories have improved mRNA delivery in mice, rats, pigs and non-human primates using lipid nanoparticles (LNPs)3,4,5,6,7,8,9,10,11,12,13,14,15,16. Independent of their chemical structure, LNPs are often discovered using a common workflow. First, dozens to hundreds of nanoparticles are made with diverse chemical compositions17. Second, drug delivery mediated by these nanoparticles are quantified in vitro one by one. Third, in vitro data are used to select a small number of nanoparticles for in vivo studies. This approach is similar to a precedent set by the short interfering RNA (siRNA) delivery field. In three representative examples18,19,20, to identify LNPs that deliver siRNA systemically, 4,736 LNPs were tested in vitro, whereas only 14 were tested in vivo.
These and other studies reveal two key lessons we applied to nebulized mRNA delivery vehicles. First, all four components often used to create LNPs—ionizable or cationic lipid18, poly(ethylene glycol) (PEG)-lipid21, cholesterol22,23 and other lipids (for example, phospholipids)8,24—influence delivery. The resulting four-dimensional chemical space may therefore encompass 1010 or more distinct LNP formulations25, even without considering active targeting ligands such as antibodies, peptides or aptamers. Second, in vitro nanoparticle delivery can poorly predict in vivo nanoparticle delivery11,26. These lessons highlight the need for an easily adopted in vivo alternative to LNP discovery that is capable of efficiently and iteratively scanning diverse chemical space.
Such an alternative to LNP discovery would be useful if applied to nebulization-mediated LNP delivery to the lung. Nebulized nanoparticles have been considered as potential therapies for genetic disease27 as well as infectious disease, since these drugs could encode nucleases, vaccines or antibodies targeting severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or its entry into human cells28. Given that LNPs can be manufactured at human scales, have been Food and Drug Administration (FDA) approved29 and have safely delivered mRNA encoding coronavirus disease 2019 (COVID-19) vaccines30,31, they constitute an alternative to large polymeric32 and polymer–peptide-based33 nanoparticles that have delivered mRNA to the lung following nebulization. For example, although large (for example, 25 kDa) polymers were shown to enable nebulized DNA delivery up to 20 years ago34,35, nebulized gene therapies using these polymers have not been approved by the FDA.
Demonstrating that an LNP can deliver mRNA to the lungs at low doses would open up a potentially clinically relevant chemical space for lung delivery. However, this task remains difficult since LNPs that deliver mRNA after systemic administration may be different from LNPs that deliver mRNA to the lungs via nebulization. For example, although state-of-the-art nebulizers are designed to gently aerosolize the therapeutic, nanoparticles still experience shearing forces that can disrupt the structure of the vehicle–drug formulation. A second reason nebulization of mRNA differs from systemic delivery is the biology of the airway. When nanoparticles are administered via nebulization, the cells, proteins, biomolecules and physical barriers with which they interact36,37 are distinct from those with which they interact in the bloodstream38. Cells in the airways are also heterogenous39, which may alter delivery.
Driven by the need for lung mRNA delivery vehicles, the lack of established LNP design principles and the absence of a workflow to identify design principles, we report an in vivo cluster-based iterative screening approach to identify LNP chemical traits that promote lung delivery. We found that (1) PEG is required for LNP formulation and (2) the combination of PEG molarity and helper lipid structure and charge influences delivery; this second fact underscores the need to understand multivariate LNP design space. Here we identify a nanoparticle named nebulized lung delivery 1 (NLD1). The structure of NLD1 is distinct from previously reported nanoparticles for nebulized delivery. Specifically, NLD1 is designed with a high amount of PEG-lipid and cationic helper lipid. While LNP-mediated nucleic acid delivery is often studied with low PEG-lipid molar ratios, we found that a greater amount of PEG within an LNP can improve mRNA delivery to the lungs via nebulization. We also found that NLD1 is more effective than clinically relevant LNPs previously optimized for systemic delivery. Finally, we demonstrate that NLD1 can be used to deliver mRNA encoding a broadly neutralizing antibody protecting mice from a lethal challenge of H1N1 influenza.
Results
On the basis of the large LNP chemical space and the poor relationship between in vitro and in vivo delivery, we designed a cluster-based workflow to meet three key criteria. We reasoned that the workflow must (1) be easily set up with equipment commonly used in nanoparticle laboratories, (2) quantify functional mRNA delivery (that is, mRNA translated into functional protein) in vivo and (3) be capable of scanning a diverse LNP chemical space iteratively. We previously reported functional in vivo LNP screening systems utilizing DNA barcodes9,10,11,40,41. Our cluster-based approach is distinct in two ways. First, it does not require next-generation sequencing, which may aid nanoparticle laboratories that do not regularly sequence DNA. Second, it uses a multistep LNP design process to select LNP formulations detailed below. Inspired by statistical design of experiment methodologies42,43, our cluster-based approach allows us to manipulate several inputs (LNP traits) to determine their effect on in vivo RNA delivery.
In this cluster-based approach (Fig. 1), we first identified several (in this schematic, three) hypotheses relating a specific LNP chemical trait to in vivo delivery. Second, we treated each hypothesis as an axis in an N-dimensional chemical space. Third, we formulated 8 to 12 LNPs at a given extreme of each axis; we termed these ‘extreme groups’. Fourth, we applied a three-tiered selection to each extreme group: did LNPs within the extreme group form small (<200 nm hydrodynamic diameter), stable (low dispersity, single peak on dynamic light scattering (DLS)) nanoparticles? If yes, did the extreme group remain stable after the LNPs were pooled together? If yes, did the pooled LNPs from the extreme group functionally deliver mRNA into the lungs following nebulization (Fig. 1a)? Fifth, we ‘expanded and combined’ promising extreme groups by formulating LNPs near the intersection of these groups. Sixth, we individually tested all these newly formulated LNPs, assaying whether they formed small, stable LNPs, and whether they delivered mRNA in the lung following nebulization (Fig. 1b). Seventh, we used all these data, including data from groups that failed, to design subsequent nanoparticles. By generating distinct hypotheses, using extreme groups to test these hypotheses, then focusing on promising chemical spaces, we scanned a diverse multidimensional chemical space in vivo, offering an alternative to traditional in vitro-based screens.

a, An individual chemical trait is treated as an axis. Multiple chemical traits, three in this example, are evaluated simultaneously. Small groups of LNPs at the extremes of this multidimensional chemical space are characterized by DLS and luciferase mRNA delivery following nebulization. b, Any promising extreme group is then locally expanded; all the LNPs within this expanded space are characterized individually. The data from this process are used to inform subsequent LNP designs.
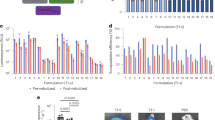
Previous data8,22,24,44,45 led us to hypothesize that four variables influenced nebulized LNP delivery: (1) the amount of PEG added to the LNP, (2) the structure of the lipid-PEG, (3) the charge of the phospholipid and (4) the presence or absence of cholesterol. We designed six extreme groups, varying molar ratios, components and charge (Fig. 2a). In all six, we formulated 8 to 12 LNPs with the compound 7C1 (Fig. 2b), an oligomer–lipid conjugate19, thereby generating 56 distinct LNPs (Supplementary Fig. 1a,b). LNPs based on 7C1 have delivered siRNA in non-human primates46.

a–e, We varied the structure and amount of PEG-lipid, the phospholipid charge and cholesterol within the LNP (a), while the 7C1 compound was kept constant (b). In this way, we created and analyzed six extreme groups (c) using LNP quality control (QC) cutoffs (d), of which three consistently led to stable LNPs as measured by the hydrodynamic diameter of all LNPs for all six groups. e, In groups D–F, we found that the diameter of the pooled LNPs was within the range of the individual LNPs making up the groups, suggesting their stability. The dotted grey line represents a 200 nm QC cutoff. f,g, Luciferase (Luc) luminescence images (f) and quantification (g) in lungs 48 h after the three groups were nebulized and administered to mice, as compared to a negative control (–Ctrl). Unless specified otherwise, *P = 0.0098, one-way analysis of variance (ANOVA), average ± s.e.m., n = 2 mice per group, biological replicates shown.
Using microfluidics47, we formulated LNPs with mRNA encoding a glycosylphosphatidylinositol-anchored nanoluciferase (AncNanoLuc) mRNA at an LNP component:mRNA mass ratio equal to 10:1. AncNanoLuc-encoding mRNA was used in place of traditional luciferase since its glycosylphosphatidylinositol anchor allows for the expressed protein to accumulate on the cell surface, which increases the sensitivity of the assay. We then analyzed all the LNPs with DLS. Extreme groups A, B and C failed our control criteria (Fig. 2c), whereas extreme groups D, E and F met the criteria (Fig. 2d). Specifically, many LNPs from extreme groups A, B and C were unstable and had hydrodynamic diameters greater than 200 nm (Fig. 2e and Supplementary Fig. 1a,b). Group A had very low percentages of PEG-lipid and groups B and C did not have PEG-lipid. We therefore identified our first design rule: PEG-lipid is critical to forming stable 7C1-based LNPs. As an additional control for LNP stability, we pooled the LNPs in extreme group D, measured the hydrodynamic diameter of the pool, and found it to be within the range of diameters making up the pool; the same control was analyzed for extreme groups E and F (Fig. 2e). Once pooled, the clusters were nebulized and administered at 43 μg mRNA per mouse (for example, 6 µg per mouse per particle on average, for n = 7 particles per mouse). Forty-eight hours later, we extracted the lungs, submerged the organs in furimazine, quantified luminescence using an IVIS in vivo imaging system, and calculated the fold change in lung luminescence flux relative to the background. Luminescence was significantly higher in lungs transfected with extreme group E, followed by D and then F (Fig. 2f,g). These results indicate that cholesterol and helper lipids are not crucial for initial LNP formulation, but they may improve functional LNP delivery.
We then expanded a combination of extreme groups D and E. Cluster D contained cationic helper lipids, whereas cluster E contained a neutral lipid and a higher percentage of PEG-lipid. We therefore created 27 LNPs using 3 charged helper lipids (1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA) and dimethyldioctadecylammonium (18:0 DDAB)), C14PEG2000, cholesterol, 7C1 and 9 different molar ratios of PEG (Fig. 3a and Supplementary Fig. 2a,b). Molar ratios of PEG varied from low (2.5, 6.5 and 15%) to high (25, 30, 35, 45, 55 and 60%). LNPs were again formulated with microfluidics to carry mRNA encoding AncNanoLuc at an LNP component:mRNA mass ratio of 10:1. Of the 27 LNPs, 16 formed stable monodisperse LNPs with hydrodynamic diameters less than 200 nm (Fig. 3b). We then analyzed whether the average hydrodynamic diameter and polydispersity index of LNPs varied with the cationic lipid (Fig. 3c and Supplementary Fig. 2c) or the lipid-PEG molar ratio (Fig. 3d and Supplementary Fig. 2d). LNPs formulated with less PEG-lipid were larger, which may be a useful way to roughly tune LNP size. We then nebulized and administered the 16 stable LNPs at the low dose of 15 μg mRNA per mouse. Forty-eight hours later, we isolated lungs and quantified luciferase luminescence. We observed a non-significant increase in luminescence when LNPs were formulated with 18:0 DDAB compared with DOTAP or DOTMA (Fig. 3e). Interestingly, when we plotted luminescence as a function of the PEG-lipid mole percentage, we observed that LNPs formulated with higher amounts of PEG tended to increase luminescence (Fig. 3f,g). Additionally, we examined the PEG-lipid mole percentage in the five top- and bottom-performing LNPs. We found that the top five LNPs contained high molar percentages of PEG, whereas the bottom five LNPs typically consisted of low molar percentages of PEG (Fig. 3h,i). The data suggest that the amount of PEG is important for formulations and that it affects delivery efficiency. These data led us to a second design rule: a combination of cationic helper lipids and high molar percentages of PEG can lead to increased mRNA delivery after nebulization (Fig. 3j).

a, LNPs were formulated with one of three cationic lipids, a PEG-lipid, cholesterol and the compound 7C1. b, LNPs with diameter less than 200 nm as measured by DLS were tested individually in mice. c,d, LNP diameter as a function of helper lipid (c) and PEG molarity (d). *P = 0.0018, two-tailed t-test. e–g, Luminescence of lungs isolated from mice relative to an untreated control (CTRL) 48 h after administration, plotted as a function of charged lipid (e) and PEG molarity (f,g). LNPs with cationic lipids, coupled with high PEG molarity, delivered more mRNA to lungs than LNPs with low PEG molarity (f), with notably high delivery with 55% PEG molarity (g). *P = 0.0346 (f), *P = 0.0201 (g), one-way ANOVA, average ± s.e.m. h–j, PEG molarity used in this study and that found in the best- and worst-performing LNPs ranked by lung luminescence. Unless specified otherwise, ****P < 0.0001, ***P = 0.0002, **P = 0.0026, *P = 0.0034, one-way ANOVA, average ± s.e.m., n = 2 mice per group, biological replicates shown. Circles represent LNPs (a–d) or biological replicates (f,g,j).
We surmised LNP interactions in the lungs can be driven by several forces including PEG-based hydrophilic interactions as well as charge48. We therefore formulated 26 additional LNPs using 2 phospholipids (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)), 2 PEG-lipids with varied tail lengths (C14PEG2000 and C18PEG2000), cholesterol and 7C1 (Fig. 4a and Supplementary Fig. 3a). Since LNPs with higher PEG molar ratios were smaller than LNPs with lower PEG molar ratios, this experimental setup also allowed us to evaluate whether LNP size, rather than LNP chemistry, caused differences in LNP delivery. Again, LNPs were formulated with AncNanoLuc mRNA at a mass ratio of 10:1, and LNPs were only nebulized if they were monodispersed with a hydrodynamic diameter less than 200 nm (Fig. 4b). We plotted the average hydrodynamic diameter and polydispersity index as a function of neutral lipid (Fig. 4c and Supplementary Fig. 3b), PEG amount (Fig. 4d and Supplementary Fig. 3c) and PEG tail length (Fig. 4e and Supplementary Fig. 3d). Notably, we observed a similar trend with LNP size; LNPs formulated with low PEG-lipid molar ratios tended to be larger than LNPs with high PEG-lipid molar ratios (Fig. 4d). We also observed a trend with polydispersity index and DSPC or low amounts of PEG-lipid (Supplementary Fig. 3b,c).

a, LNPs were formulated with one of two neutral lipids, one of two distinct PEG-lipids, cholesterol and the compound 7C1. b, LNPs with hydrodynamic diameter less than 200 nm as measured by DLS were tested individually in mice. c–e, LNP diameter as a function of helper lipid (c), PEG molarity (d) and PEG-lipid tail length (e). f–i, Luminescence of lungs isolated from mice relative to an untreated control 48 h after administration, plotted as a function of neutral helper lipid (f), PEG-lipid tail length (g) and PEG molarity, defined as either high and low (h) or by mole percent (i). *P = 0.024 (g), **P = 0.0089 (h), *P = 0.020 (i), one-way ANOVA, average ± s.e.m. LNPs with neutral lipids, coupled with low PEG molarity, delivered more mRNA to lungs than LNPs with high PEG molarity. j–l, PEG molarity used in this study (j), the PEG molarity found in the best- and worst-performing LNPs (k), and lung luminescence (l). For l, unless specified otherwise, ****P < 0.0001, ***P = 0.0005, **P = 0.002, *P = 0.018, *P = 0.027, *P = 0.010, *P = 0.012, *P = 0.048 (*P values arranged from left to right of l), one-way ANOVA, average ± s.e.m., n = 2 mice per group, biological replicates shown.
LNPs that passed the quality control assay were nebulized at a dose of 13.5 μg mRNA per mouse. Forty-eight hours later, we isolated lungs and quantified luminescence. Consistent with our previous results (Fig. 3e), we did not observe substantial differences when delivery was plotted as a function of these helper lipids (Fig. 4f). In addition, we again observed that delivery changed with PEG; specifically, LNPs formulated with a C18 tail tended to have greater mRNA expression than LNPs formulated with a C14 tail (Fig. 4g). Moreover, LNPs formulated with lower molar percentages of amounts of PEG, specifically LNPs containing a 15% PEG molar amount, exhibited higher luciferase expression (Fig. 4h,i). We then analyzed the five top- and bottom-performing LNPs, ranked by luminescence. We found that the top five LNPs contained lower amounts of PEG, while the bottom five LNPs contained higher amounts of PEG (Fig. 4j,k). Additionally, more of the top five LNPs contained PEG with a C18 tail, while all bottom five LNPs contained PEG with a C14 tail (Supplementary Fig. 3e). We again found that the molar percentage of the components influenced the efficiency of an LNP (Fig. 4l). These data led us to the preliminary conclusion that LNP chemistry played a more important role in nebulized delivery than LNP size within the ranges we tested. Future work is needed to understand whether LNP size influences delivery across larger ranges. Together, these data suggest a strong correlation between PEG-lipid ratio and helper lipid choice, and therefore led us to a third design rule: LNPs formulated with a neutral phospholipid lipid require less PEG than LNPs formulated with a cationic helper lipid.
Using our three design rules (Fig. 5a), we selected an LNP named NLD1 for further analysis (Fig. 5b). NLD1 formed small LNPs that were stable for 4 d when stored at 4 °C in 1× PBS (Fig. 5c). We formulated NLD1 to carry AncNanoLuc mRNA, nebulized it and administered it to mice at a dose of 20 µg per mouse. We compared mRNA delivery to three controls: (1) 7C3, a previously reported LNP that also uses the 7C1 lipid, (2) cKK-E12, which delivers RNA to the liver in non-human primates in low doses49 and (3) MC3, which has been used in an FDA-approved drug29 (Supplementary Fig. 4a). They therefore provide an opportunity to evaluate whether design rules for systemic mRNA delivery can be applied to nebulized delivery. Forty-eight hours after administering the LNPs, we quantified luminescence and found it to be significantly higher in lungs treated with NLD1 than in lungs in all three controls (Fig. 5d and Supplementary Fig. 4b,c). As an additional control, we evaluated whether MC3- and cKK-E12-based LNPs outperformed NLD1 when formulated with the NLD1 molar ratio. The control LNPs did not form stable nanoparticles, demonstrating that NLD1 delivers mRNA to the lungs more efficiently. We evaluated delivery to off-target organs, and observed no luminescence in other tissues after NLD1 treatment (Supplementary Fig. 4d,e).

a, LNP delivery to the lung can be improved by carefully selecting the type and molar amount of PEG-lipid. b, NLD1 was designed with a cationic lipid and high amount of PEG-lipid. c, NLD1 forms small, stable LNPs over time, average ± s.t.d., n = 3 replicates per group. d, NLD1 delivers more mRNA to lungs at low doses in vivo than leading LNPs previously optimized for systemic mRNA delivery. ***P < 0.0007, one-way ANOVA, average ± s.e.m., n = 5 mice per group, biological replicates shown. e,f, A total of 547 inflammatory genes were examined after exposure to NLD1. NLD1 was well tolerated in vivo (e) as compared to an LPS control (f). Each grey or purple dot represents a gene, and light grey dotted vertical lines indicate >2-fold change in gene expression. The dotted horizontal line indicates a P value of 0.05. g,h, NLD1 carrying AncNanoLuc was administered at a dose of 20 µg per mouse. Protein expression was quantified (g) and imaged (h) over several days. ****P < 0.0001, *P = 0.021, one-way ANOVA, average ± s.e.m., n = 3 mice per group, biological replicates shown. i, NLD1 therapeutic treatment regimen for H1N1 study. Mice were treated with two NLD1 treatments (total dose, 100 µg per mouse) before administering 1.5× LD50 dose of PR8 virus. j,k, Mouse weights were taken daily for up to 14 d post infection. All mice treated with NLD1 recovered on the basis of weights. Out of 6 control mice treated with H1N1, 5 mice lost 15% or more of body weight and were euthanized. n = 6 mice per group, biological replicates shown.
We then evaluated the importance of LNP biophysical stability during the nebulization process. Specifically, we hypothesized that LNPs that survive nebulization ‘least damaged’ may deliver mRNA more effectively. We therefore measured the stability of NLD1, 7C3, cKK-E12 and MC3 before and after nebulization. By comparing the relative size of different DLS peaks, we found that NLD1 was more stable than 7C3, cKK-E12 and MC3 after nebulization (Supplementary Fig. 5a–d). We compared LNP morphology before and after nebulization using transmission electron microscopy (TEM) and found the results to be consistent with our DLS analysis (Supplementary Fig. 5e). These data suggest that design rules for LNP systemic delivery may not select LNPs that are sufficiently stable for nebulization. Taken together, these results underscore the value of designing a system to quickly uncover design rules specific for nebulization.
To understand the biodistribution profile of NLD1 in the lungs, we performed fluorescence in situ hybridization (FISH) to visualize the delivered mRNA. We nebulized mice with 100 µg NLD1 carrying AncNanoLuc mRNA. After 4 h, animals were killed, lungs were isolated and tissues were prepared for imaging. We observed broad delivery of mRNA throughout lung sections, with a uniform punctate pattern (Supplementary Fig. 6a). The probes displayed high specificity for AncNanoLuc mRNA with minimal background staining (Supplementary Fig. 6b). Treated lungs displayed increased mRNA density at the periphery of the lung sections in the more terminal alveolar spaces. In contrast, we observed decreased mRNA density towards the centre of the sections nearer to larger air spaces.
Next we analyzed mRNA uptake in four of the major epithelial cell types in the lung50. We included probes against AncNanoLuc and two markers for bronchial epithelial cells (Supplementary Fig. 6c). We used probes against Foxj1 and Scgb1a1 mRNA to mark ciliated and club bronchial epithelial cells, respectively51. We observed mRNA delivery to these cell types. In separate staining panels, we included probes against Pdpn and Sftpc mRNA to mark alveolar type I and II cells, respectively (Supplementary Fig. 6d,e). We detected uptake of AncNanoLuc mRNA in these cell types throughout the lung as well. While we only included probes against epithelial cells, there is probable delivery to other cell types, such as alveolar macrophages. Future studies are needed to fully characterize mRNA uptake across more diverse cell types. However, taken with the biodistribution data, we believe these images demonstrate that NLD1 delivers mRNA broadly throughout the lung and results in uptake by the four major epithelial cell subtypes. Both of these properties are critical to achieving a strong therapeutic effect in the lung and represent opportunities for further optimization.
We analyzed NLD1 in vivo tolerability by measuring how the expression of 547 inflammatory genes (Supplementary Table 1) changed after exposure to NLD1. Mice were treated with 50 µg mRNA via nebulization; 4 h later, lungs were isolated and gene expression was measured. We chose this timepoint because RNA therapies elicit strong early immune responses that typically resolve by later timepoints such as 24 h. Thus, we were more likely to observe an immune response at an early timepoint. Relative to untreated mice, only 6 of 547 genes significantly (P < 0.05) increased by more than twofold (Fig. 5e), demonstrating that NLD1 was well tolerated, even at early timepoints. As control, we separately administered mild-dose lipopolysaccharides (LPS), and found that 86 genes significantly increased by twofold (Fig. 5f). Additionally, no significant weight loss was observed in mice 24 h after exposure to NLD1 (Supplementary Fig. 7a).
We then evaluated whether NLD1 efficiently delivered mRNA encoding for a therapeutic antibody. We chose mRNA encoding membrane-anchored FI6 (aFI6), an antibody that binds haemagglutinin, neutralizing 16 subtypes of influenza A viruses52. To optimize our dosing regimen, we evaluated the kinetics of membrane-anchored-protein expression following NLD1 nebulization. After formulating AncNanoLuc into NLD1, we measured luminescence at timepoints between 6 h and 10 d. We observed peak luminescence 2 d after nebulization and lower luminescence up to 7 d after nebulization (Fig. 5g,h). We then investigated whether NLD1 could be readministered and found that luminescence increased with the number of administrations (Supplementary Fig. 7b). On the basis of these data, we designed a treatment regimen for influenza A virus subtype H1N1 (Fig. 5i). Specifically, we nebulized mRNA encoding aFI6, and administered it 3 d and 2 d before inoculating mice with influenza A/Puerto Rico/8/1934 (PR8) at 1.5× LD50, where LD50 is the dose at which 50% of the control animals die after exposure. We then monitored disease progression by measuring mouse weights over time. Substantial weight loss post treatment with influenza is considered a marker to predict eventual death and trigger euthanasia53. As expected with the administered viral dose, five of the six control mice died after progressively losing weight (Fig. 5j,k). In contrast, all six animals treated with NLD1 survived. These results demonstrate the rapid therapeutic translation of this cluster-based screening system to treat a dangerous infectious disease.
Discussion
Nebulized mRNA delivery is well positioned to treat diverse lung diseases54. However, nebulized RNA therapies will require clinically relevant delivery. The need for delivery is supported by a recent nebulized gene therapy clinical trial that, unfortunately, did not result in sufficient efficacy27; notably, the authors highlighted the need for improved delivery.
Concurrent with these advances in lung biology have been improvements in LNP design for systemic RNA delivery, which led to an FDA-approved RNA drug29. Unlike early LNPs, which had poor therapeutic windows and required high doses for effective delivery, LNPs reported as early as 2012 (ref. 55) are scalable to human doses and have clinically relevant therapeutic windows56. However, most LNPs have been optimized for systemic delivery. This led to two key questions: First, can LNPs be redesigned for nebulized lung mRNA delivery? Second, how similar or dissimilar will design rules for nebulized LNPs be to design rules for systemic LNPs? By testing our NLD1 against LNPs previously optimized for systemic delivery, our initial data suggest that LNP design rules for nebulized delivery can be distinct from design rules for systemic delivery. It is too early to understand whether this conclusion is consistent across all LNP chemistries; however, these data support a more important general concept: answering these two key questions requires new LNP discovery workflows. Specifically, on the basis of data demonstrating that many LNP chemical traits influence delivery and that in vitro delivery assays do not predict in vivo delivery, we reasoned that an ideal way to discover LNPs for nebulized delivery must be easy to set up, enable chemically diverse LNPs to be tested quickly, and be performed directly in vivo. This in vivo screening approach identified a nebulized LNP that produced a therapeutically efficacious level of protein. However, future studies should be conducted to understand whether NLD1-based therapies can treat viral challenges that occur on different timescales relative to the NLD1 treatment. Although we did not test other nanoparticle classes (for example, peptides or inorganics), we believe this workflow may be used to study them as well.
By testing a series of chemically distinct structures at low doses in vivo, we identified several design rules for nebulized LNPs. Thousands of additional LNPs will need to be tested before we can fully understand whether these specific rules are applicable to all LNPs. However, the data provide compelling evidence that PEG molarity and structure are critical when designing LNPs. For example, using the same 7C1 lipid, we found a 28-fold difference in delivery between the best- and worst-performing LNPs simply by changing the formulation ratio. Additionally, PEG density can alter PEG configuration57,58. Thus, PEG-lipid conformation may affect the efficiency of mRNA delivery to the lungs; future studies should explore the relationship between PEG conformation and nebulized delivery. We also found that the inclusion of a cationic lipid might be important for lung delivery. This hypothesis is consistent with recent work8. Using a method known as selective organ targeting, the researchers demonstrate that the inclusion of charged phospholipids could shift LNP delivery away from hepatocytes and facilitate lung delivery8.
One additional unanswered question we hope to address is the mechanism by which changing the PEG alters nebulized lung delivery. The relationship between PEG density and systemic delivery of nanoparticles has been studied. Many clinically relevant LNP formulations contain PEG-lipids29,49,59 to shield them from opsonization or increase circulation time45,57,60,61. In addition, nanoparticles developed for lung delivery have been formulated with PEG-lipids to stabilize the particles and reduce clearance44. However, there remains an important opportunity to understand how PEG density influences nebulized lung delivery. Our first hypothesis is that the harsh conditions of nebulization predominate; specifically, all LNP formulations are damaged during nebulization, and thus the LNPs that survive this process with the least damage deliver mRNA effectively. This hypothesis is consistent with our results that NLD1 was more stable than several control LNPs, which it outperformed in nebulization experiments. This hypothesis also aligns with our results demonstrating that LNPs with high PEG molarity and cationic lipids delivered more mRNA in vivo than LNPs with low PEG molarity; we reasoned that LNPs damaged during nebulization could aggregate, and therefore the combination of steric (PEG) and electrostatic (cationic) interactions, which promote self-assembly, decreased this aggregation. However, our data demonstrating that neutral LNPs deliver more mRNA at low PEG molar amounts do not support this hypothesis. A second hypothesis is that lung biology predominates; more specifically, stable LNP formulations survive nebulization, but are differentially affected by mucus and other physiological barriers within the lung. The most likely hypothesis is that both nebulization and lung biology alter delivery, and the extent to which one predominates over the other varies with the LNP. This is difficult to deconvolute in vivo; we are currently attempting to design experiments to test these hypotheses.
It is important to acknowledge several additional limitations of this study. First, our results focused on 7C1-containing LNPs, two PEG-lipids and several helper lipids. In future iterations, we will include more diverse ionizable lipids, PEG-lipids and helper lipids, at additional molar ratios. Further work should be done to understand other relationships between LNP composition and nebulized delivery. Second, it is possible that the protein expression kinetics we observed may vary with the mRNA payload. Additionally, one unexplored question in the field is whether protein expression kinetics can be altered by immune response. More extensive studies need to be done to understand the relationship between payload and delivery. Third, these studies were limited to mice. The anatomy of a mouse lung may not be an optimal model for nanoparticle delivery in a human lung. It is possible that these results may not be replicated in larger animals such as non-human primates. Fourth, our data do not exclude the possibility that LNP size may play a critical role in delivery for larger nanoparticles. Similarly, it may be possible that zeta potential influences LNP delivery. Fifth, several other factors are involved in pulmonary RNA delivery in diseased states. Specifically, cystic fibrosis causes severe mucus barriers within the lungs62. The mucus penetration of an LNP formulation will be important for the treatment of cystic fibrosis63. Finally, while we demonstrated that nebulized NLD1 could efficiently deliver therapies before an infection, future studies will need to assess whether this vehicle and construct can be used after an infection. Despite these limitations, we believe these data provide evidence that LNP design parameters depend on the route of administration, that a cluster-based approach can efficiently identify nanoparticles that deliver drugs in vivo, and that an mRNA encoding an antibody can protect against an otherwise lethal H1N1 flu challenge.
Methods
Synthesis of anchored nanoluciferase-encoding mRNA
Messenger RNA was synthesized as described previously64. Briefly, the anchored nanoluciferase sequence was ordered as a DNA gBlock (Integrated DNA Technologies) containing a 5′ untranslated region with Kozak sequence, a 3′ untranslated region derived from the mouse alpha globin sequence, and extensions to allow for Gibson assembly. The sequence was human codon optimized using the Integrated DNA Technologies website. The sequences of the glycosylphosphatidylinositol anchor and nanoluciferase have been previously stated64. The gBlock was then cloned into a PCR-amplified pMA7 vector through Gibson assembly using NEBuilder (New England BioLabs, NEB) with three molar excess of insert. All reaction transcripts were purified with 0.8% agarose gel before assembly reaction. Subsequent plasmids from each colony were Sanger-sequenced to ensure desired sequence fidelity. Plasmids were linearized with NotI-HF (NEB) overnight at 37 °C. Linearized templates were purified using ammonium acetate (Thermo Fisher Scientific) precipitation before being rehydrated with nuclease-free water. In vitro transcription was performed overnight at 37 °C using the HiScribe T7 kit (NEB) following the manufacturer’s instructions (N1-methyl-pseudouridine modified). The RNA product was treated with DNase I (Aldevron) for 30 min to remove the template, and purified by lithium chloride precipitation (Thermo Fisher Scientific). RNA was heat-denatured at 65 °C for 10 min before being capped with a Cap1 structure using guanylyl transferase (Aldevron) and 2′-O-methyltransferase (Aldevron). Transcripts were then polyadenylated enzymatically (Aldevron). Messenger RNA was then purified by lithium chloride precipitation, treated with alkaline phosphatase (NEB), and purified again. Concentrations were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific). mRNA stock concentrations were 3–5 mg ml–1. Purified RNA products were analyzed by gel electrophoresis to ensure purity.
Nanoparticle synthesis
7C1 and cKK-E12 were synthesized as previously described19,49. Detailed synthesis procedures are in Supplementary Fig. 8. d-Lin-MC3-DMA (MC3) was purchased from MedKoo Biosciences.
Nanoparticle formulation
Nanoparticles were formulated using a microfluidic device as previously described19. Luciferase mRNAs were diluted in 10 mM citrate buffer (Teknova). 7C1, PEG-lipids (1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000]), cholesterols and helper lipids (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, 1,2-distearoyl-sn-glycero-3-phosphocholine, 1,2-dioleoyl-3-trimethylammonium-propane, 1,2-di-O-octadecenyl-3-trimethylammonium propane and dimethyldioctadecylammonium) were diluted in 100% ethanol. All PEGs, cholesterols and helper lipids were purchased from Avanti Lipids. Citrate and ethanol phases were combined in a microfluidic device using syringes (Hamilton Company) at a flow rate of 3:1.
Nanoparticle characterization
The diameter and dispersity of the LNPs were measured using DLS (DynaPro Plate Reader II, Wyatt). LNPs were diluted in sterile 1× PBS and analyzed. To avoid using unstable LNPs and enable sterile purification using a 0.22 μm filter, LNPs were included only if they met three criteria: diameter >20 nm, diameter <200 nm and correlation function with 1 inflection point. For screens, particles that met these criteria were pooled into respective groups. Particles were dialysed in 20 kD dialysis cassettes (Thermo Fisher Scientific). The nanoparticle concentration was determined using NanoDrop (Thermo Fisher Scientific).
Animal experiments
All animal experiments were performed in accordance with the Georgia Institute of Technology’s Institutional Animal Care and Use Committee. All animals were housed in the Georgia Institute of Technology Animal Facility. BALBc mice (BALB/cJ, 000651; Jackson Laboratories) were used in all experiments, with n = 2 to 6 mice per group.
Nebulization
Mice were loaded into a custom-built nose-only exposure system constructed of a clear PVC tee and animal restraints (CODA Small Mouse Holder, Kent Scientific). The custom built nose-only exposure systems were connected using a custom 3D-printed nosecone (3D Printing Tech) made of a flexible thermoplastic polyurethane material. The nebulizer (Aeroneb, Kent Scientific) was then placed on the upward facing port of the tee. Doses were added dropwise to the nebulizer at a rate of 50 µl per mouse per droplet. After each individual droplet was nebulized, the clear tee was inspected until the vaporized dose had cleared (approximately 15–45 s per drop). Droplets were added until the desired dose per animal was achieved. After the vapour had cleared following the last droplet, the mice were removed from the restraints.
Whole-organ imaging
Tissues were isolated 48 h after administration of LNPs, unless otherwise noted. To measure luminescence, mice were killed and organs were collected; organs were submerged in Nano-Glo luciferase assay substrate (Promega) for 5 min before being placed on solid black paper for imaging. Luminescence was measured using an IVIS imaging system (PerkinElmer) and quantified using LivingImage software (PerkinElmer).
TEM
The nanoparticles were loaded onto formvar-coated nickel grids (Electron Microscopy Sciences), followed by negative staining using a 2% phosphotungstic acid solution (Sigma). The morphology of the nanoparticles was imaged using HT7700 TEM (Hitachi) at 120 kV, coupled with a digital micrograph camera and software suite (Gatan).
Toxicity study
Mice were treated with 50 µg mRNA encapsulated in NLD1 or 5 µg LPS. Four hours later, tissues were isolated and flash frozen on dry ice. RNA was extracted and gene expression was examined using the nanoString nCounter FLEX. Gene expression profiles were analyzed with nanoString nSolver software. Tissues were handled and analyzed through the Emory Integrated Genomics Core at Emory University.
Tissue immunostaining
Mice were treated with 100 µg AnNanoLuc encapsulated in NLD1 or 1× PBS via nose-cone nebulization. Four hours later, tissues were isolated and fixed in 1 ml paraformaldehyde (4%) for 24 h. Tissues were paraffin embedded and sectioned through the Emory Winship Cancer Institute Cancer Tissue and Pathology Facility.
mRNA FISH analysis
Delivered AncNanoLuc mRNA and endogenous mRNA were visualized in tissues using RNAscope Multiplex Fluorescent Reagent Kit v2 (Advanced Cell Diagnostics, 323136) according to the manufacturer’s instructions. A custom probe set was designed against the AncNanoLuc mRNA sequence (ACD 879571). Foxj1 was used as a marker for ciliated bronchial epithelial cells (ACD 317091-C2), Scgb1a1 as a marker for club cells (ACD 420351-C3), Pdpn as a marker for alveolar type I cells (ACD 437771-C2), and Sftpc as a marker for alveolar type II cells (ACD 314101-C2). Images for cell-type staining were acquired using a Zeiss Plan-Apo 40×1.3 NA oil objective on an UltraVIEW spinning disk confocal microscope equipped with a Hamamatsu Flash 4.0v2 CMOS camera. Images for biodistribution analysis were acquired using a Zeiss Plan-Apo 20×0.8 NA air objective. Images were captured and processed using Volocity software (PerkinElmer).
H1N1 flu study
mRNA encoding for aFI6 was encapsulated in NLD1. Mice were dosed with 50 µg nebulized aFI6 mRNA per dose at 3 and 2 d before infection. Control mice were mock dosed with an equal volume of PBS. Mice were anaesthetized with isoflurane and infected with 1.5× LD50 of influenza H1N1 PR8, diluted in DMEM. For survival curves, mice weights were monitored until 14 d post infection.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. Data on the expression of inflammatory genes are available as Supplementary Information. The raw and analyzed datasets generated during the study are too large to be publicly shared, but they are available for research purposes from the corresponding authors on reasonable request.
References
Sahin, U., Kariko, K. & Tureci, O. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Hajj, K. A. & Whitehead, K. A. Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2, 17056 (2017).
Zhu, X. et al. Systemic mRNA therapy for the treatment of Fabry disease: preclinical studies in wild-type mice, Fabry mouse model, and wild-type non-human primates. Am. J. Hum. Genet. 104, 625–637 (2019).
Sabnis, S. et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 26, 1509–1519 (2018).
Bahl, K. et al. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol. Ther. 25, 1316–1327 (2017).
Richner, J. M. et al. Modified mRNA vaccines protect against Zika virus infection. Cell 168, 1114–1125.e10 (2017).
Liang, F. et al. Efficient targeting and activation of antigen-presenting cells in vivo after modified mRNA vaccine administration in rhesus macaques. Mol. Ther. 25, 2635–2647 (2017).
Cheng, Q. et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 15, 313–320 (2020).
Paunovska, K. et al. Nanoparticles containing oxidized cholesterol deliver mRNA to the liver microenvironment at clinically relevant doses. Adv. Mater. 31, e1807748 (2019).
Lokugamage, M. P. et al. Mild innate immune activation overrides efficient nanoparticle-mediated RNA delivery. Adv. Mater. 32, e1904905 (2019).
Sago, C. D. et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl Acad. Sci USA 115, E9944–E9952 (2018).
Miao, L. et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 37, 1174–1185 (2019).
Kranz, L. M. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016).
Zhang, X. et al. Biodegradable amino-ester nanomaterials for Cas9 mRNA delivery in vitro and in vivo. ACS Appl. Mater. Interfaces 9, 25481–25487 (2017).
Fenton, O. S. et al. Customizable lipid nanoparticle materials for the delivery of siRNAs and mRNAs. Angew. Chem. Int. Ed. 57, 13582–13586 (2018).
Kaczmarek, J. C. et al. Optimization of a degradable polymer-lipid nanoparticle for potent systemic delivery of mRNA to the lung endothelium and immune cells. Nano Lett. 18, 6449–6454 (2018).
Zhang, Y., Sun, C., Wang, C., Jankovic, K. E. & Dong, Y. Lipids and lipid derivatives for RNA delivery. Chem. Rev. https://doi.org/10.1021/acs.chemrev.1c00244 (2021).
Akinc, A. et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 26, 561–569 (2008).
Dahlman, J. E. et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat. Nano 9, 648–655 (2014).
Siegwart, D. J. et al. Combinatorial synthesis of chemically diverse core–shell nanoparticles for intracellular delivery. Proc. Natl Acad. Sci. USA 108, 12996–13001 (2011).
Mui, B. L. et al. Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol. Ther. Nucleic Acids 2, e139 (2013).
Patel, S. et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 11, 983 (2020).
Paunovska, K. et al. Analyzing 2000 in vivo drug delivery data points reveals cholesterol structure impacts nanoparticle delivery. ACS Nano 12, 8341–8349 (2018).
Ball, R. L., Hajj, K. A., Vizelman, J., Bajaj, P. & Whitehead, K. A. Lipid nanoparticle formulations for enhanced co-delivery of siRNA and mRNA. Nano Lett. 18, 3814–3822 (2018).
Lokugamage, M. P., Sago, C. D. & Dahlman, J. E. Testing thousands of nanoparticles in vivo using DNA barcodes. Curr. Opin. Biomed. Eng. 7, 1–8 (2018).
Paunovska, K. et al. A direct comparison of in vitro and in vivo nucleic acid delivery mediated by hundreds of nanoparticles reveals a weak correlation. Nano Lett. 18, 2148–2157 (2018).
Alton, E. et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 3, 684–691 (2015).
Dhama, K. et al. COVID-19, an emerging coronavirus infection: advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Hum. Vaccin. Immunother. 16, 1232–1238 (2020).
Akinc, A. et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 14, 1084–1087 (2019).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2020).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Patel, A. K. et al. Inhaled nanoformulated mRNA polyplexes for protein production in lung epithelium. Adv. Mater. 31, e1805116 (2019).
Guan, S. et al. Self-assembled peptide–poloxamine nanoparticles enable in vitro and in vivo genome restoration for cystic fibrosis. Nat. Nanotechnol. 14, 287–297 (2019).
Densmore, C. L. et al. Aerosol delivery of robust polyethyleneimine–DNA complexes for gene therapy and genetic immunization. Mol. Ther. 1, 180–188 (2000).
Rudolph, C. et al. Methodological optimization of polyethylenimine (PEI)-based gene delivery to the lungs of mice via aerosol application. J. Gene Med. 7, 59–66 (2005).
Yin, B. et al. Intrapulmonary cellular-level distribution of inhaled nanoparticles with defined functional groups and its correlations with protein corona and inflammatory response. ACS Nano 13, 14048–14069 (2019).
Raesch, S. S. et al. Proteomic and lipidomic analysis of nanoparticle corona upon contact with lung surfactant reveals differences in protein, but not lipid composition. ACS Nano 9, 11872–11885 (2015).
Monopoli, M. P., Aberg, C., Salvati, A. & Dawson, K. A. Biomolecular coronas provide the biological identity of nanosized materials. Nat. Nanotechnol. 7, 779–786 (2012).
Montoro, D. T. et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018).
Lokugamage, M. P., Sago, C. D., Gan, Z., Krupczak, B. R. & Dahlman, J. E. Constrained nanoparticles deliver siRNA and sgRNA to T cells in vivo without targeting ligands. Adv. Mater. 31, e1902251 (2019).
Sago, C. D. et al. Nanoparticles that deliver RNA to bone marrow identified by in vivo directed evolution. J. Am. Chem. Soc. 140, 17095–17105 (2018).
Kauffman, K. J. et al. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 15, 7300–7306 (2015).
Jones, B. & Nachtsheim, C. J. A class of three-level designs for definitive screening in the presence of second-order effects. J. Qual. Technol. 43, 1–15 (2011).
Kaczmarek, J. C. et al. Polymer-lipid nanoparticles for systemic delivery of mRNA to the lungs. Angew. Chem. Int. Ed. 55, 13808–13812 (2016).
Kumar, V. et al. Shielding of lipid nanoparticles for siRNA delivery: impact on physicochemical properties, cytokine induction, and efficacy. Mol. Ther. Nucleic Acids 3, e210 (2014).
Khan, O. F. et al. Endothelial siRNA delivery in nonhuman primates using ionizable low-molecular weight polymeric nanoparticles. Sci. Adv. 4, eaar8409 (2018).
Chen, D. et al. Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J. Am. Chem. Soc. 134, 6948–6951 (2012).
Huckaby, J. T. & Lai, S. K. PEGylation for enhancing nanoparticle diffusion in mucus. Adv. Drug Deliv. Rev. 124, 125–139 (2018).
Dong, Y. et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl Acad. Sci. USA 111, 3955–3960 (2014).
Raredon, M. S. B. et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 5, eaaw3851 (2019).
Strickland, D. H. et al. Persistent and compartmentalised disruption of dendritic cell subpopulations in the lung following influenza A virus infection. PLoS ONE 9, e111520 (2014).
Tiwari, P. M. et al. Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection. Nat. Commun. 9, 3999 (2018).
Gonzalez, A. J., Ijezie, E. C., Balemba, O. B. & Miura, T. A. Attenuation of influenza A virus disease severity by viral coinfection in a mouse model. J. Virol. 92, e00881-18 (2018).
Sahu, I., Haque, A., Weidensee, B., Weinmann, P. & Kormann, M. S. D. Recent developments in mRNA-based protein supplementation therapy to target lung diseases. Mol. Ther. 27, 803–823 (2019).
Jayaraman, M. et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. 51, 8529–8533 (2012).
Adams, D. et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21 (2018).
Hak, S. et al. The effect of nanoparticle polyethylene glycol surface density on ligand-directed tumor targeting studied in vivo by dual modality imaging. ACS Nano 6, 5648–5658 (2012).
Georgiev, G. A., Sarker, D. K., Al-Hanbali, O., Georgiev, G. D. & Lalchev, Z. Effects of poly (ethylene glycol) chains conformational transition on the properties of mixed DMPC/DMPE-PEG thin liquid films and monolayers. Colloids Surf. B 59, 184–193 (2007).
Semple, S. C. et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 28, 172–176 (2010).
Suk, J. S., Xu, Q., Kim, N., Hanes, J. & Ensign, L. M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 99, 28–51 (2016).
Pelaz, B. et al. Surface functionalization of nanoparticles with polyethylene glycol: effects on protein adsorption and cellular uptake. ACS Nano 9, 6996–7008 (2015).
Rubin, B. K. Mucus structure and properties in cystic fibrosis. Paediatr. Respir. Rev. 8, 4–7 (2007).
da Silva Sanchez, A., Paunovska, K., Cristian, A. & Dahlman, J. E. Treating cystic fibrosis with mRNA and CRISPR. Hum. Gene Ther. 31, 940–955 (2020).
Lindsay, K. E. et al. Aerosol delivery of synthetic mRNA to vaginal mucosa leads to durable expression of broadly neutralizing antibodies against HIV. Mol. Ther. 28, 805–819 (2020).
Acknowledgements
We thank the Emory Integrated Genomics Core (EIGC) at Emory University and the Emory Winship Cancer Institute Cancer Tissue and Pathology Core (CTP). J.E.D. thanks T. E. Shaw. The work was funded by Emily’s Entourage (awarded to J.E.D.), the National Institutes of Health (UG3-TR002855, awarded to J.E.D. and P.J.S.) and DARPA (PREPARE grant number HR00111920008, awarded to P.J.S. and J.E.D.).
Author information
Authors and Affiliations
Contributions
M.P.L., D.V., J.B., P.J.S. and J.E.D. conceived the experiments. M.P.L., D.V., J.B., Y.K., P.J.S. and J.E.D. designed the experiments. M.P.L., D.V., J.B., M.Z.C.H., L.R., E.S.E., H.E.P., H.N. and J.-K.Y. performed the experiments. M.P.L. and J.E.D. wrote the initial draft, which was edited by all authors. P.J.S. and J.E.D. oversaw the research.
Corresponding authors
Ethics declarations
Competing interests
J.E.D. is a consultant for Beam Therapeutics and GV. The other authors declare no competing interests.
Additional information
Peer review information Nature Biomedical Engineering thanks Dan Peer, Holun Wong and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary figures.
Supplementary Table 1
Expression of inflammatory genes.
Rights and permissions
About this article

Cite this article
Lokugamage, M.P., Vanover, D., Beyersdorf, J. et al. Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs. Nat Biomed Eng 5, 1059–1068 (2021). https://doi.org/10.1038/s41551-021-00786-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-021-00786-x
This article is cited by
-
Translational medicine for acute lung injury
Journal of Translational Medicine (2024)
-
Combinatorial development of nebulized mRNA delivery formulations for the lungs
Nature Nanotechnology (2024)
-
High-throughput barcoding of nanoparticles identifies cationic, degradable lipid-like materials for mRNA delivery to the lungs in female preclinical models
Nature Communications (2024)
-
Strategies for non-viral vectors targeting organs beyond the liver
Nature Nanotechnology (2024)
-
Nano–Bio Interactions: Exploring the Biological Behavior and the Fate of Lipid-Based Gene Delivery Systems
BioDrugs (2024)
