
Abstract
Computer-aided discovery of vaccine targets has become a cornerstone of rational vaccine design. In this article, I discuss how Machine Learning (ML) can inform and guide key computational steps in rational vaccine design concerned with the identification of B and T cell epitopes and correlates of protection. I provide examples of ML models, as well as types of data and predictions for which they are built. I argue that interpretable ML has the potential to improve the identification of immunogens also as a tool for scientific discovery, by helping elucidate the molecular processes underlying vaccine-induced immune responses. I outline the limitations and challenges in terms of data availability and method development that need to be addressed to bridge the gap between advances in ML predictions and their translational application to vaccine design.
Similar content being viewed by others

Introduction
Vaccine design is rapidly progressing from empirical to more systematic, rational strategies that benefit from computational predictions to assist the identification of pathogen regions targeted by the immune system (epitopes)1. Examples are reverse vaccinology approaches for the design of protein subunit vaccines2, which start from the genetic sequence of the pathogen and screen the possible antigens by their potential immunogenic and protective efficacy to select a few main targets. An accurate selection of targets is essential to imparting specific yet sufficiently immunogenic stimuli, while potentially avoiding antigens that do not elicit protective immunity. Since identifying epitope regions experimentally is resource and time-consuming, predictions in silico play the fundamental role of narrowing down the number of candidate targets to carry forward to in vitro and in vivo testing. As such, they will be key to rapid and cost-effective manufacturing of next-generation viral vectored or nucleic acid-based vaccines, first commercially developed during the recent Sars-Cov-2 pandemic3.
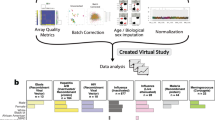
Computational screening of putative targets can be performed via several bioinformatic tools (see for example refs. 4,5), made available on the Immune Epitope Data Base (IEDB)6 and other web servers7,8,9. The methods primarily gaining momentum and prominence among these tools are the ones from Machine Learning (ML), the ensemble of algorithms and model architectures to learn from data in such a way as to better analyze them and make new predictions (see Box 1 for the basic ML terminology). Several ML-based reverse vaccinology pipelines have been developed7,8,9,10,11,12,13,14,15, with promising applications to the prediction of bacterial protective antigens7,10,11,12 and Sars-Cov-2 antigens8,13,14,15. ML can assist several stages of vaccine design16, but its application is particularly key to a fast and accurate target selection during the initial phase (Fig. 1a). Here ML algorithms serve for the identification and optimization of B and T cell epitopes, and can inform the study of correlates of protection by helping assess quality and specificity of vaccine-induced cellular and humoral responses. Important questions in this regard concern which antibodies and T Cell Receptors (TCRs) bind to epitopes and trigger specific and high-magnitude responses, but also which of them can confer cross-variant immunity, a crucial question to formulate broadly protective vaccines for viruses undergoing fast antigenic drift like coronaviruses17. ML algorithms for epitope discovery, immunogen design, and prediction of epitope-paratope interactions have witnessed massive progress in recent years, spurred by fast-growing data availability and the latest developments in ML for protein modeling, standing out as illustrative examples of the potential advantages of ML in rational vaccine design.

Schematic of the rational vaccine design process (a) and machine learning applications to key tasks in vaccine target selection: B and T cell epitope discovery and immunogen design (b, d); characterization of correlates of protection through quantitative modeling of epitope-paratope interactions (c, e). Structures' images obtained with Mol*199.
Despite the success of conventional vaccination strategies, the molecular and cellular processes contributing to the efficacy and long-term protection of several vaccines are still not fully understood. In this regard, ML is emerging also as a tool for scientific discovery that, while delivering useful predictions for rational vaccine design, provides new methods for investigation in systems immunology and proteomics and can thus improve our understanding of immune responses across individuals.
In this article, I describe the current trends in ML methods for the discovery of B and T cell epitopes and for characterizing the response by the adaptive immune system to those epitopes at the molecular level. While comprehensive reviews of such methods are provided elsewhere18,19,20,21,22,23,24, my aim here is to describe the basic ideas, model architectures, and types of data involved in recent developments of ML in this context, as well as to discuss the prediction tasks and the biological insights made possible by them. I conclude with a brief overview of other ML predictions that are relevant to vaccine design (in vaccine construction and preclinical validation of selected vaccine targets), and with an outlook on current challenges and important directions for future work.
Machine learning in immunology
The development of ML methods for immunology has been fueled by the production of large-scale immune repertoire and immunopeptidomic datasets, and their systematic collection and annotation in specialized databases6,25,26. These data provide information on the central proteins involved in immune responses (antibodies, TCRs, antigens), represented in terms of their sequence and/or structure (see Box 2 for a description of protein representations that are relevant to modeling immune protein data).
Computational techniques of ML applied to large immunological datasets can detect statistical patterns reflecting structural and functional properties, and can leverage them to learn models of the mapping between a given input (like a protein sequence) to the structural or functional property (like the protein’s binding specificity). Learning (or training) a model consists of iteratively adjusting its parameter values on the available training data (Box 1) in such a way as to achieve a certain prediction task, a procedure which is typically expressed mathematically as the optimization of an appropriately defined objective function. Once trained, the model can be evaluated on new data, enabling novel predictions and insights fully in silico.
Model training can be performed in a supervised way (Box 1), like for classification tasks (e.g., classifying epitope vs non-epitope protein sites) and regression tasks (e.g., predicting the antigen-antibody binding affinity); or in an unsupervised way, like for clustering tasks (e.g., grouping TCR sequences with similar binding motifs). Hence ML is appealing for its predictive and exploratory power, which helps build accurate prediction models and facilitates the inspection and discovery of biologically meaningful features.
The ML predictions related to epitope discovery, immunogen design, and prediction of epitope-paratope interactions are typically formulated in terms of ‘scores’, quantifying for example the probability that a given residue belongs to a conformational epitope (Fig. 1b) or the probability of peptide presentation and immunogenicity (Fig. 1d). Assigning these scores enables a fast ranking of candidate targets and the subsequent prioritization of a few. It also accelerates additional in silico studies relying on more computationally intensive methods, like molecular dynamics.
Several ML architectures have been applied in this context (Box 3), which differ by mode of learning supported (e.g., supervised vs unsupervised), type of prediction (e.g., regression vs clustering), expressive power (the ability to capture non-linear relationships and correlations in the data) and interpretability of the predictions obtained. In general, the choice of a certain ML architecture (e.g., a transformer vs a convolutional neural network) and of its specific structure (e.g., the number of its internal layers, setting the number of parameters to learn) is motivated by the specific prediction task to achieve and by the type and quantity of data available for training (for example, more parameters increase the model’s expressive power but may lead to overfitting). I will provide an illustration of these model selection aspects while introducing ML approaches to predicting B and T cell epitopes and epitope-paratope interactions.
B and T cell epitope discovery
Prediction of B cell epitopes
Broadly speaking, the ML methods used for linear and conformational B cell epitope prediction are trained in a supervised way to discriminate epitope sites from generic ones that are typically not targeted by B cells, outputting an epitope likelihood score for each site27,28,29,30,31,32,33,34,35,36 (Fig. 1b). B cell epitopes are predominantly conformational, hence their prediction is better supported by methods trained on protein structures (Box 2), which can exploit information on the antigen surface topology in addition to the biochemical composition provided by the sequence.
In general, ML for B cell epitope discovery builds upon feature-based ML, which performs a key preliminary step of feature selection and engineering (Box 2). The intuition behind this is that only a few sequence and structure properties contribute to determine whether a residue is an antibody binding site. Indeed, residues’ physico-chemical properties have been suggested to favor the maturation of high-affinity antibodies, and have been used for epitope identification also before the advent of ML37,38, along with conformational properties such as flexibility39, residue protrusion40,41, and surface accessibility42. Feature selection enables as well to reduce the dimensionality of the input data (otherwise specified by thousands of atomic coordinates), with gains in computational efficiency. In conformational epitope discovery, these features typically consist of physico-chemical attributes (e.g., hydrophobicity and electrostatic potential28,33), high-level geometric properties (e.g., type of secondary structure28,33, solvent accessibility and average curvature of the molecular surface28), evolutionary information (e.g., conservation28,33), and specific combinations of amino acids in pairs or triplets29.
Graph-based representations (Box 2) of epitope regions have also been used in this context along with residue physico-chemical properties29,33. ML approaches based on graph-theoretical descriptors have been successful at protein design43,44, identifying interaction sites45,46,47,48,49, and predicting the effect of mutations50: all these works provide additional examples of feature selection and learning strategies that could be adapted to the epitope identification problem as this field progresses and new data become available. The motivation for developing graph-based approaches to epitope identification is that epitope regions exhibit distinctive signatures (in terms of residue packing as well as type and topological arrangement of bonds) that can be conveniently summarized by a graph representation29,33. An advantage of graph-based ML is that it can leverage efficient algorithms from the well-established field of graph theory51. The challenge however remains of determining the appropriate scale for constructing the graph (e.g., atom vs residue level), and the information to embed in the definition of graph links (e.g., whether weighting them by geometrical characteristics of the modeled protein region29). The design of graph-based descriptors, and more generally feature engineering, depend on our understanding of the most relevant features, which can render the predictions prone to bias due to over-reliance on certain properties commonly associated to the functional behavior of interest (e.g., a protrusive instead of planar surface for epitopes29). Even if correlations of epitope propensity to chemical and geometrical features have been established, an open question is how they should be combined when used as inputs of ML algorithms to achieve an accurate epitope prediction. At present, there are no general guiding principles to address this problem, which is mainly dealt with by careful and potentially very time-consuming work of systematic feature elimination and search over feature combinations.
A new approach that has led to substantial gains in performance at B cell epitope identification is the one of learning protein representations tailored to the B cell epitope prediction task (Box 2). Ref. 35 has pioneered this approach, using deep learning to build representations of spatio-chemical arrangements of residues’ neighborhoods that are informative about protein-protein binding and epitope recognition (see also section Interpretable machine learning approaches). Another approach recently proposed36 is to appeal to residue-specific representations extracted by protein language models (Box 2), learnt in such a way as to embed contextual information (the rest of the sequence and the backbone structure52), and use them as information-rich inputs to train a ML epitope predictor. The key idea behind this approach is that the unsupervised learning of language models from massive protein datasets discovers inter-residue dependencies that are not captured by handcrafted features, and that can be leveraged for the downstream task of B cell epitope prediction, reaching a performance AUROC ~ 0.8 36 (Box 1).
In general, efforts of structural characterization of the targeted protein, already pursued through comparative protein structure modeling53 and protein-protein docking54,55,56,57, can serve to optimize the antigen-antibody interaction surface1,58 (Fig. 1b), and to identify amino acid substitutions conferring enhanced conformational stability and expression (for example the 2 proline mutations at positions 986 and 987 for the Sars-Cov-2 spike protein, included in several COVID-19 vaccines59). ML has the potential to assist this task by identifying residues most involved in conformational variation60, whose mutations can be further studied via molecular dynamics, or by predicting free energy changes upon residue mutations61,62,63. While the performance of the later approaches seems stagnating63, recent progress in deep learning-based protein design holds promise to be useful at proposing expression and stability-enhancing mutations64.
On the other hand, ML predictors of B cell epitopes that are sequence-based27,30,31,32 (Box 2) are more convenient than structure-based ones, due to their higher computational speed. Despite having typically lower performance compared to structure-based ones (AUROC slightly above 0.75 for the example of state-of-the-art method of ref. 31), they enjoy a wider and more flexible scope of application given the large number of protein sequences available compared to structures. They are better-suited for linear B cell epitopes, but they are potentially useful also for conformational ones by capturing, thanks to the context-aware representations from protein language models (Box 2–3), functional dependencies between amino acids far apart along the sequence but proximal in the 3D structure31.
ML methods like AlphaFold65,66,67, trRosetta68, and RoseTTAFold69 can bridge this scale gap between sequence and structure data availability by enabling predictions of protein structure from sequence alone with unprecedented accuracy. Predictors of protein structure have huge potential still to be fully explored for the design of immunogens guided by structural insights59,70, as well as for antibody and TCR engineering. Antibody-specific predictors have been proposed71,72,73,74 based on deep learning architectures similar to AlphaFold, TrRosetta and RoseTTAFold. A specialized version of AlphaFold has been developed to study the structural interactions of the molecular complexes antigen-TCR75. In addition, ML-predicted structures are used for the complementary task of data augmentation, i.e., to enlarge the available training and test sets36,74,76. However, paratope, epitope, and in general functional site identification remains challenging even with the availability of these methods; for instance, the prediction of epitope-paratope binding sites by Alphafold-Multimer67 (the AlphaFold method tailored to protein complexes) was found to be inaccurate35,67.
Prediction of antigen presentation
Protein targets of T cells are presented on the cell surface as short linear epitopes by the Human Leukocyte Antigen (HLA) complexes, with the epitopes of killer T cells presented in the context of HLA class I (HLA-I) molecules and the ones of helper T cells presented by HLA class II (HLA-II). Antigen presentation is the most selective step determining what pathogenic protein regions are likely to be targeted by T cells, hence its computational prediction is key to filtering effectively candidate targets for vaccine design (Fig. 1d). For example, the proteome of SARS-CoV-2 harbors ~ 104 potential 9-mer HLA-I antigens. Bioinformatic analyses typically seek for ~ 1% of these peptides as predicted presented antigens per HLA allele77, corresponding to general estimates of the viral peptidome fraction that binds to HLAs78.
Figure 2 illustrates how the different ML concepts and methods in Box 1 and Box 3 have been adapted to the prediction of HLA-I antigen presentation (see also refs. 18,19 for comprehensive reviews). Existing ML predictors range from unsupervised clustering methods to perform binding motif deconvolution from unannotated eluted ligand data, like MixMHCp and MixMHCpred79,80,81 (Fig. 2a), to feed-forward neural networks trained in a supervised way to predict peptide presentation from known peptide-HLA pairs, like MHCflurry82,83 and the NetMHC and NetMHCpan suites84,85,86,87,88 (Fig. 2c). An alternative approach is RBM-MHC89, which addresses the problem of assigning antigens to their respective HLA-I molecule in newly produced or custom immunopeptidomic samples by resorting to a semi-supervised strategy (Box 1). The ML architecture here (a Restricted Boltzmann Machine, RBM, see Box 3) internally transforms sequence data onto a lower-dimensional representation, which facilitates the task of annotating antigens by their HLA-I type, since in this representation space antigens cluster by their HLA-binding motifs. Such a cluster structure enables to build an accurate predictor of HLA specificity using only a small amount of HLA-annotated antigen data from public databases (Fig. 2b). In addition, MixMHCp and RBM-MHC (Fig. 2a, b) learn generative models (Box 3), i.e., they estimate the probability distribution describing the immunopeptidomic data, assuming a different parametric form for such a distribution (respectively, a mixture of probabilistic independent-site models and an RBM). This peptide sequence probability can be used as a probabilistic score of presentation to distinguish presentable from generic non presentable sequences.

Examples of predictors of HLA class I antigen presentation that are based on different types of ML methods: a MixMHCp79,81 is an unsupervised method using a mixture of probabilistic independent-site models to perform clustering of peptides and binding motif deconvolution; b RBM-MHC89 is a semi-supervised method relying on a dimensionality reduction step (performed through an RBM model) to leverage small amounts of antigens labeled by their HLA specificity to train an HLA-type classifier; c NetMHCpan87,88 is based on a supervised feed-forward neural network trained on antigen and HLA sequences to predict peptide binding affinity (from affinity data) and a score of peptide elution (from mass spectrometry eluted data).
The data used to train these methods are HLA-antigen binding assays and eluted peptidomic data obtained via mass spectrometry, to a large extent publicly available in the database IEDB6. Recently there has been a shift towards an increasing use of eluted data from mass spectrometry83,88,90,91,92,93,94, which allow one to machine-learn information about all the steps of HLA-mediated processing and presentation83,92,94, and not only peptide binding affinity to the presenting HLA. For instance, the most recent versions of NetMHCpan have been tailored to integrate both data types to boost performance87,88 (Fig. 2c).
Currently, HLA polymorphism remains an unmet challenge for HLA-I presentation prediction. Most of the methods achieve near-perfect prediction for common HLA alleles, but perform poorly for rarer alleles. Improving the accuracy of predictors across all HLAs is key to ensuring high HLA coverage of vaccines across human populations. This problem has motivated the development of methods that use information on the HLA sequence to deliver HLA pan-specific predictions, like MHCflurry 2.083 and NetMHCpan methods87,88 (Fig. 2c), and methods that can be easily re-trained by the user on newly available HLA-specific datasets89.
Predicting the presentation by HLA-II is much more challenging due to limited data availability and the diversity of allele-specific binding motifs. The data used for training the predictors are still limited to a few alleles (mainly from the genetic locus HLA-DR), and precisely the increased quantity of data on HLA-II presented peptides has been key to the latest improvements in prediction performance91,93,95,96,97, especially for less well-characterized alleles97. Binding-motif diversity is two-fold: first, alternative binding modes for the same HLA allele, including binding in the reverse peptide orientation, have been documented96; second, HLA-II presented peptides exhibit substantial variability in length (12-25 amino acids, compared to 8-14 for HLA-I), with multiple peptides of different length sharing a similar binding core at a variable starting position. To deal with this difficulty, state-of-the-art methods91,93,95 implement a dynamical search for the binding core within each peptide, either by scoring different sliding motifs along peptides93,95, or by appealing to the ability of Convolutional Neural Networks (CNNs, Box 3) to detect features regardless of their exact location91. Currently positive hits are distinguished from negative ones with AUROC in the range 0.8-0.85 at best, indicating that there is still room for improvement in performance.
Prediction of antigen immunogenicity
Only a subset of HLA-presented antigens is immunogenic, so a few computational and ML methods have been proposed to predict which presented antigens tend to promote a T cell response and are likely to be immunodominant. Predictors of T cell epitope immunogenicity typically compare presented antigens that are immunogenic to non-immunogenic ones to estimate scores of immunogenicity, both for HLA-I98,99,100,101,102,103,104 and HLA-II peptides101,105. Such scores can be predicted based on the single-site amino acid enrichment in immunogenic vs non-immunogenic antigens98,99 or by supervised ML methods that are trained to discriminate them, using only sequence information100,101,103,104,105 or including also the peptide-HLA complex structure102. In these studies, the propensity to TCR binding has been correlated to physico-chemical properties of the peptide’s side chains facing out from HLA binding groove, such as hydrophobicity and aromaticity, and based on this observation some predictors select a priori peptide positions98,99,103,104 or amino acid properties100 deemed to be important to immunogenicity. The ML approach we have recently proposed106 models immunogenicity by learning the statistical differences in amino acid composition between immunogenic and presented-only antigens, avoiding the need for data validated as non-immunogenic, and recovering, instead of assuming a priori, the peptide positions and properties more frequently involved in TCR response.
The prediction of T cell epitope immunogenicity is of particular interest in pipelines of neoantigen discovery for the design of T cell-based anti-cancer vaccines107,108,109. Prediction methods here need to take into account immunogenicity-determining factors specific to immunity in cancer, such as low cross-reactivity with self-antigens and clonality of mutations. A recent large-scale validation of existing predictors used for neoantigen discovery has highlighted the need for substantial improvement in their performance110.
Indeed, in general, immunogenicity prediction methods have maximal AUROCs ~ 0.7103,106 (hence lower than for B cell epitope prediction), and in particular the performance becomes poor beyond a few immunodominant epitopes presented by common HLAs21. A main shortcoming is that the biological parameters determining immunogenicity, and hence to account for in a ML model, remain to be understood. For example, there is no consensus regarding whether high affinity and stability of binding to the HLA is correlated to high immunogenicity111, observed in some settings112,113 but not in others114. A paratope-agnostic identification of epitope sites, which side-steps the details of specific epitope-paratope interactions, has clear advantages in a translational setting of vaccine or therapy design but has also limited predictive power, because sequence and structure of the target protein are not the only determinants of a positive immune response. Modeling epitope-paratope interactions is hence crucial to improve epitope prediction, as well as to characterize more globally correlates of immune response upon vaccination (Fig. 1c, e).
Epitope-paratope interactions
Another area relevant to rational vaccine design is modeling through ML the specificity of epitope recognition both by TCRs (as reviewed in refs. 22,115), and antibodies (as reviewed in refs. 23,24). These ML models are trained on: (i) sets of TCR/antibody only (Fig. 3a, b); (ii) TCR/antibody-antigen binding pairs (Fig. 3c, d).

Scheme of ML methods to predict epitope-paratope interactions for B and T cells, organized in terms of type of input: applicable to TCR/antibody sets only (a, b) vs TCR/antibody-antigen pairs (c, d); sequence (a, c) vs structure-based (b, d). Structures' images obtained with Mol*199.
Examples of ML models of type (i) are the ones built for in silico paratope identification in antibodies, based on the assumption that the position of the paratope is largely antigen-independent. ML methods here are trained to classify an antibody residue as part or not of the paratope, estimating for each residue a probabilistic score of belonging to it76,116,117,118; their performance is currently quite high (AUROC above 0.9, see one of the latest comparisons in ref. 76). In silico paratope identification is relevant especially to antibody design, since it helps propose putatively binding-improving mutations72. Other main examples of ML models of type (i) are generative models (Box 3) learnt from TCR/antibody sequences (Fig. 3a), which estimate a sequence probability distribution119,120,121,122,123,124,125,126. Generative models are generally of great interest to the field of molecular design: sampling from the learnt distribution allows one to generate putatively functional synthetic data, for instance antibodies with optimized binding properties124,125,126. Several ML models of type (i) are trained on sets of TCR sequences binding to the same antigen, predicting scores to classify new TCRs as specific or unspecific to the corresponding antigen119,120,121,122,127,128,129,130 (Fig. 3a). Some of these approaches can also detect recurrent amino acid motifs in TCRs that are the statistical signature of antigen-binding specificity120,122,128, similarly to the clustering methods designed for binding motif discovery131,132,133 (Fig. 1e).
Models of type (ii) attempt to model the specific interactions involved in epitope-paratope binding. They can typically predict binding scores, that are able to discriminate epitope-paratope binding pairs from non-binders134,135,136,137,138,139,140,141,142,143. These predictions are useful to characterize antigen specificity of unseen TCRs134,135,136,137,138,139,140,143,144,145,146, to identify paratope and epitope sites147, and to accelerate further analyses through docking algorithms, e.g. by improving the selection of docking poses142,148. Such binding predictions can inform vaccine design, because they enable the screening in silico of putative antigen targets against large sets of TCRs and antibodies, thus helping characterize them in terms of elicited response, dominance and prevalence.
Similarly to conformational epitope discovery, structure-based methods for epitope-paratope interactions (Fig. 3b, d, Box 2) generally rely on a first step of feature selection, which extracts and embeds into feature variables their physico-chemical and geometrical properties118,143,147,148,149, including graph-based representations of the interface regions76,147,148; ML predictors of epitope-paratope binding are then trained on these features.
Epitope-paratope interactions are mediated by binding motifs that vary position and composition-wise across antibody-antigen pairs, as a consequence also of the variability in length of the Complementarity Determining Regions (CDRs). Identifying such motifs calls for prediction tools that are able to leverage information from residue neighborhoods and detect spatially localized features independently of their exact position. This type of prediction resembles the object recognition task in computer vision, where the state-of-the-art ML tools are CNNs (Box 3). CNNs have become a main trend in ML architectures for structure-based epitope-paratope binding142,147 along with neural networks designed to process graph-shaped inputs76,148. The richness of structural information enables the prediction of antigen specificity in TCRs with a performance comparable to sequence-based methods, despite the smaller training datasets143. It enables also to model the mapping between the antibody-antigen complex structure and its binding affinity148,149 (Fig. 3d), with a performance, estimated through the correlation coefficient between true and predicted affinity values, of up to 0.79149.
Sequence-based methods (Fig. 3a, c, Box 2) appeal as well to deep CNNs116,117,128,134,135,137,138,140,141,144,145, while in general spanning a variety of ML architectures, from decision trees and random forests129,150,151 to networks based on the attention mechanism122,134,139,140 (Box 3). Most recently, sequence-based methods have benefitted from the breakthroughs in ML for natural language processing, with several methods for epitope-paratope interactions and paratope prediction directly using language model ML architectures116,121,123,124,125,126,130,136,137,146 (Box 3). These architectures capture potentially long-range dependencies between residues along the sequence, resulting in representations of each protein site capable of incorporating the effect of the physico-chemical context152,153.
A recent public benchmark of sequence-based methods to predict TCR-epitope specificity has flagged up a few important trends115. Firstly, data set the performance to a larger extent than the particular model architecture. Indeed, the generalization power of different methods is consistent across antigens, with typical AUROCs in the range 0.7–0.9, and is correlated to the heterogeneity in sequence composition of TCRs binding to the same antigen. Secondly, predicting antigen specificity based on the TCR sequence similarity provides already a good baseline performance, in line with the observation of enriched sequence motifs in TCR sets with a given antigen specificity. Finally, the gain of deep learning over simpler models seems modest with the available data. Given that training deep learning models is data-demanding, tests on larger datasets are needed to clarify this point.
Interpretable machine learning approaches
The need to better understand the molecular basis of epitope immunogenicity and epitope-paratope binding specificity highlights the importance to be able to extract biological insights from ML models. ML approaches that are explainable in terms of biological modes of action are increasingly recognized as a priority in immunology154,155, and more generally for ML applications of biomedical and clinical relevance156. Figure 4 introduces, in the form of graphical sketches, examples of ways in which the predictions from ML models can be made biologically interpretable, and how they have been employed in epitope discovery and epitope-paratope interaction studies.

a Feature importance; b interpretability pipelines; c attention maps; d weights visualization; e learnable spatio-chemical filters.
Decision trees (Box 3) have been used for a variety of predictions relevant to immunology30,101,149,157, including classifying TCRs into specific binders of an epitope or non-binders129,150,151. The model ‘decides’ whether a TCR is binder or non-binder through a series of splits in the space of sequence features (e.g., average and positional physico-chemical properties), which are determined by whether a given feature is higher or lower than a threshold. Such decision rules take into account one feature at a time, hence the importance of each feature to the final prediction can be evaluated (Fig. 4a). Based on this analysis, ref. 150 finds that the average basicity of the CDR3 (on the β chain), as well as basicity and hydrophobicity of the amino acids in the CDR3β center, play an important role at discriminating epitope-specific from unspecific sequences.
Explainable predictions can be obtained in a model-agnostic fashion by applying interpretability pipelines, for example the one estimating ‘anchors’158. An anchor is an explanation that ‘anchors’ the model’s prediction locally to specific data attributes and formulated as an if-then rule. Once applied to models for classification of TCRs by epitope specificity159, anchors recapitulate the presence of specific amino acids in certain positions of epitope-specific TCR sequences (Fig. 4b), for example polar amino acids like serine (S) in the CDR3β binding to the peptide KLGGALQAK159.
The attention mechanism typical of transformers has been increasingly explored as tool to gain interpretability in protein language models160 (Fig. 4c, Box 3). For each sequence, an attention map describes how relevant each residue on the horizontal axis is in the prediction of all the other residues (vertical axis), detecting in this way structurally and functionally important residues that exhibit correlations with the other ones. In transformer models of antibody sequences, attention concentrates on sites in contact or belonging to the paratope123 (Fig. 4c).
ML architectures with a limited amount of parameters are more amenable to the inspection of the biological information learnt, for instance by direct visualization of their parameters. A point in case is the RBM architecture used to predict antigen presentation in ref. 89, whose main parameters are the sets of weights connecting the input layer to the only hidden layer (Box 3, Fig. 4d). The visualization of weights entering one hidden unit highlighted the existence of two distinct binding motifs within antigens of the same HLA type89, which correspond to two alternative and structurally validated HLA-binding modes81. Antigens bearing the different binding motifs can be readily distinguished via the projection of the data onto this set of weights (it is indeed this projection onto one or more sets of weights that defines the coordinates of the model’s representation space, where antigens cluster by sequence motifs connected to their HLA binding properties, see Fig. 2b).
Finally, so-called ‘geometric’ deep learning161 is another approach with potential for interpretability, as it models the underlying regularities of the data and leverages them for prediction. The protein binding site prediction method in ref. 35 implements this approach through the convolution (Box 3) of geometric representations of protein regions at the atomic and amino acid scale with learnable filters. Visualizing the parts of these representations that most contribute to each filter’s output results in interpretable spatio-chemical patterns, defined by sets of biochemical attributes (like specific amino acids and their degree of solvent exposure) along with their spatial coordinate (Fig. 4e). Such patterns highlight co-determinants of protein-protein binding like coordination number and electrostatic potential, generating insight also into the physico-chemical principles underlying antibody-epitope binding. For instance, ref. 35 detects a pattern at the amino acid scale positively correlated with epitope probability consisting of an exposed, charged amino acid close to a disulfide bond: this is a structural motif that confers stability, and hence plausibly facilitates high-affinity antibody binding.
These examples show that there exist interpretability strategies that are model-specific, relying on specific building blocks of a given ML architecture (like attention maps, Fig. 4c), and model-agnostic ones (like anchors, Fig. 4b), which are more broadly applicable to ML models to explain their output. In all cases, domain expertise has been essential to assess the biological relevance of the patterns learnt from immunological data. Strategies like the ones discussed, combined to domain expertise, point towards the feasibility of intepretable ML for molecular biology, and provide the basis for further work in this direction.
Computational and ML tools in vaccine design beyond epitope prediction
Epitope identification is the most important prediction in rational vaccine design, yet it is only the starting point of the elaborate and challenging process of vaccine design (Fig. 1a). After epitope prediction has returned a set of vaccine candidates, additional computational methods and analyses are needed, first of all for the evaluation of structural and functional features of the candidate targets. This might further inform their selection and optimization along with the ML-enabled prediction of epitope-paratope interactions already discussed. Such evaluation steps (see refs. 13,15,162,163,164 for examples) consist of: structural modeling (e.g. with the tool165), to ensure surface accessibility of the predicted epitopes; molecular docking and molecular dynamics (e.g. with the tool166), to probe the stability and affinity of the binding between vaccine targets and immune receptors; screening of the targets’ similarity to the host proteome and of allergenicity (e.g. with the tools167,168), to discard the targets that can potentially trigger auto-immune reactions and side effects; evaluation of population coverage of the selected epitopes (e.g. with the tool169), as well as their degree of conservation, since targeting conserved regions might increase cross-variant protection; an assessment of biochemical properties such as solubility (e.g. with the tools170,171) that are key to the delivery and molecular mode of action of the selected targets; computer simulations of the immune response elicitable (e.g. with the tool172), to optimize vaccine dosage, formulation, and schedule. (The analysis resource section of IEDB6 makes available a number of computational tools for these tasks). ML is emerging as a technology that can assist also several of the evaluation steps, starting from structural characterization through the ground-breaking new ML methods for protein structure prediction65,66,67,68,69, as mentioned above. ML is increasingly used in drug design to predict computationally a number of molecular properties (for example solubility173,174); as such, it can guide the selection of adjuvants in vaccine construction164,175 or help predict mRNA stability to optimize mRNA-based vaccines’ intracellular delivery176.
Immune simulation approaches are moving toward combining the digital twins technology with ML177, an arena where ML can serve to incorporate proficiently pharmacokinetic and molecular binding data in the digital twin’s parameters to calibrate. In silico clinical trials178 are another set of computational models and simulation techniques to assist the assessment of safety and efficacy profiles in vaccine design. It is increasingly recognized that in silico clinical trials can be empowered by ML for tasks like: data augmentation (by generating synthetic patients to complement small-size cohorts178); outcome and response prediction156 (by detecting patterns in electronic records on previous trials and harnessing them for prediction); automation and optimization of participant recruitment, data collection and management, and trial monitoring156,179,180. These example tasks illustrate how ML could inform the design and planning of actual clinical trials to help improve their feasibility, efficiency, and success rate, albeit more work is needed for the large-scale deployment of such techniques.
Finally, a complementary and much needed scope of use for ML is to predict regions of the viral genome prone to harbor mutations, in such a way as to anticipate new variants before they emerge and design vaccine strategies robust to them. Approaches combining mathematical modeling and statistical learning have been developed to detect high-mutability regions181,182 and to model the fitness gain and potential for immune evasion conferred by mutations183,184. ML will contribute to boost their accuracy and applicability, by enhancing our understanding of epitope determinants in protein structure and sequence space and of the impact of mutations on epitope-paratope interactions.
Limitations, challenges, and perspectives
In this perspective, I have discussed the type of predictions and methods by which ML can inform and guide vaccine target selection, mainly the tasks of B and T cell epitope discovery and the prediction of epitope-paratope interactions. There is a series of limitations and challenges, both at the level of datasets and methodology, that, once overcome, could pave the way to the wide application in rational vaccine design of the latest developments in this field.
Data availability and quality
A key aspect to consider is that type, quality, and quantity of training data are crucial to the predictive power of any ML approach.
The main bottleneck preventing major leaps forward in the predictive performance of both structure- and sequence-based epitope-paratope interaction models is the scarcity of data to use as training sets. Training data should be seen as realizing a sampling of the full space of sequences and structures to model, and this sampling should be ideally exhaustive, or at least representative, of the modeled space and consistent. Major challenges are the extreme diversity of both the epitope and paratope sequence spaces to sample185,186, the cross-reactivity of epitope-paratope interactions, especially for T cells187,188, and their conformational diversity, with multiple binding modes even for the same target189. Assays sampling epitope-paratope binding pairs in a high-throughput fashion are lacking, already at the sequence level, and the available sequence data often consist only of a single chain. Available structural data, as mentioned, are even sparser. By way of example, solved structures of antibodies to date amount to a few thousands (7967 on the SabDab database26) and the ones of TCRs to a few hundreds (605 on the STCRDab database190). Ad hoc ML strategies can mitigate in part the problem of scarcity of data on epitope-paratope interactions by modeling them as particular instances of general protein-protein interactions, for which more data are available. Based on this assumption, a ML strategy pursued is to pre-train a model on these data, and next to fine-tune the model’s parameters on the epitope-paratope datasets134,147.
In addition to the limited amount of data, there is the problem that the aggregation of data from heterogeneous experimental assays can become a noise source, and the one of sampling biases. Antigens of biomedical interest that give rise to positive responses tend to be over-represented111,191, making it difficult to label with confidence ‘negative’ examples36,192; such a redundancy at the antigen level leads to models that are prone to overfitting and with imbalanced performance across epitopes (and concomitantly HLA types). Biases in the data can further propagate when new targets tested are chosen based on predictors trained on biased data, as it has been discussed in relation to peptide-HLA binding affinity assays193. On the other hand, mass spectrometry techniques used to map HLA-bound peptides suffer from technical biases in the detection of some amino acids, e.g. cysteine19,193.
An area requiring a concerted effort of the immunology community is thus the production, curation, and dissemination to ML experts of high-quality and internally consistent datasets. Efforts of method development need as well to be cognizant of existing biases, for example by including corrections for biased amino acid detection by mass spectrometry to improve performance for cystein-containing peptides89. Another promising avenue to resolve the lack of truly negative examples is to rely on ML approaches trained on positives vs unlabeled examples36 or positives only106,119,120,146. Bayesian inference has also been proposed to take into account biases and uncertainty in database annotation on T cell epitope immunogenicity and include systematically information on the number of responders to a given epitope194. Given the importance of the training data in setting performance, methods should be designed in such a way as to make re-training on newly produced data feasible and straightforward.
Prediction performance and method integration
The advent of ML algorithms for the tasks discussed has led to better performance compared to more traditional bioinformatic approaches, yet there is still substantial room for improvement.
Controlled comparisons carried out in the literature are helping elucidate the entity of the improvements brought along by ML over bioinformatic approaches based on motifs, sequence similarity, or selected biophysical properties. The rather simple, linear motifs describing peptide-HLA-I binding preferences are well characterized by matrix-based models scoring independently every peptide position79,81, which have then comparable performance to neural network methods at scoring HLA-I presentation18,19,88; relatedly, the later methods tend to rely on shallow networks (typically limited to one hidden layer). To predict the immunogenicity of HLA-I-presented epitopes, we found that ML tools perform better than matrix-based ones, but also in this case the optimal predictor of immunogenicity is given by a shallow, as opposed to a deep, network106. ML methods give the best performance at predicting TCR specificity to HLA-I epitopes, but the difference compared to predictions based on TCR sequence similarity alone is rather modest115. On the other hand, a deep architecture is seen to have evident advantage over shallow and matrix-based models when predicting scores for HLA-II-presented epitopes195. Also for conformational B cell epitope prediction, large gains have been reported recently thanks to deep learning35,36, for instance compared to naive predictors scoring residues based on relative surface accessibility36. Hence, the need for training deep architectures, which enable to model highly non-linear input-output relationships but are data-demanding, is more or less clear depending on the prediction task. To bring clarity in this regard, regular, systematic benchmarks of the available methods on independent datasets and according to uniform assessment criteria are pivotal (see for example ref. 115), to recognize strengths and limitations in performance and to formulate recommendations for the next developments. IEDB performs automatic benchmarks of new predictors of HLA-I and II antigen presentation on the data that become available, in order to recommend methods and metrics for prediction, a procedure that, despite its pitfalls19, could serve as an example to follow.
One of the crucial problems performance-wise is the low precision of the final epitope identification, due to false positives, which can slow down and hamper the downstream steps of in silico, in vitro, and in vivo validation. For B cell epitopes, state-of-the-art methods35,36 assign to epitopes on average a score higher than ~ 70% of the scores for the same protein, indicating that many false positives do occur among the highest-ranked epitope residues. For class I T cell epitopes with well-characterized HLAs, with the best performing methods88 one has > 99% chance of identifying a presented antigen taking the top scoring peptide among all the possible ones from the proteome of interest; it is rather the subsequent prediction of immunogenic antigens among the presented ones that suffers from low precision, as reported in benchmarks with experimentally tested targets21.
The prediction of immunogenicity of candidate targets is particularly challenging, and ultimately can be validated only by experimental tests and clinical trials, being it an intrinsically multifactorial and multiscale effect. Firstly, protein-protein interactions are dynamic and susceptible to the cellular environment; a first step to account for these aspects is to complement ML predictions by molecular docking simulations of the interactions mediating the adaptive response (peptide docking to the HLA57,196, docking of TCRs to the peptide-HLA complex56, antigen-antibody docking54,55). Furthermore, immune activation and effector function are dependent on co-stimulatory signals, and more generally on the context at the cell and tissue level. For example, high antigen expression levels have been suggested to compensate for weak HLA-antigen binding, thus including cell type and tissue-specific information on antigen abundance has resulted in improved predictions of T cell epitopes92,195,197. Protection eventually depends on many factors, like innate control, infective dose, as well as the genetic and environmental factors that shape the individual immune repertoires (age, previous exposure, etc.). ML predictions should be therefore interpreted as inherently probabilistic, i.e., they come with an uncertainty stemming from the variety of factors that contribute to a positive response and are not included in the models.
Suboptimal precision implies more permissive prediction thresholds to ensure that a sufficient number of true epitopes is recovered, hence epitope prediction can result in hundreds of candidates to analyze and test, while only 10-30 subunits are necessary for the final construction of a multi-epitope vaccine13,15. Post-epitope prediction evaluations can be time-consuming for this reason, and because usually their steps are not integrated and automated. A strategy for higher-efficiency vaccine design recently proposed15 compresses epitope identification and property evaluation in one step by training a deep neural network to directly predict vaccine subunits with the desired properties, which results in fewer candidates to further evaluate. Indeed, more rapid screening of possible targets requires the development of frameworks that can perform and combine multiple predictions, similarly to approaches in drug discovery integrating ML models and docking simulations198. The design of such target selection pipelines integrating different prediction steps will benefit from ML methods that are clearly documented in terms of scope, modes, and optimal conditions of use. Working with standardized input formats and output metrics would be also important to save efforts of data pre-processing and post-processing and to facilitate method integration. For the future development of rational vaccine technologies, two of the most pressing needs are hence: increased precision of epitope prediction, to reliably narrow down target selection to fewer candidates; integrated frameworks connecting the bioinformatic and ML software necessary for ML-assisted epitope prediction and the subsequent evaluations, possibly developed within a user-friendly infrastructure that is easy to access and implement either as a web server or a downloadable package. Such improvements are essential to reducing the time, manual work, and resources involved in vaccine target selection and validation, thus they are prerequisites to the flexibility and scalability sought-after in personalized neoantigen discovery and in the adaptation of vaccines to newly-emerged viral strains.
Data availability
No datasets were generated or analyzed in this article.
Code availability
No new algorithms or code were developed or used for this article.
References
He, L. & Zhu, J. Computational tools for epitope vaccine design and evaluation. Curr. Opin. Virol. 11, 103–112 (2015).
Sette, A. & Rappuoli, R. Reverse vaccinology: developing vaccines in the era of genomics. Immunity 33, 530–541 (2010).
Kyriakidis, N. C. et al. SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. npj Vaccines 6, 1–17 (2021).
Soria-Guerra, R. E., Nieto-Gomez, R., Govea-Alonso, D. O. & Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J. Biomed. Inform. 53, 405–414 (2015).
Srivastava, S., Chatziefthymiou, S. D. & Kolbe, M. Vaccines Targeting Numerous Coronavirus Antigens, Ensuring Broader Global Population Coverage: Multi-epitope and Multi-patch Vaccines. In Vaccine Design: Methods and Protocols, Volume 1. Vaccines for Human Diseases. Methods in Molecular Biology. (ed. Thomas, S.) 149–175 (Springer US, 2022).
Vita, R. et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019).
Dimitrov, I., Zaharieva, N. & Doytchinova, I. Bacterial immunogenicity prediction by machine learning methods. Vaccines 8, 709 (2020).
Ong, E. et al. Vaxign2: the second generation of the first web-based vaccine design program using reverse vaccinology and machine learning. Nucleic Acids Res. 49, W671–W678 (2021).
Herrera-Bravo, J. et al. VirVACPRED: a web server for prediction of protective viral antigens. Int. J. Pept. Res. Ther. 28, 35 (2021).
Bowman, B. N. et al. Improving reverse vaccinology with a machine learning approach. Vaccine 29, 8156–8164 (2011).
Heinson, A. I. et al. Enhancing the biological relevance of machine learning classifiers for reverse vaccinology. Int. J. Mol. Sci. 18, 312 (2017).
Ong, E. et al. Vaxign-ML: supervised machine learning reverse vaccinology model for improved prediction of bacterial protective antigens. Bioinformatics 36, 3185–3191 (2020).
Ong, E., Wong, MU., Huffman, A. & He, Y. COVID-19 coronavirus vaccine design using reverse vaccinology and machine learning. Front. Immunol. 11, 1581 (2020).
Yarmarkovich, M., Warrington, J. M., Farrel, A. & Maris, J. M. Identification of SARS-CoV-2 vaccine epitopes predicted to induce long-term population-scale immunity. Cell Rep. Med. 1, 100036 (2020).
Yang, Z., Bogdan, P. & Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 11, 3238 (2021).
Mohanty, E. & Mohanty, A. Role of artificial intelligence in peptide vaccine design against RNA Viruses. Inf. Med. Unlocked 26, 100768 (2021).
Swadling, L. et al. Pre-existing polymerase-specific T cells expand in abortive seronegative SARS-CoV-2. Nature 601, 110–117 (2022).
Mei, S. et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Brief. Bioinform. 21, 1119–1135 (2019).
Nielsen, M., Andreatta, M., Peters, B. & Buus, S. Immunoinformatics: predicting peptide–MHC binding. Annu. Rev. Biomed. Data Sci. 3, 191–215 (2020).
Kar, P., Ruiz-Perez, L., Arooj, M. & Mancera, R. L. Current methods for the prediction of T-cell epitopes. Pept. Sci. 110, e24046 (2018).
Buckley, P. R. et al. Evaluating performance of existing computational models in predicting CD8+ T cell pathogenic epitopes and cancer neoantigens. Brief. Bioinform. 23, bbac141 (2022).
Lee, C. H. et al. Predicting cross-reactivity and antigen specificity of T cell receptors. Front. Immunol. 11, 565096 (2020).
Norman, R. A. et al. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief. Bioinform. 21, 1549–1567 (2020).
Kim, J., McFee, M., Fang, Q., Abdin, O. & Kim, P. M. Computational and artificial intelligence-based methods for antibody development. Trends Pharmacol. Sci. 44, 175–189 (2023).
Shugay, M. et al. VDJdb: a curated database of t-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 46, D419–D427 (2018).
Dunbar, J. et al. SAbDab: the structural antibody database. Nucleic Acids Res. 42, D1140–1146 (2014).
Saha, S. & Raghava, G. P. S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65, 40–48 (2006).
Rubinstein, N. D., Mayrose, I. & Pupko, T. A machine-learning approach for predicting B-cell epitopes. Mol. Immunol. 46, 840–847 (2009).
Zhao, L., Wong, L., Lu, L., Hoi, S. C. & Li, J. B-cell epitope prediction through a graph model. BMC Bioinform. 13, S20 (2012).
Jespersen, M. C., Peters, B., Nielsen, M. & Marcatili, P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 45, W24–W29 (2017).
Clifford, J. N. et al. BepiPred-3.0: improved B-cell epitope prediction using protein language models. Protein Sci.: Publ. Protein Soc. 31, e4497 (2022).
Liu, T., Shi, K. & Li, W. Deep learning methods improve linear B-cell epitope prediction. BioData Mining 13, 1 (2020).
da Silva, B. M., Myung, Y., Ascher, D. B. & Pires, D. E. V. epitope3D: a machine learning method for conformational B-cell epitope prediction. Brief. Bioinform. 23, bbab423 (2022).
Shashkova, T. I. et al. SEMA: antigen B-cell conformational epitope prediction using deep transfer learning. Front. Immunol. 13, 960985 (2022).
Tubiana, J., Schneidman-Duhovny, D. & Wolfson, H. J. ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction. Nat. Methods 19, 730–739 (2022).
Høie, M. H. et al. DiscoTope-3.0 - improved B-celL epitope prediction using AlphaFold2 modeling and inverse folding latent representations. bioRxiv https://doi.org/10.1101/2023.02.05.527174 (2023).
Parker, J. M., Guo, D. & Hodges, R. S. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 25, 5425–5432 (1986).
Kolaskar, A. S. & Tongaonkar, P. C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 276, 172–174 (1990).
Karplus, P. A. & Schulz, G. E. Prediction of chain flexibility in proteins. Naturwissenschaften 72, 212–213 (1985).
Thornton, J. M., Edwards, M. S., Taylor, W. R. & Barlow, D. J. Location of ’continuous’ antigenic determinants in the protruding regions of proteins. EMBO J. 5, 409–413 (1986).
Ponomarenko, J. et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 9, 514 (2008).
Emini, E. A., Hughes, J. V., Perlow, D. S. & Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 55, 836–839 (1985).
Ingraham, J., Garg, V. K., Barzilay, R. & Jaakkola, T. Generative Models for Graph-Based Protein Design. NIPS 2019 (2019).
Strokach, A., Becerra, D., Corbi-Verge, C. & Kim, P. M. Fast and flexible protein design using deep graph neural networks. Cell Syst. 11, 402–411.e4 (2020).
Fout, A., Byrd, J., Shariat, B. & Ben-Hur A. Protein interface prediction using graph convolutional networks. In: Advances in Neural Information Processing Systems. vol. 30 (Curran Associates, Inc., 2017).
Yuan, Q., Chen, J., Zhao, H., Zhou, Y. & Yang, Y. Structure-aware protein–protein interaction site prediction using deep graph convolutional network. Bioinformatics 38, 125–132 (2021).
Abdollahi, N., Tonekaboni, S. A. M., Huang, J., Wang, B. & MacKinnon, S. NodeCoder: a graph-based machine learning platform to predict active sites of modeled protein structures. arXiv https://doi.org/10.48550/arXiv.2302.03590 (2023).
Cha, M. et al. Unifying structural descriptors for biological and bioinspired nanoscale complexes. Nat. Comput. Sci. 2, 243–252 (2022).
Roche, R., Moussad, B., Shuvo, M. H. & Bhattacharya, D. E(3) equivariant graph neural networks for robust and accurate protein-protein interaction site prediction. PLoS Comput. Biol. 19, e1011435 (2023).
Ferreira, M. V., Nogueira, T., Rios, R. A., Lopes, T. J. S. A graph-based machine learning framework identifies critical properties of FVIII that lead to Hemophilia A. Front. Bioinform. 3, 1152039 (2023).
Zhou, J. et al. Graph neural networks: a review of methods and applications. AI Open 1, 57–81 (2020).
Hsu, C. et al. Learning inverse folding from millions of predicted structures. In: Proceedings of the 39th International Conference on Machine Learning. p. 8946–8970 (PMLR, 2022).
Muhammed, M. T. & Aki-Yalcin, E. Homology modeling in drug discovery: overview, current applications, and future perspectives. Chem. Biol. Drug Des. 93, 12–20 (2019).
Ambrosetti, F., Jiménez-García, B., Roel-Touris, J. & Bonvin, A. M. J. J. Modeling antibody-antigen complexes by information-driven docking. Structure 28, 119–129.e2 (2020).
Schoeder, C. T. et al. Modeling immunity with rosetta: methods for antibody and antigen design. Biochemistry 60, 825–846 (2021).
Peacock, T. & Chain, B. Information-driven docking for TCR-pMHC complex prediction. Front. Immunol. 12, 686127 (2021).
Atanasova, M. & Doytchinova, I. Docking-based prediction of peptide binding to MHC proteins. Methods Mol. Biol. 2673, 237–249 (2023).
Dormitzer, P. R., Ulmer, J. B. & Rappuoli, R. Structure-based antigen design: a strategy for next generation vaccines. Trends Biotechnol. 26, 659–667 (2008).
Higgins, M. K. Can we AlphaFold our way out of the next pandemic? J. Mol. Biol. 433, 167093 (2021).
Pavlova, A. et al. Machine learning reveals the critical interactions for SARS-CoV-2 spike protein binding to ACE2. J. Phys. Chem. Lett. 12, 5494–5502 (2021).
Benevenuta, S., Pancotti, C., Fariselli, P., Birolo, G. & Sanavia, T. An antisymmetric neural network to predict free energy changes in protein variants. J. Phys. D: Appl. Phys. 54, 245403 (2021).
Li, B., Yang, Y. T., Capra, J. A. & Gerstein, M. B. Predicting changes in protein thermodynamic stability upon point mutation with deep 3D convolutional neural networks. PLoS Comput. Biol. 16, e1008291 (2020).
Pucci, F., Schwersensky, M. & Rooman, M. Artificial intelligence challenges for predicting the impact of mutations on protein stability. Curr. Opin. Struct. Biol. 72, 161–168 (2022).
Dauparas, J. et al. Robust deep learning–based protein sequence design using proteinMPNN. Science 378, 49–56 (2022).
Senior, A. W. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. biorxiv https://doi.org/10.1101/2021.10.04.463034 (2022).
Du, Z. et al. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 16, 5634–5651 (2021).
Baek, M. et al. Accurate prediction of protein structures and interactions using a 3-track neural network. Science 373, 871–876 (2021).
Hederman, A. P. & Ackerman, M. E. Leveraging deep learning to improve vaccine design. Trends Immunol. 44, 333–344 (2023).
Ruffolo, J. A., Guerra, C., Mahajan, S. P., Sulam, J. & Gray, J. J. Geometric potentials from deep learning improve prediction of CDR H3 loop structures. Bioinformatics 36, i268–i275 (2020).
Ruffolo, J. A., Sulam, J. & Gray, J. J. Antibody structure prediction using interpretable deep learning. Patterns 3, 100406 (2022).
Abanades, B., Georges, G., Bujotzek, A. & Deane, C. M. ABlooper: fast accurate antibody cdr loop structure prediction with accuracy estimation. Bioinformatics 38, 1877–1880 (2022).
Ruffolo, J. A., Chu, L. S., Mahajan, S. P. & Gray, J. J. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Nat. Commun. 14, 2389 (2023).
Bradley, P. Structure-based prediction of T cell receptor: peptide-MHC interactions. eLife 12, e82813 (2023).
Chinery, L., Wahome, N., Moal, I. & Deane, C. M. Paragraph—antibody paratope prediction using graph neural networks with minimal feature vectors. Bioinformatics 39, btac732 (2023).
Grifoni, A. et al. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 27, 671–680.e2 (2020).
Vitiello, A. & Zanetti, M. Neoantigen prediction and the need for validation. Nat. Biotechnol. 35, 815–817 (2017).
Bassani-Sternberg, M. & Gfeller, D. Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide–HLA interactions. J. Immunol. 197, 2492–2499 (2016).
Bassani-Sternberg, M. et al. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput. Biol. 13, e1005725 (2017).
Gfeller, D. et al. The length distribution and multiple specificity of naturally presented HLA-I ligands. J. Immunol. 201, 3705–3716 (2018).
O’Donnell, T. J. et al. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 7, 129–132.e4 (2018).
O’Donnell, T. J., Rubinsteyn, A. & Laserson, U. MHCflurry 2.0: improved pan-allele prediction of MHC class I-presented peptides by incorporating antigen processing. Cell Syst. 11, 42–48.e7 (2020).
Nielsen, M. et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE 2, e796 (2007).
Lundegaard, C. et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC Class I affinities for peptides of length 8–11. Nucleic Acids Res. 36, W509–W512 (2008).
Andreatta, M. & Nielsen, M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 511–517 (2016).
Jurtz, V. et al. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368 (2017).
Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454 (2020).
Bravi, B. et al. RBM-MHC: a semi-supervised machine-learning method for sample-specific prediction of antigen presentation by HLA-I alleles. Cell Syst. 12, 195–202.e9 (2021).
Abelin, J. G. et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 46, 315–326 (2017).
Abelin, JG. et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity 51, 766–779.e17 (2019).
Sarkizova, S. et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 38, 199–209 (2020).
Racle, J. et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 37, 1283–1286 (2019).
Lawrence, P. J. & Ning, X. Improving MHC class I antigen-processing predictions using representation learning and cleavage site-specific kernels. Cell Rep. Methods 2, 100293 (2022).
Reynisson, B. et al. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. J. Proteome Res. 19, 2304–2315 (2020).
Racle, J. et al. Machine learning predictions of MHC-II specificities reveal alternative binding mode of class II epitopes. Immunity 56, 1359–1375.e13 (2023).
Nilsson, J. B. et al. Machine learning reveals limited contribution of trans-only encoded variants to the HLA-DQ immunopeptidome. Commun. Biol. 6, 1–13 (2023).
Calis, J. J. A. et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 9, e1003266 (2013).
Trolle, T. & Nielsen, M. NetTepi: an integrated method for the prediction of T cell epitopes. Immunogenetics 66, 449–456 (2014).
Chowell, D. et al. TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Natl Acad. Sci. USA 112, E1754–E1762 (2015).
Ogishi, M. & Yotsuyanagi, H. Quantitative prediction of the landscape of T cell epitope immunogenicity in sequence space. Front. Immunol. 10, 827 (2019).
Riley, T. P. et al. Structure based prediction of neoantigen immunogenicity. Front. Immunol. 10, 2047 (2019).
Schmidt, J. et al. Prediction of neo-epitope immunogenicity reveals TCR recognition determinants and provides insight into immunoediting. Cell Rep. Med. 2, 100194 (2021).
Gfeller, D. et al. Improved predictions of antigen presentation and TCR recognition with MixMHCpred2.2 and PRIME2.0 reveal potent SARS-CoV-2 CD8+ T-cell epitopes. Cell Syst. 14, 72–83.e5 (2023).
Dhanda, S. K. et al. Predicting HLA CD4 immunogenicity in human populations. Front. Immunol. 9, 1369 (2018).
Bravi, B. et al. A transfer-learning approach to predict antigen immunogenicity and T-cell receptor specificity. eLife 12, e85126 (2023).
Finotello, F., Rieder, D., Hackl, H. & Trajanoski, Z. Next-generation computational tools for interrogating cancer immunity. Nat. Rev. Genet. 20, 724–746 (2019).
Roudko, V., Greenbaum, B. & Bhardwaj, N. Computational prediction and validation of tumor-associated neoantigens. Front. Immunol. 11, 27 (2020).
Roesler, A. S. & Anderson, K. S. Beyond sequencing: prioritizing and delivering neoantigens for cancer vaccines. Methods Mol. Biol. 2410, 649–670 (2022).
Wells, D. K. et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell 183, 818–834.e13 (2020).
Schaap-Johansen, A. L., Vujović, M., Borch, A., Hadrup, S. R. & Marcatili, P. T cell epitope prediction and its application to immunotherapy. Front. Immunol. 12, 2994 (2021).
Croft, N. P. et al. Most viral peptides displayed by class I MHC on infected cells are immunogenic. Proc. Natl Acad. Sci. USA 116, 3112–3117 (2019).
Bjerregaard, A. M. et al. An analysis of natural T cell responses to predicted tumor neoepitopes. Front. Immunol. 8, 1566 (2017).
Kristensen, N. P. et al. Neoantigen-reactive CD8+ T cells affect clinical outcome of adoptive cell therapy with tumor-infiltrating lymphocytes in melanoma. J. Clin. Investig. 132, e150535 (2022).
Meysman, P. et al. Benchmarking solutions to the T-cell receptor epitope prediction problem: IMMREP22 workshop report. ImmunoInformatics 9, 100024 (2023).
Liberis, E., Veličković, P., Sormanni, P., Vendruscolo, M. & Liò, P. Parapred: antibody paratope prediction using convolutional and recurrent neural networks. Bioinformatics 34, 2944–2950 (2018).
Ambrosetti, F. et al. proABC-2: PRediction of AntiBody contacts v2 and its application to information-driven docking. Bioinformatics 36, 5107–5108 (2020).
Daberdaku, S. & Ferrari, C. Antibody interface prediction with 3D zernike descriptors and SVM. Bioinformatics 35, 1870–1876 (2019).
Isacchini, G., Walczak, A. M., Mora, T. & Nourmohammad, A. Deep generative selection models of T and B cell receptor repertoires with soNNia. Proc. Natl Acad. Sci. USA. 118, e2023141118 (2021).
Bravi, B. et al. Probing T-cell response by sequence-based probabilistic modeling. PLoS Comput. Biol. 17, e1009297 (2021).
Wu, K. et al. TCR-BERT: learning the grammar of T-cell receptors for flexible antigen-binding analyses. The 2022 ICML Workshop on Computational Biology (2022).
Sidhom, J. W., Larman, H. B., Pardoll, D. M. & Baras, A. S. DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nat. Commun. 12, 1605 (2021).
Leem, J., Mitchell, L. S., Farmery, J. H. R., Barton, J. & Galson, J. D. Deciphering the language of antibodies using self-supervised learning. Patterns 3, 100513 (2022).
Akbar, R. et al. In silico proof of principle of machine learning-based antibody design at unconstrained scale. mAbs 14, 2031482 (2022).
Saka, K. et al. Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci. Rep. 11, 5852 (2021).
Shin, J. E. et al. Protein design and variant prediction using autoregressive generative models. Nat. Commun. 12, 2403 (2021).
Jokinen, E., Huuhtanen, J., Mustjoki, S., Heinonen, M. & Lähdesmäki, H. Predicting recognition between T cell receptors and epitopes with TCRGP. PLoS Comput. Biol. 17, e1008814 (2021).
Zhang, W. et al. A framework for highly multiplexed dextramer mapping and prediction of T cell receptor sequences to antigen specificity. Sci. Adv. 7, eabf5835 (2021).
Gielis, S. et al. Detection of enriched T cell epitope specificity in full T cell receptor sequence repertoires. Front. Immunol. 10, 2820 (2019).
Croce, G. et al. Deep learning predictions of TCR-epitope interactions reveal epitope-specific chains in dual alpha T cells. bioRxiv https://doi.org/10.1101/2023.09.13.557561 (2023).
Dash, P. et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017).
Glanville, J. et al. Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017).
Mayer-Blackwell, K. et al. TCR meta-clonotypes for biomarker discovery with Tcrdist3 Enabled identification of public, HLA-restricted clusters of SARS-CoV-2 TCRs. eLife 10, e68605 (2021).
Weber, A., Born, J. & Rodriguez Martínez, M. TITAN: T-cell receptor specificity prediction with bimodal attention networks. Bioinformatics 37, i237–i244 (2021).
Moris, P. et al. Current challenges for unseen-epitope TCR interaction prediction and a new perspective derived from image classification. Brief. Bioinform. 22, bbaa318 (2021).
Springer, I., Tickotsky, N. & Louzoun, Y. Contribution of T cell receptor alpha and beta CDR3, MHC typing, V and J genes to peptide binding prediction. Front. Immunol. 12, 664514 (2021).
Lu, T. et al. Deep learning-based prediction of the T cell receptor–antigen binding specificity. Nat. Mach. Intell. 3, 864–875 (2021).
Xu, Z. et al. DLpTCR: an ensemble deep learning framework for predicting immunogenic peptide recognized by T cell receptor. Brief. Bioinform. 22, bbab335 (2021).
Grazioli, F. et al. Attentive variational information bottleneck for TCR–peptide interaction prediction. Bioinformatics 39, btac820 (2023).
Gao, Y. et al. Pan-peptide meta learning for T-cell receptor–antigen binding recognition. Nat. Mach. Intell. 5, 236–249 (2023).
Huang, Y., Zhang, Z. & Zhou, Y. AbAgIntPre: a deep learning method for predicting antibody-antigen interactions based on sequence information. Front. Immunol. 13, 1053617 (2022).
Schneider, C., Buchanan, A. & Taddese, B. DLAB: deep learning methods for structure-based virtual screening of antibodies. Bioinformatics 38, 377–383 (2022).
Milighetti, M., Shawe-Taylor, J. & Chain, B. Predicting T cell receptor antigen specificity from structural features derived from homology models of receptor-peptide-major histocompatibility complexes. Front. Physiol. 12, 730908 (2021).
Montemurro, A. et al. NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCRα and β sequence data. Commun. Biol. 4, 1–13 (2021).
Jensen, M. F. & Nielsen, M. NetTCR 2.2 - improved TCR specificity predictions by combining pan- and peptide-specific training strategies, loss-scaling and integration of sequence similarity. bioRxiv https://doi.org/10.1101/2023.10.12.562001 (2023).
Meynard-Piganeau, B., Feinauer, C., Weigt, M., Walczak, A. M. & Mora, T. TULIP — a transformer based unsupervised language model for interacting peptides and T-cell receptors that generalizes to unseen epitopes. bioRxiv https://doi.org/10.1101/2023.07.19.549669 (2023).
Pittala, S. & Bailey-Kellogg, C. Learning context-aware structural representations to predict antigen and antibody binding interfaces. Bioinformatics 36, 3996–4003 (2020).
Myung, Y., Pires, D. E. V. & Ascher, D. B. CSM-AB: graph-based antibody-antigen binding affinity prediction and docking scoring function. Bioinformatics 38, 1141–1143 (2022).
Yang, Y. X., Wang, P. & Zhu, B. T. Binding affinity prediction for antibody-protein antigen complexes: a machine learning analysis based on interface and surface areas. J. Mol. Graph. Model. 118, 108364 (2023).
De Neuter, N. et al. On the feasibility of mining CD8+ T cell receptor patterns underlying immunogenic peptide recognition. Immunogenetics 70, 159–168 (2018).
Tong, Y. et al. SETE: sequence-based ensemble learning approach for TCR epitope binding prediction. Comput. Biol. Chem. 87, 107281 (2020).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Bepler, T. & Berger, B. Learning the protein language: evolution, structure, and function. Cell Syst. 12, 654–669.e3 (2021).
Dens, C., Bittremieux, W., Affaticati, F., Laukens, K. & Meysman, P. Interpretable deep learning to uncover the molecular binding patterns determining TCR–epitope interaction predictions. ImmunoInformatics 11, 100027 (2023).
Rodríguez Martínez, M., Barberis, M. & Niarakis, A. Computational modelling of immunological mechanisms: from statistical approaches to interpretable machine learning. ImmunoInformatics. 12, 100029 (2023).
Askin, S., Burkhalter, D. & Calado, G. Artificial intelligence applied to clinical trials: opportunities and challenges. Health Technol. 13, 203–213 (2023).
Olimpieri, P. P., Chailyan, A., Tramontano, A. & Marcatili, P. Prediction of site-specific interactions in antibody-antigen complexes: The proABC method and server. Bioinformatics 29, 2285–2291 (2013).
Ribeiro, M. T., Singh, S. & Guestrin, C. Anchors: high-precision model-agnostic explanations. Proc. AAAI Conf. Artif. Intell. 32 https://ojs.aaai.org/index.php/AAAI/article/view/11491 (2018).
Papadopoulou, I., Nguyen, A. P., Weber, A. & Martínez, M. R. DECODE: a computational pipeline to discover T cell receptor binding rules. Bioinformatics 38, i246–i254 (2022).
Vig, J. et al. BERTology meets biology: interpreting attention in protein language models. In 9th International Conference on Learning Representations (ICLR, 2021).
Bronstein, MM., Bruna, J., Cohen, T., Veličković, P. Geometric deep learning: grids, groups, graphs, geodesics, and gauges. arXiv https://doi.org/10.48550/arXiv.2104.13478 (2021).
Malone, B. et al. Artificial intelligence predicts the immunogenic landscape of SARS-CoV-2 leading to universal blueprints for vaccine designs. Sci. Rep. 10, 22375 (2020).
Samad, A. et al. Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J. Biomol. Struct. Dyn. 40, 14–30 (2022).
Thomas, S., Abraham, A., Baldwin, J., Piplani, S. & Petrovsky, N. Artificial intelligence in vaccine and drug design. Methods Mol. Biol. 2410, 131–146 (2022).
Källberg, M. et al. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522 (2012).
Kozakov, D. et al. The ClusPro web server for protein–protein docking. Nat. Protoc. 12, 255–278 (2017).
Kim, C. K. et al. AllergenPro: an integrated database for allergenicity analysis and prediction. Bioinformation 10, 378–380 (2014).
Dimitrov, I., Naneva, L., Doytchinova, I. & Bangov, I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30, 846–851 (2014).
Bui, H. H. et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 7, 153 (2006).
Gasteiger, E. et al. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook. Springer Protocols Handbooks. (ed. Walker, J. M.) 571–607 (Humana Press, 2005).
Magnan, C. N., Randall, A. & Baldi, P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 25, 2200–2207 (2009).
Rapin, N., Lund, O., Bernaschi, M. & Castiglione, F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 5, e9862 (2010).
Khurana, S. et al. DeepSol: a deep learning framework for sequence-based protein solubility prediction. Bioinformatics 34, 2605–2613 (2018).
Ansari, M. & White, A. D. Serverless prediction of peptide properties with recurrent neural networks. J. Chem. Inf. Model. 63, 2546–2553 (2023).
Hioki, K. et al. Machine learning-assisted screening of herbal medicine extracts as vaccine adjuvants. Front. Immunol. 13, 847616 (2022).
Wayment-Steele, H. K. et al. Deep learning models for predicting RNA degradation via dual crowdsourcing. Nat. Mach. Intell. 4, 1174–1184 (2022).
Zohdi, T. I. Machine-learning and digital-twins for rapid evaluation and design of injected vaccine immune-system responses. Comput. Methods Appl. Mech. Eng. 401, 115315 (2022).
Pappalardo, F., Russo, G., Tshinanu, F. M. & Viceconti, M. In silico clinical trials: concepts and early adoptions. Brief. Bioinform. 20, 1699–1708 (2019).
Chaudhari, N., Ravi, R., Gogtay, N. J. & Thatte, U. M. Recruitment and retention of the participants in clinical trials: challenges and solutions. Perspect. Clin. Res. 11, 64–69 (2020).
Weissler, E. H. et al. The role of machine learning in clinical research: transforming the future of evidence generation. Trials 22, 537 (2021).
Jain, S., Xiao, X., Bogdan, P. & Bruck, J. Generator based approach to analyze mutations in genomic datasets. Sci. Rep. 11, 21084 (2021).
Rodriguez-Rivas, J., Croce, G., Muscat, M. & Weigt, M. Epistatic models predict mutable sites in SARS-CoV-2 proteins and epitopes. Proc. Natl Acad. Sci. USA 119, e2113118119 (2022).
Łuksza, M. & Lässig, M. A predictive fitness model for influenza. Nature 507, 57–61 (2014).
Barton, J. P. et al. Relative rate and location of intra-host HIV evolution to evade cellular immunity are predictable. Nat. Commun. 7, 11660 (2016).
Paul, S. et al. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J. Immunol. 191, 5831–5839 (2013).
Mora, T. & Walczak, A. M. How many different clonotypes do immune repertoires contain. Curr. Opin. Syst. Biol. 18, 104–110 (2019).
Mason, D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 19, 395–404 (1998).
Birnbaum, M. E. et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157, 1073–1087 (2014).
Bradley, P. & Thomas, P. G. Using T cell receptor repertoires to understand the principles of adaptive immune recognition. Annu. Rev. Immunol. 37, 547–570 (2019).
Leem, J., de Oliveira, S. H. P., Krawczyk, K. & Deane, C. M. STCRDab: the structural T-cell receptor database. Nucleic Acids Res. 46, D406–D412 (2018).
Hudson, D., Fernandes, RA., Basham, M., Ogg, G. & Koohy, H. Can we predict T cell specificity with digital biology and machine learning? Nat. Rev. Immunol. 23, 1–11 (2023).
Dalsass, M., Brozzi, A., Medini, D. & Rappuoli, R. Comparison of open-source reverse vaccinology programs for bacterial vaccine antigen discovery. Front. Immunol. 10, 113 (2019).
Gfeller, D. & Bassani-Sternberg, M. Predicting antigen presentation—what could we learn from a million peptides. Front. Immunol. 9, 1716 (2018).
Li, G., Iyer, B., Prasath, V. B. S., Ni, Y. & Salomonis, N. Deepimmuno: deep learning-empowered prediction and generation of immunogenic peptides for T-cell immunity. Brief. Bioinform. 22, bbab160 (2021).
Chen, B. et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 37, 1332–1343 (2019).
Rigo, M. M. et al. DockTope: a web-based tool for automated pMHC-I modelling. Sci. Rep. 5, 18413 (2015).
Koşaloğlu-Yalçin, Z. et al. Combined assessment of MHC binding and antigen abundance improves T cell epitope predictions. iScience 25, 103850 (2022).
Batra, R. et al. Screening of therapeutic agents for COVID-19 using machine learning and ensemble docking studies. J. Phys. Chem. Lett. 11, 7058–7065 (2020).
Sehnal, D., Rose, A. S., Koča J., Burley, S. K. & Velankar, S. Mol*: towards a common library and tools for web molecular graphics. in Proceedings of the Workshop on Molecular Graphics and Visual Analysis of Molecular Data. MolVA ’18. Brno, Czech Republic. p. 29–33 (Eurographics Association, 2018).
Bengio, Y., Courville, A. & Vincent, P. Representation learning: a review and new perspectives. IEEE Trans. Pattern Anal. Mach. Intell. 35, 1798–1828 (2013).
Detlefsen, N. S., Hauberg, S. & Boomsma, W. Learning meaningful representations of protein sequences. Nat. Commun. 13, 1914 (2022).
Kingma, D. P. & Welling, M. Auto-encoding variational Bayes. arXiv https://doi.org/10.48550/arXiv.1312.6114 (2014).
Goodfellow, I. J. et al. Generative adversarial nets. in Proceedings of the 27th International Conference on Neural Information Processing Systems - Volume 2. NIPS’14. p. 2672–2680 (MIT Press, 2014).
Sohl-Dickstein, J., Weiss, E., Maheswaranathan, N. & Ganguli, S. Deep unsupervised learning using nonequilibrium thermodynamics. in Proceedings of the 32nd International Conference on Machine Learning. p. 2256–2265 (PMLR, 2015).
Acknowledgements
B.B. wishes to thank Kevin Michalewicz, Leo Swadling, Haowen Zhao for their feedback and careful reading of the manuscript, as well as the anonymous referees for their suggestions and for pointing out several useful references.
Author information
Authors and Affiliations
Contributions
B.B., as the sole author, fulfills all authorship criteria.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bravi, B. Development and use of machine learning algorithms in vaccine target selection. npj Vaccines 9, 15 (2024). https://doi.org/10.1038/s41541-023-00795-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-023-00795-8
