
Abstract
High-grade serous ovarian carcinoma (HGSC) is the most lethal gynecologic malignancy due to the lack of reliable biomarkers, effective treatment, and chemoresistance. Improving the diagnosis and the development of targeted therapies is still needed. The molecular pathomechanisms driving HGSC progression are not fully understood though crucial for effective diagnosis and identification of novel targeted therapy options. The oncogene CTCFL (BORIS), the paralog of CTCF, is a transcriptional factor highly expressed in ovarian cancer (but in rarely any other tissue in females) with cancer-specific characteristics and therapeutic potential. In this work, we seek to understand the regulatory functions of CTCFL to unravel new target genes with clinical relevance. We used in vitro models to evaluate the transcriptional changes due to the presence of CTCFL, followed by a selection of gene candidates using de novo network enrichment analysis. The resulting mechanistic candidates were further assessed regarding their prognostic potential and druggability. We show that CTCFL-driven genes are involved in cytoplasmic membrane functions; in particular, the PI3K-Akt initiators EGFR1 and VEGFA, as well as ITGB3 and ITGB6 are potential drug targets. Finally, we identified the CTCFL targets ACTBL2, MALT1 and PCDH7 as mechanistic biomarkers to predict survival in HGSC. Finally, we elucidated the value of CTCFL in combination with its targets as a prognostic marker profile for HGSC progression and as putative drug targets.
Similar content being viewed by others


Introduction
High-grade serous carcinoma (HGSC) is the most common type of ovarian cancer (OC) and the most lethal gynecologic malignancy1. The main reasons for the high mortality is the late diagnosis due to the lack of reliable biomarkers (60% of the tumors are detected once they have metastasized)2 and lack of effective treatment; i.e., current treatment involves surgical resection and standard chemotherapy, which has several side effects and tumors usually become resistant to it3. Thus, improving the diagnosis and the development of novel targeted therapies for HGSC is an ongoing research task since few previously proposed targeted therapies have been tested4. Furthermore, OC is a highly heterogeneous disease and the molecular mechanisms that drive OC progression and chemoresistance are not fully understood. Therefore, the identification of molecular pathway activity aberration in OC is a crucial first step in the development of effective diagnosis and novel targeted therapy options. Recent genome-wide studies show that, at the genetic level, the most frequent alterations in HGSC are in the P53 pathway, including mutations in the TP53 gene5. At the transcriptional level, Immuno, hormone-related, and MAPK signaling pathways are deregulated in specific clusters of HGSC patients6.
Furthermore, integrative omic analyses of OC tumors indicate that the protein CTCFL is a relevant molecular driver of OC7,8. CTCFL (BORIS) is the paralog of the CTCF gene, which encodes a ubiquitous well-known transcription factor (TF) with an 11-zinc-finger DNA-binding domain that recognizes binding sites (BSs) in the genome and participates in the establishment of chromatin organization and transcriptional regulation9,10. Likewise, CTCFL is a transcriptional regulator that competes for the same BSs with CTCF11. CTCFL has gained interest because omics data show that it is a cancer-specific protein: it is highly expressed in OC but absent in healthy normal tissue in women, except in testis (www.proteinatlas.org/ENSG00000124092-CTCFL)9,12,13. Due to its particular tissue specificity, CTCFL is classified as a cancer-testis antigen (CTA), a family of proteins with therapeutic importance in cancer14,15. Notably, CTCFL is considered an oncogene16 and appears to be a master TF that maintains a stemness state in cancer17 through the transcriptional regulation of several well-characterized oncogenes and also other CTAs in several cancer types18,19,20,21.
Integrative OC-specific omic analyses have found that CTCFL is a molecular marker of HGSC in three different levels: DNA-methylation, gene expression, and DNA copy number22. Also, CTCFL plays an important role in the progression of HGSC; for instance, Hillman et al. demonstrated that CTCFL expression could be a key factor of initiation8 and additional studies indicate that CTCFL can contribute to OC progression through different mechanisms; such as androgen receptor-associated pathway7, dysregulation of hTERT telomerase23 and it is associated with poor prognosis and advanced stage24.
Together, the above makes CTCFL an exciting and promising unique target for OC treatment or biomarker development25,26. Despite the notable improvements in the study of CTCFL in cancer, a detailed examination of the transcriptional effects of CTCFL expression in OC remains to be fully characterized, as well as the extent to which it influences other cancer-related processes and their potential application for the development of mechanistic therapies for OC.
In this work, we study the regulatory functions of CTCFL in OC at the transcriptional level to identify mechanistic targets with clinical relevance in OC. We used in vitro models to evaluate the transcriptional changes due to knockdown and overexpression of CTCFL; followed by a selection of gene candidates, the identification of CTCFL-DNA-binding sites and de novo pathway enrichment analysis for the identification of potential OC driver mechanisms controlled by CTCFL. Finally, the expression level of candidate genes were utilized as endophenotypic marker profiles to assess their prognostic power as molecular signature and their druggability potential.
Results
CTCFL-regulated genes participate in cell motility, membrane transport, and extracellular matrix-related processes
In this study, we aim to identify genes regulated by CTCFL that are of clinical importance in OC, either because they are associated with prognosis or because they could be useful as targeted therapy. To address this goal, we used an OC-derived cell line model (OVCAR3) and OC tumor samples (TCGA) to follow three main analysis steps (Fig. 1a). Characterization of CTCFL transcript and protein levels was previously published in7, showing that it is an HGSOC-derived cell line with high levels of CTCFL, making it an appropriate model for the following experiments. First, we evaluated the transcriptional profiles of OVCAR3 cells with knockdown and overexpression of CTCFL to identify the differentially expressed genes (DEGs) due to the presence of CTCFL. Following, we selected gene candidates by examining the DEGs with two complementary analyses, the identification of CTCFL binding sites (BSs) and de novo pathway enrichment analysis. Finally, the expression level of candidate genes (33 genes + CTCFL) was further analyzed in normal (GTEx) and ovarian tumor (TCGA) samples to assess their prognostic and druggable potential.

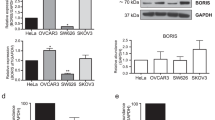
a Schematic representation of the workflow followed in this study. RNA-seq data were obtained from OVCAR3 cell lines with CTCFL overexpressed and underexpressed from Salgado-Albarran et al.7 and subjected to differential expression analysis. Next, CTCFL BS identification and network enrichment analysis was performed to obtain a candidate set of genes. Finally, the candidate gene list was evaluated in OC-derived tumor samples and normal tissue. b Experimental validation of CTCFL overexpression at the protein level by Western Blot. The upper panel shows the protein abundance obtained by densitometry. BORIS band is 65 kDa. (*) indicate nonspecific bands. c Transcript levels of CTCFL and CTCF in each experimental condition. d Change in gene expression and statistical confidence of CTCFL and CTCF in each experimental condition vs control. e Venn diagram with the number of DEGs found in each and shared between experimental conditions. f Relative gene expression (row z-score of normalized reads) of the 149 DEGs found in both experimental conditions classified by the direction of gene expression change. Gene names in red indicate previously reported CTCFL transcriptional targets7,31,37,89.
OC shows the highest expression of CTCFL among cancer types; however, the downstream genes and pathways deregulated by it are not fully described in OC. In order to obtain a more detailed landscape of the transcriptional changes occurring due to CTCFL expression and identify potential prognostic and druggable targets for treatment, we performed a differential expression analysis with RNA-seq data obtained from two cellular conditions: CTCFL knockdown cells (CTCFL-KD) previously characterized7 and the overexpression of CTCFL (CTCFL-OE). The increase in the protein levels of the experimental conditions was verified by Western Blot (Fig. 1b and Supplementary Fig. 1a) and at the transcript level (Fig. 1c), where CTCFL transcripts show a 2.99 log2 fold increase and a 4.46 log2 fold decrease (Fig. 1d) in the corresponding conditions. Furthermore, we also confirm that CTCF expression levels did not vary (Fig. 1d); thus, confirming that the experimental approach to modify CTCFL expression was specific.
Once the experimental conditions were validated at the expression and protein levels, two differential expression analyses were performed with the transcriptional data: CTCFL-OE vs Control and CTCFL-KD vs Control. A comparative examination of the identified DEGs shows that 86 DEGs are found when CTCFL is overexpressed only, 2713 DEGs when CTCFL is decreased and 149 DEGs are found in both conditions (Fig. 1e). PCA plot of the samples (Supplementary Fig. 1b) shows that the difference in the number of DEGs found in each condition is due to little change in OE compared to KD, and not because of sample variation within a condition. A possible explanation for this is the fact the OVCAR3 is a cancer cell line that already expresses CTCFL; thus, the exogenous expression might not have a big effect on transcription because target binding sites are already occupied, while the decrease using KD leads to great differences in expression even with small changes in CTCFL levels.
Functional enrichment analysis of the DEG found with the overexpression of CTCFL (86 genes) shows that the proteins encoded by them are localized mainly in the cytoplasmic membrane and cell periphery compartment (Supplementary Fig. 1c). Notably, among the deregulated genes localized in these compartments are membrane transporters; such as members of the solute carrier family (SLC26A7, SLC8A1, SLC6A12, SLC7A2, and SLC03A1) and ATP-binding cassette (ABCC6). Likewise, we identified cytoskeleton-related proteins (SGIP1, MAPT, PACSIN1, ACTG1, and VIM), cell adhesion-related (SCUBE1, ICAM5, and BVES), motility-related (ENPP2, CSPG4, and C5AR1), metalloproteinases (ADAM11), the insulin receptor (INSR), and one of its known ligands (IGF1) (Supplementary Fig. 1d).
Likewise, when evaluating the DEGs after the knockdown of CTCFL (2713 genes), components of the cell membrane, cell periphery, and cytoskeleton stand out for being differentially expressed (Supplementary Fig. 2a). In particular, similar to the observed in the opposite experimental condition, 50 members of the solute carrier family are differentially expressed (Supplementary Fig. 2b) and cytoskeleton-related proteins (122 DEGs), including ACTB and ACTR2 (Supplementary Fig. 2c). Together, the latter indicates that, while the majority of the genes affected by the presence and absence of CTCFL are different, at the functional level, many of these genes belong to the same protein families or participate in the same cellular processes; i.e., cell motility (cytoskeleton), and membrane transport, which are well-known mechanisms associated with the development of metastases27,28 and tumor resistance to drugs29,30; suggesting that CTCFL could play an important role in OC metastases.
After the examination of the DEGs uniquely identified in the experimental conditions, we also explored the DEGs found in both conditions: CTCFL overexpression and underexpression (149 genes). In this case, we also found previously validated targets of CTCFL (Supplementary Fig. 3a, b), such as TGFB131, FN17, and the H19 gene, which has been widely studied as a target of CTCFL and CTCF given its DNA-methylation dependent transcriptional regulation32,33.
In addition to the already-known targets, we further identified relevant cancer-associated genes; such as, FGFR1, VEGFA, WNT9A, and FZD4 (Fig. 1f). Moreover, we found that similar to the described above, the DEGs are mainly involved in the extracellular matrix or cell motility-related processes, such as actin (ACTBL2), kinesins (KIF1A and KIF3C), laminin (LAMC3 and LAMA), microtubule-associated (MAP2 and MAP1B) and solute carrier family genes (SLC7A8, SLC38A4, and SLC6A9) (Fig. 1f and Supplementary Fig. 3c).
Together, these results show that, in general, the cellular effect due to the presence of CTCFL is similar, being the most relevant effects, those associated with extracellular matrix functions, cell motility, and cytoplasmic membrane transporters. Furthermore, these cellular effects are achieved through diverse deregulated genes between experimental conditions.
CTCFL-induced protein interaction network is composed of key PI3K-Akt signaling pathway and extracellular matrix genes
After examining the DEGs found in each experimental condition, we aimed to select relevant genes with potential mechanistic molecular importance in OC by analyzing the shared DEGs with two approaches: the identification of CTCFL BSs and de novo protein pathway enrichment analysis.
First, we searched BSs in the promoters of the 149 DEGs using the reported DNA motif of CTCFL (Jaspar and Factorbook) and the HMMer tools for motif search (Fig. 2a)34,35,36. A total of 48 differentially expressed genes showed at least one CTCFL BSs in its promoter (Supplementary Data 2). Next, these genes were further analyzed by selecting the ones that show occupancy of CTCFL and CTCF using publicly available ChIP-seq data from Pugacheva et al.11, where six genes (Fig. 2b) showed ChIP-seq peak that matches the BSs identified (VEGFA, IL12A, WSCD2, RBM20, TGFB1, and MAP1B). Notably, the CTCFL gene contains a CTCFL BSs in its own promoter, in line with previous reports showing a CTCF-controlled transcription37. Furthermore, we also identified TGFB1, which was previously validated as a direct target of CTCFL31. Together, these results support our analyses and allow us to propose new previously uncharacterized CTCFL targets. In this regard, VEGFA is a previously uncharacterized target of CTCFL that plays a key role in angiogenesis38. We found VEGFA differentially expressed and its promoter is occupied by both CTCFL and CTCF in OC cell lines; thus, it is a potential direct target of CTCFL in OC. The latter is of great clinical relevance given the availability of precision therapies using VEGFA pathways as targets in other cancer types39 and growing evidence shows that this could be a promising target in OC40.

a Schematic representation of the workflow followed for the identification of DEGs with CTCFL BS. Sequences flanking the TSS of DEGs found in both experimental conditions were retrieved and used as input for motif identification with HMMer using the reported CTCFL motifs in Jaspar and Factorbook. Next, genes with a predicted BS and experimentally validated CTCFL and CTCF occupancy (ChIP-seq) were selected. b Visual representation of CTCFL and CTCF occupancy in predicted CTCFL BSs of the genes VEGFA, IL12A, WSCD2, RBM20, TGFB1, MAP1B, and CTCFL. c Protein interaction network enriched with CTCFL-deregulated genes found in vitro. d Functional enrichment analysis of genes in the protein interaction network using KEGG, Reactome, and Wikipathway databases.
Next, in order to identify an underlying relevant protein pathway affected by the presence of CTCFL we obtained the largest interaction network enriched with the DEGs using KeyPathwayMiner41, allowing one exception (which is relevant to build the network and might have a mechanistic relevance) (Fig. 2c). The resulting network thus contains 32 CTCFL-driven DEGs and one exception. Functional evaluation genes in the network show that they participate primarily in the PI3k-Akt signaling pathway (FGFR1, ITGB3, ITGB6, FN1, VEGFA, NTF3, and IL7R) and are also involved in an extracellular matrix organization (NCAM1, ITGB3, ITGB6, FN1, COL26A1, and CAST) (Fig. 2d).
Notably, regarding the PI3K-Akt pathway, the proteins identified correspond mainly to the cell membrane receptors (FGFR1, ITGB3, ITGB6, and IL7R) and their ligands (VEGFA). Additionally, when examining protein complexes in the CORUM database42, we found that well-characterized functional protein complexes are deregulated by CTCFL; such as the ITGA2B-ITGB3-FN1-TGM2 complex (CORUM 2376) known to participate in cell adhesion43, where three out of the four members of the complex are affected; and the ITGB6-FYN-FN1 complex (CORUM 2351), which is involved in focal adhesion44. The above is of great importance in OC since established evidence shows that this pathway is significantly deregulated in OC5,45,46,47. Notably, FN1 seems to play a special role in the downstream effects of CTCFL deregulation through the alteration of signaling pathways (PI3K-Akt), as a key component of the extracellular matrix and through their interaction with VEGFA and TGFB1, which are not only differentially expressed but also have CTCFL and CTCF occupancy in their promoter.
Together, this data provides more detailed insights into the cellular effects occurring due to the transcriptional changes driven by CTCFL. In particular, these results indicate that CTCFL could be an important factor driving changes in the extracellular matrix components, which in turn, lead to changes in cell migration. Furthermore, our data suggest that the PI3K-Akt signaling pathway might play an important role in OC patients with a high expression of CTCFL. Finally, FN1 appears to be one of the main effectors of CTCFL-driven downstream changes in OC tumor cells.
CTCFL-induced protein interaction network is deregulated and associated with survival in OC
Following the identification of a relevant interaction network enriched with DEGs due to the presence of CTCFL (33 genes), we aimed to evaluate whether these play a relevant role in OC patients, either by being associated with the survival and progression prognosis or by being druggable targets for treatment.
To address this question, we evaluated the expression profile of the 34 genes (33 previously found plus CTCFL) in the TCGA (Serous Cystadenocarcinoma) and GTEX (Normal Tissue) datasets. We found that 27 out of the 34 genes are differentially expressed (Fig. 3a) between tumor and normal tissue, including VEGFA and IL7R, and being CTCFL, KLK5, and ITGB6 the most upregulated and TGFBR3 the top downregulated genes (Supplementary Fig. 4a). Furthermore, genes that belonged to the ITGB6-FYN-FN1 and ITGA2B-ITGB3-FN1-TGM2 complex (ITGB3, ITGB6, and FN1) are also differentially expressed; as well as PI3K-Akt pathway genes (FGFR1). This indicates that the genes deregulated by CTCFL are not only involved in relevant cancer-associated processes (such as signaling pathways and extracellular component organization as described above), but they are also differentially expressed in OC patients.

a Relative expression of the 34 CTCFL-regulated candidate genes in normal and tumor samples (left panel) and their corresponding fold change (right panel). Genes are grouped into DEGs (p-adj < 0.05) or not DEGs (p-adj ≥ 0.05). b Survival analysis with Cox PH model using four survival data: overall survival (OS), disease-specific survival (DSS), and progression-free survival (PFI). c Average Concordance Index (CI) obtained with the gene expression of the 34 candidate genes using the random survival forest (RSF) model. The dashed horizontal line indicates the average CI of random sets of genes for each survival dataset. d Kaplan–Meier plots of MALT1 (top relevant gene found by OS and DSS RSF models) in combination with CTCFL expression. e CTCFL-regulated protein interaction network found in vitro depicting the druggable targets that are differentially expressed in tumor samples versus normal tissue (TCGA vs GTEx). Red and blue nodes indicate genes upregulated and downregulated in tumor vs normal, respectively. Targeting drugs are shown in red boxes, red drug labels indicate drugs currently clinically used for OC treatment.
After comparing the expression profile of the selected genes in tumor versus normal samples, we aimed to evaluate whether they have prognostic potential by doing a Cox proportional hazards (CPH) regression analysis and a Random Survival Forest (RSF) analysis. We evaluated four different survival data types: overall survival (OS), disease-specific survival (DSS), disease-free interval (DFI), and progression-free interval (PFI).
The CPH analysis (Fig. 3b) shows that 8 genes are associated with the survival of patients; particularly, IL7R, PCDH7, COL26A, and NFASC are associated with a protective effect (low expression is associated with poor prognosis), whereas TGFB1, ACTBL2, CAST, and FN1 are associated with an increased risk (high expression is associated with poor prognosis). Also, we evaluated the potential to predict survival with RSF, a nonparametric ensemble method for the prediction of survival with the advantage that it does not depend on model assumptions48. We trained and further applied the model to the four datasets using the 34 candidate genes as features and we also evaluated randomly selected genesets of the same size. We found that the candidate set of genes found by our previous network analysis is capable of predicting the survival of patients with significantly higher accuracy (Concordance Index, CI) than the random genes for OS, DSS and DFI datasets (Fig. 3c). The feature importance (contribution of each gene to the prediction of survival) for each of these models shows that the top predictor gene is MALT1 for OS and DSS, PCDH7 for PFI, and ACTBL2 for DFI (Supplementary Fig. 4b). Notably, MALT1 was not previously found by the CPH analysis, and Kaplan–Meier plots also show the relevance of MALT1 in combination with CTCFL expression in the survival of OC patients (Fig. 3d). Together, this suggests that the selected genes are not only CTCFL-driven genes involved in cancer pathways and deregulated in OC patients; but they are also significantly associated with the survival of these patients.
Once we addressed the relevance of candidate genes in the prognosis of OC patients, we finally evaluated whether these genes are potential druggable targets. We used the CoVex platform49,50 to obtain drugs targeting the genes in the DEG-enriched network (Fig. 3e). We found three drugs, out of the 15 total approved drugs for OC (Altretamine, Gemcitabine, Carboplatin, Topotecan, Thiotepa, Niraparib, Paclitaxel, Cisplatin, Olaparib, Hydroxyurea, Trabectedin, Melphalan, Bevacizumab, Doxorubicin, and Rucaparib); such as the first-line chemotherapy drugs Paclitaxel and Docetaxel and the antiangiogenic drug Bevacizumab. The most identified drugs are tyrosine kinase inhibitors (TKI), which target mainly EPHA7, FGFR1, and ITGB3 (Fig. 3d). The latter is of clinical relevance since ITGB3 appears to be a key connector gene in the CTCFL-DEG-enriched network, it participates in the PI3K-Akt pathway and it is differentially expressed in OC patients. Despite the importance as a prognostic marker of MALT1, no approved targeting drugs were found; thus, its therapeutic potential remains to be explored and it could be a good candidate for drug development. Together, these results provide mechanistic insights into ovarian cancer-associated transcriptional changes while also being potential markers of survival and targets for precision medicine therapies; in particular, in patients with CTCFL overexpression.
Discussion
CTCFL has been widely reported overexpressed in several cancer types16,17,51 and proposed as a promising therapeutic target given its patterns of expression in human tissues. Previous research suggests that the oncogenic functions of CTCFL are due to the promotion of a stemness state17,52,53 by regulating several processes, including apoptosis23,54,55,56 and epithelial to mesenchymal transition (EMT)52. In ovarian cancer patients, CTCFL is overexpressed specifically in HGSC patients22 and it is associated with poor prognosis24. However, little is known about the molecular mechanisms that are driven by CTCFL expression. As the lack of therapeutic alternatives and prognostic biomarkers still is a massive problem in OC, the identification of such CTCFL-driven mechanisms can contribute to the development of systems medicine approaches and more complex prognostic biomarkers in OC. Here we show that the underexpression and overexpression of CTCFL produce changes in cellular processes related mainly to the PI3K-Akt signaling pathway and extracellular matrix-related genes. Furthermore, these genes, together with CTCFL, are good mechanistic predictors of survival and can also be targeted by drugs currently being used or tested in other cancer types.
The PI3K-Akt pathway is the second most frequently altered pathway after p535 and is proposed as a useful approach for therapeutic intervention in OC57, given its association with chemotherapy resistance58,59,60 and poor prognosis in OC61. We found several membrane receptors and their ligands, members of the PI3K-Akt pathway, as the key deregulated genes by CTCFL; most notably, EGFR1, ITGB3, ITGB6, and FN1 are deregulated by CTCFL in vitro and are also deregulated in OC patients. EGFR, ITGB3, and ITGB6 are membrane receptors that have AKT as a downstream target, and thus the inhibition of these could be a useful approach for OC therapy in patients showing chemotherapy resistance. Importantly, to our knowledge, no PI3K-Akt targeting drugs have been approved for the treatment of OC to date. However, preclinical evidence shows that the inhibition of EGFR-AKT signaling affects OC cell growth62,63. Further studies have been carried out to evaluate the performance of molecular inhibitors targeting PI3K or AKT in preclinical models46,64 and some inhibitors are downstream mTOR blockers with varying effectiveness65. Thus, the use of the RTK inhibitors (the upstream initiators of the PI3K-Akt pathway) to target the CTCFL-deregulated genes; such as EGFR1, could aid in improving the effectiveness of OC therapy. Furthermore, we found VEGFA, a key regulator of angiogenesis, deregulated by CTCFL in vitro and affected in OC patients. Targeting VEGFA in OC has been widely attempted with antibody-based therapies, such as Bevacizumab, which is FDA approved for the treatment of resistant OC66,67; however little therapeutic benefit has been observed68. Thus, the use of antiangiogenic therapies or anti-PI3K-Akt signaling pathways, in patients with overexpression of CTCFL or in combination with anti-CTCFL immunotherapy could be a promising therapeutic approach for drug-resistant tumors, since monotherapy in solid tumors appears to be ineffective69.
In addition to the described above, we identified several membrane transporters members of the Solute Carrier Family (SLC) and the ATP-binding cassette (ABC) differentially expressed in CTCFL-deregulated cells. SLC and ABC genes have been widely described as key mechanisms of resistance to Paclitaxel and Cisplatin, the current first chemotherapy option for OC patients29. We identified more than 50 members of the SLC and ABC family of transporters deregulated by CTCFL, thus suggesting that OC patients with deregulated levels of CTCFL protein could be associated with either resistance or sensitivity to chemotherapy. Previous studies in a neuroblastoma model indicate that the overexpression of CTCFL is related to a resistant phenotype, through the regulation of epigenetic processes70 and through the alteration of the ALK gene (whose downstream signal is the PI3K-Akt pathway as well)71. Also, previous studies suggest a role of CTCFL in drug resistance through the maintenance of a stemness state17. The latter supports our results that indicate that CTCFL can, not only serve as a therapeutic target but also could be associated with tumor chemoresistance in OC. However, further analyses are required in OC to validate the potential use of CTCFL as a predictor of chemoresistance.
In addition to the potential role as a therapeutic target, we found that CTCFL expression levels, in combination with the expression of other identified genes in our protein interaction network, provide a good prediction of survival. Notably, in patients with high expression of CTCFL, low levels of MALT1 indicate a good prognosis (OS and DSS). Previous studies report that MALT1 is a key regulator of chemoresistance in other cancer types72. This supports the idea that MALT1 may be utilized as a biomarker of chemoresistance in combination with CTCFL.
As our analysis is mainly based on gene expression data, we cannot be fully certain that our marker genes’ expression is reflected in the abundance of the corresponding proteins. However, our results, together with the fact that CTCFL itself is a cancer biomarker and a target for immunotherapy26, suggest that the combinatorial use of CTCFL and its target genes have high utility as mechanistic predictors for survival and metastasis.
Taken together, our study elucidates the molecular mechanisms driven by the oncogene CTCFL in ovarian cancer, which may further be utilized as prognostic biomarkers as well as for targeted therapy and drug development. In particular, we found PI3K-Akt initiators; such as the RTKs EGFR1 and VEGFA, and the integrins ITGB3 and ITGB6, to be potential drug targets in ovarian cancer patients with high CTCFL expression. We also identified the additional CTCFL targets ACTBL2, MALT1, and PCDH7 to be predictive for ovarian cancer treatment outcomes.
Methods
Cell lines
The human epithelial ovarian cancer cell line OVCAR3 (ATCC HTB-161) was obtained from ATCC (Manassas, VA, USA) and subcultured in a complete growth medium contained RPMI-1640 Medium (Catalog No. 30-2001; ATCC), supplemented with 20% fetal bovine serum (Catalog No. 10437028; Gibco) and 0.01 mg/ml bovine insulin (Catalog No. I0516-5ML; Sigma). OVCAR3 cells were subcultured at 37 °C in a 5% CO2 incubator.
Cell transfections
The plasmid pCpGfree-vitroHBORIS contains CTCFL ORF encoding the canonic protein of 663 amino acids. pCpGfree-vitroHLacZ plasmid was used as a negative control. Both constructs were kindly donated by Prof. Lobanenkov and Dr. Pugacheva11. pCpGfree-vitroHBORIS and pCpGfree-vitroHLacZ (Catalog No.cat. no. pcpgvth-lz; InvivoGen) plasmids were cloned in E. coli GT115 (Catalog No. gt115-11; InvivoGen) and Hygromycin B (Catalog No. 10 843 555 001; Sigma) was used as selection antibiotic. Plasmid purification was performed with QIAEX II Gel Extraction Kit (Catalog No. 20021: QIAGEN). OVCAR3 cells grown to 70% density were transiently transfected with pCpGfree-vitroHBORIS (CTCFL-OE) or pCpGfree-vitroHLacZ (Control) plasmids, using Xfect Transfection Reagent (Catalog No. 631317; Clontech) and 2 µg of plasmid, transfections were performed overnight. Cells were collected 48 h post-transfection for RNA and protein extraction. Three technical replicates were pooled together to obtain one biological replicate. Three independent biological replicates were performed.
Protein extraction and western blot
Collected cells were washed with ice-cold PBS solution and resuspended in RIPA buffer with protease inhibitor cocktail cOmplete (Catalog No. 4693132001; Roche). Protein quantification was performed with DC protein assay (Catalog No. 500-0116; Bio-Rad) and the signal was obtained with iMark™ Microplate Absorbance Reader (Catalog No. 1681130; Bio-Rad). Next, 10 µg of total protein per sample was prepared for electrophoresis in 10% bis-acrylamide SDS-PAGE gel. Proteins were transferred to a PVDF membrane overnight. Antibodies used for immunodetection are Anti-CTCFL (Catalog No. HPA001472; SIGMA Aldrich) dilution 1;10, Anti-GAPDH (Catalog No. SC-25778; Santa Cruz Bt.) dilution 1:2500, and HRP anti-rabbit (Catalog No. SC-2004; Santa Cruz Bt) dilution 1:5000. CTCFL abundance was determined by standard densitometry analysis using ImageJ software (NIH, USA) (Fig. 1a and Supplementary Fig. 1a). Signal normalization was performed using GAPDH (Catalog No. SC-47724; Santa Cruz Bt). Gels are derived from the same experiment and were processed in parallel.
RNA extraction and RNA-sequencing
Total RNA from CTCFL-OE and Controls was extracted using TRIzol Reagent (Catalog No. 15596026; Invitrogen) and Direct-zol RNA Miniprep (Catalog No. R2051; Zymo). RNA from CTCFL-KD was obtained from Knockdown experiments validated and published previously7. The integrity and quality of RNA was analyzed with TapeStation 2200 (Agilent, USA). Sequencing was performed by Unidad de Secuenciación (INMEGEN, Mexico City). mRNA for NGS sequencing was quantified with Qubit 4 (Thermo Fisher Scientific, USA) and libraries were prepared with TruSeq Stranded mRNA Library Prep (Illumina). Sequencing was performed in a NextSeq 500 platform (Illumina) with 25 M reads per sample, paired-end, and 75 bp read length.
Differential expression analysis
Initial quality assessment and trimming of low-quality reads was performed with FastQC v0.10.173 and Trimmomatic v0.2774. Alignment to the genome was performed using STAR75 and the Homo sapiens reference transcriptome (GRCh38). Counts were obtained with FeatureCounts from the R package Rsubread v1.32.476. Differential expression analysis was performed with DESeq2 v1.22.277, genes were selected with |Fold Change | > 2 and FDR p-adj < 0.1 (for CTCFL-OE vs Control) and FDR p-adj < 0.05 (for CTCFL-KD vs Control). Differentially expressed genes are shown in Supplementary Data 1. Differentially expressed genes identified in each comparison were used for Functional Enrichment Analysis with gProfiler a cutoff of p-adj < 0.001 (for CTCFL-OE vs Control) and p-adj < 0.05 (for CTCFL-KD vs Control).
CTCFL binding site identification
Sequences flanking the TSS (2 Kbp) of DEGs identified in CTCFL-OE and CTCFL-KD were obtained using Biomart78. HMMER 3.2.1 software36 was used to search for CTCFL BSs using the known motifs reported in Jaspar database34 (profile MA1102.1) and Factorbook35. Clustal Omega software79 was used for the alignment of known motifs. For regions with more than one identified BS, only the site with the best e-value was selected. Two independent searches with HMMER were performed, one for each CTCFL motif (Jaspar and Factorbook). Genes with identified BS by both independent searches and position in the same genomic coordinate were selected. (Supplementary Data 2).
ChIP-seq data integration
WIG formatted ChIP-seq data for CTCFL and CTCF in OVCAR8 cell lines was obtained from Pugacheva et al.11 available in GEO database (GEO accession GSE70764)80. Coordinates were converted to GRCh38/hg38 genome assembly using the liftOver tool from UCSC Genome Browser81. Visualization of ChIP-seq peaks was performed with IGV software82.
Network analysis and target-drug associations
Differentially expressed genes identified in CTCFL-OE and CTCFL-KD were used as active genes to create a custom indicator matrix as input for de novo pathway enrichment with KeyPathwayMiner83 using a physical interaction network obtained from BioGrid and the parameters K = 1, L = 0. Cytoscape was used for network visualization84.
To retrieve drugs targeting proteins in the network, gene names were mapped to UniprotIDs and input into CoVex49,50; followed by the MuST algorithm with default parameters (except including non-seed viral proteins=IGNORE) and the Closeness centrality algorithm with default parameters and result size = 500.
Expression analysis in tumor and normal ovarian samples
For differential expression analysis of tumor vs normal ovarian samples, RSEM_expected counts were downloaded from the TCGA, TARGET, and GTEx dataset from the UCSC Toil RNA-seq Recompute85 available through the Xena Browser86. A total of 88 normal (GTEx) and 419 neoplastic (TCGA) ovarian samples were retained after removing recurrent tumor samples. Counts were subjected to differential expression analysis using DEseq277. Differentially expressed genes were selected with abs(Fold Change) > 1.5 and p-adj < 0.05.
For survival analysis, gene expression (RSEM tpm) of the TCGA, TARGET, and GTEx datasets was downloaded from the UCSC Toil RNA-seq Recompute85. A total of 419 neoplastic (TCGA) ovarian samples were retained after removing recurrent tumor samples. Four different outcome labels were used, producing four separate datasets: overall survival (OS, 417 samples), disease-specific survival (DSS, 387 samples), disease-free interval (DFI, 202 samples), and progression-free interval (PFI, 417 samples).
Preprocessing, Kaplan–Meier estimation, Cox proportional hazard model (CPH), and random survival forest (RSF) were applied to all datasets. For the preprocessing, the datasets were checked for high-correlating features (Pearson correlation coefficient >95%), where zero genes showed high correlation, thus all genes were retained. For Kaplan–Meier estimation and CPH Model, the lifelines package87 was used; in particular, for CPH, a five-times repeated fivefold cross-validation was used and the concordance index (c-index) was used as an evaluation metric. RSF analysis was performed with the scikit-survival package88 by splitting the dataset into 80% train and 20% test. On the training set, a randomized hyperparameter search was performed with a five-times repeated fivefold cross-validation and evaluated using the c-index. The hyperparameters of the best-performing model of the randomized search were used to train the actual model on the whole training set, which was evaluated on the 20% unseen test data. Finally, the feature importance of the model was computed.
Data availability
The datasets generated and analysed during the current study are available in the GEO repository, GSE166767.
References
Matulonis, U. A. et al. Ovarian cancer. Nat. Rev. Dis. Prim. 2, 16061 (2016).
Cancer of the Ovary - Cancer Stat Facts. SEER https://seer.cancer.gov/statfacts/html/ovary.html (2019).
Pokhriyal, R., Hariprasad, R., Kumar, L. & Hariprasad, G. Chemotherapy resistance in advanced ovarian cancer patients. Biomark. Cancer 11, 1179299X19860815 (2019).
Coward, J. I., Middleton, K. & Murphy, F. New perspectives on targeted therapy in ovarian cancer. Int. J. Women’s Health 7, 189–203 (2015).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337.e10 (2018).
Zhang, Z. et al. Molecular subtyping of serous ovarian cancer based on multi-omics data. Sci. Rep. 6, 26001 (2016).
Salgado-Albarrán, M. et al. The epigenetic factor BORIS (CTCFL) controls the androgen receptor regulatory network in ovarian cancer. Oncogenesis 8, 41 (2019).
Hillman, J. C. et al. BORIS expression in ovarian cancer precursor cells alters the CTCF cistrome and enhances invasiveness through GALNT14. Mol. Cancer Res. 17, 2051–2062 (2019).
Loukinov, D. I. et al. BORIS, a novel male germ-line-specific protein associated with epigenetic reprogramming events, shares the same 11-zinc-finger domain with CTCF, the insulator protein involved in reading imprinting marks in the soma. Proc. Natl Acad. Sci. USA 99, 6806–6811 (2002).
Phillips, J. E. & Corces, V. G. CTCF: master weaver of the genome. Cell 137, 1194–1211 (2009).
Pugacheva, E. M. et al. Comparative analyses of CTCF and BORIS occupancies uncover two distinct classes of CTCF binding genomic regions. Genome Biol. 16, 161 (2015).
Marshall, A. D., Bailey, C. G. & Rasko, J. E. J. CTCF and BORIS in genome regulation and cancer. Curr. Opin. Genet. Dev. 24, 8–15 (2014).
Uhlen, M. et al. A pathology atlas of the human cancer transcriptome. Science 357, eaan2507 (2017).
Odunsi, K. et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. 63, 6076–6083 (2003).
Scanlan, M. J., Gure, A. O., Jungbluth, A. A., Old, L. J. & Chen, Y.-T. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol. Rev. 188, 22–32 (2002).
Martin-Kleiner, I. BORIS in human cancers - a review. Eur. J. Cancer 48, 929–935 (2012).
Soltanian, S. & Dehghani, H. BORIS: a key regulator of cancer stemness. Cancer Cell Int. 18, 154 (2018).
Vatolin, S. et al. Conditional expression of the CTCF-paralogous transcriptional factor BORIS in normal cells results in demethylation and derepression of MAGE-A1 and reactivation of other cancer-testis genes. Cancer Res. 65, 7751–7762 (2005).
Hong, J. A. et al. Reciprocal binding of CTCF and BORIS to the NY-ESO-1 promoter coincides with derepression of this cancer-testis gene in lung cancer cells. Cancer Res. 65, 7763–7774 (2005).
Bhan, S. et al. BORIS binding to the promoters of cancer testis antigens, MAGEA2, MAGEA3, and MAGEA4, is associated with their transcriptional activation in lung cancer. Clin. Cancer Res. 17, 4267–4276 (2011).
Risinger, J. I. et al. Global expression analysis of cancer/testis genes in uterine cancers reveals a high incidence of BORIS expression. Clin. Cancer Res. 13, 1713–1719 (2007).
Engqvist, H. et al. Integrative genomics approach identifies molecular features associated with early-stage ovarian carcinoma histotypes. Sci. Rep. 10, 7946 (2020).
Renaud, S. et al. BORIS/CTCFL-mediated transcriptional regulation of the hTERT telomerase gene in testicular and ovarian tumor cells. Nucleic Acids Res. 39, 862–873 (2011).
Woloszynska-Read, A. et al. Coordinated cancer germline antigen promoter and global DNA hypomethylation in ovarian cancer: association with the BORIS/CTCF expression ratio and advanced stage. Clin. Cancer Res. 17, 2170–2180 (2011).
Okabayashi, K. et al. Cancer-testis antigen BORIS is a novel prognostic marker for patients with esophageal cancer. Cancer Sci. 103, 1617–1624 (2012).
Loukinov, D. Targeting CTCFL/BORIS for the immunotherapy of cancer. Cancer Immunol. Immunother. 67, 1955–1965 (2018).
Fife, C. M., McCarroll, J. A. & Kavallaris, M. Movers and shakers: cell cytoskeleton in cancer metastasis. Br. J. Pharm. 171, 5507–5523 (2014).
Kleiner, D. E. & Stetler-Stevenson, W. G. Matrix metalloproteinases and metastasis. Cancer Chemother. Pharmacol. 43, S42–S51 (1999).
Li, Q. & Shu, Y. Role of solute carriers in response to anticancer drugs. Mol. Cell Ther. 2, 15 (2014).
Sun, Y.-L., Patel, A., Kumar, P. & Chen, Z.-S. Role of ABC transporters in cancer chemotherapy. Chin. J. Cancer 31, 51–57 (2012).
Janssen, S. M. et al. BORIS/CTCFL promotes a switch from a proliferative towards an invasive phenotype in melanoma cells. Cell Death Disco. 6, 1 (2020).
Szabó, P. E., Tang, S.-H. E., Silva, F. J., Tsark, W. M. K. & Mann, J. R. Role of CTCF binding sites in the Igf2/H19 imprinting control region. Mol. Cell. Biol. 24, 4791–4800 (2004).
Schultz, B. et al. A common polymorphism within the IGF2 imprinting control region is associated with parent of origin specific effects in infantile hemangiomas. PLoS ONE 10, e0113168 (2015).
Fornes, O. et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92 (2020).
Wang, J. et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22, 1798–1812 (2012).
Wheeler, T. J. & Eddy, S. R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489 (2013).
Renaud, S. et al. Expression of the CTCF-paralogous cancer-testis gene, brother of the regulator of imprinted sites (BORIS), is regulated by three alternative promoters modulated by CpG methylation and by CTCF and p53 transcription factors. Nucleic Acids Res. 35, 7372–7388 (2007).
Claesson-Welsh, L. & Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 273, 114–127 (2013).
Comunanza, V. & Bussolino, F. Therapy for cancer: strategy of combining anti-angiogenic and target therapies. Front Cell Dev. Biol. 5, 101 (2017).
Kim, J. Y., Cho, C. H. & Song, H. S. Targeted therapy of ovarian cancer including immune check point inhibitor. Korean J. Intern. Med. 32, 798–804 (2017).
Alcaraz, N. et al. Robust de novo pathway enrichment with KeyPathwayMiner 5. F1000Res. 5, 1531 (2016).
Giurgiu, M. et al. CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res. 47, D559–D563 (2019).
Akimov, S. S., Krylov, D., Fleischman, L. F. & Belkin, A. M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 148, 825–838 (2000).
Li, X. et al. Alphavbeta6-Fyn signaling promotes oral cancer progression. J. Biol. Chem. 278, 41646–41653 (2003).
Ediriweera, M. K., Tennekoon, K. H. & Samarakoon, S. R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 59, 147–160 (2019).
Mabuchi, S., Kuroda, H., Takahashi, R. & Sasano, T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 137, 173–179 (2015).
Li, H., Zeng, J. & Shen, K. PI3K/AKT/mTOR signaling pathway as a therapeutic target for ovarian cancer. Arch. Gynecol. Obstet. 290, 1067–1078 (2014).
Ishwaran, H., Kogalur, U. B., Blackstone, E. H. & Lauer, M. S. Random survival forests. Ann. Appl. Stat. 2, 841–860 (2008).
Matschinske, J. et al. Individuating possibly repurposable drugs and drug targets for COVID-19 treatment through hypothesis-driven systems medicine using CoVex. Assay. Drug Dev. Technol. 18, 348–355 (2020).
Sadegh, S. et al. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat. Commun. 11, 3518 (2020).
Klenova, E. M., Morse, H. C. 3rd, Ohlsson, R. & Lobanenkov, V. V. The novel BORIS + CTCF gene family is uniquely involved in the epigenetics of normal biology and cancer. Semin. Cancer Biol. 12, 399–414 (2002).
Garikapati, K. R. et al. Down-regulation of BORIS/CTCFL efficiently regulates cancer stemness and metastasis in MYCN amplified neuroblastoma cell line by modulating Wnt/β-catenin signaling pathway. Biochem. Biophys. Res. Commun. 484, 93–99 (2017).
Alberti, L., Losi, L., Leyvraz, S. & Benhattar, J. Different effects of BORIS/CTCFL on stemness gene expression, sphere formation and cell survival in epithelial cancer stem cells. PLoS ONE 10, e0132977 (2015).
Macaluso, M. et al. Integrating role of T antigen, Rb2/p130, CTCF and BORIS in mediating non-canonical endoplasmic reticulum-dependent death pathways triggered by chronic ER stress in mouse medulloblastoma. Cell Cycle 11, 1841–1850 (2012).
Zhang, Y. et al. Brother of Regulator of Imprinted Sites (BORIS) suppresses apoptosis in colorectal cancer. Sci. Rep. 7, 40786 (2017).
Alberti, L., Renaud, S., Losi, L., Leyvraz, S. & Benhattar, J. High expression of hTERT and stemness genes in BORIS/CTCFL positive cells isolated from embryonic cancer cells. PLoS ONE 9, e109921 (2014).
Gasparri, M. L. et al. PI3K/AKT/mTOR pathway in ovarian cancer treatment: are we on the right track? Geburtshilfe Frauenheilkd. 77, 1095–1103 (2017).
Liu, R. et al. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 11, 797 (2020).
Pogge von Strandmann, E., Reinartz, S., Wager, U. & Müller, R. Tumor–host cell interactions in ovarian cancer: pathways to therapy failure. Trends Cancer Res. 3, 137–148 (2017).
Yang, J. et al. Adipocytes promote ovarian cancer chemoresistance. Sci. Rep. 9, 13316 (2019).
Bai, H. et al. The PI3K/AKT/mTOR pathway is a potential predictor of distinct invasive and migratory capacities in human ovarian cancer cell lines. Oncotarget 6, 25520–25532 (2015).
Loganathan, S., Kandala, P. K., Gupta, P. & Srivastava, S. K. Inhibition of EGFR-AKT axis results in the suppression of ovarian tumors in vitro and in preclinical mouse model. PLoS ONE 7, e43577 (2012).
Shao, G. et al. Inactivation of EGFR/AKT signaling enhances TSA-induced ovarian cancer cell differentiation. Oncol. Rep. 37, 2891–2896 (2017).
Fruman, D. A. & Rommel, C. PI3K and cancer: lessons, challenges and opportunities. Nat. Rev. Drug Discov. 13, 140–156 (2014).
Behbakht, K. et al. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: a Gynecologic Oncology Group study. Gynecol. Oncol. 123, 19–26 (2011).
Claussen, C., Rody, A. & Hanker, L. Treatment of recurrent epithelial ovarian cancer. Geburtshilfe Frauenheilkd. 80, 1195–1204 (2020).
Aghajanian, C. et al. Final overall survival and safety analysis of OCEANS, a phase 3 trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent ovarian cancer. Gynecol. Oncol. 139, 10–16 (2015).
Lim, D. et al. Angiogenesis and vasculogenic mimicry as therapeutic targets in ovarian cancer. BMB Rep. 53, 291–298 (2020).
Hanker, A. B., Kaklamani, V. & Arteaga, C. L. Challenges for the clinical development of PI3K inhibitors: strategies to improve their impact in solid tumors. Cancer Disco. 9, 482–491 (2019).
Debruyne, D. N. et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature 572, 676–680 (2019).
American Association for Cancer Research. Resistance to ALK inhibitors in neuroblastoma is regulated by BORIS. Cancer Discov. 9, 1335–1335 (2019).
McAuley, J. R. et al. MALT1 is a critical mediator of PAR1-driven NF-κB activation and metastasis in multiple tumor types. Oncogene 38, 7384–7398 (2019).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Durinck, S., Spellman, P. T., Birney, E. & Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 (2009).
Sievers, F. & Higgins, D. G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27, 135–145 (2018).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 41, D991–D995 (2013).
Kuhn, R. M., Haussler, D. & Kent, W. J. The UCSC genome browser and associated tools. Brief. Bioinform. 14, 144–161 (2013).
Robinson, J. T., Thorvaldsdóttir, H., Wenger, A. M., Zehir, A. & Mesirov, J. P. Variant review with the integrative genomics viewer. Cancer Res. 77, e31–e34 (2017).
List, M. et al. KeyPathwayMinerWeb: online multi-omics network enrichment. Nucleic Acids Res. 44, W98–W104 (2016).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Vivian, J. et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 35, 314–316 (2017).
Goldman, M. J. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020).
Davidson-Pilon, C. et al. CamDavidsonPilon/lifelines: v0.25.6. Zenodo https://doi.org/10.5281/zenodo.4136578 (2020).
Pölsterl, S. scikit-survival: a library for time-to-event analysis built on top of scikit-learn. J. Mach. Learn. Res. 21, 1–6 (2020).
Nguyen, P. et al. CTCFL/BORIS is a methylation-independent DNA-binding protein that preferentially binds to the paternal H19 differentially methylated region. Cancer Res. 68, 5546–5551 (2008).
Acknowledgements
This work was supported by Apoyo para proyectos de investigación científica, desarrollo tecnológico e innovación en salud ante la contingencia por COVID-19, CONACyT [00312021 to ES-R], Fondo CB-SEP-CONACyT [284748 to ES-R] and PROMEP [250690 to ES-R], and CONACyT [290041 to RG-B]. JB’s work was supported by his VILLUM Young Investigator Grant No. 13154. This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreements No. 826078 (JB and JS) and No. 777111 (JB and JS). This work was also in parts supported by the German Federal Ministry of Education and Research(BMBF) within the framework of the *e:Med *research and funding concept (*grant 01ZX1908A*). This publication reflects only the authors’ view, and the European Commission is not responsible for any use that may be made of the information it contains. MS-A is a doctoral student from “Programa de maestría y doctorado en Ciencias Bioquímicas, UNAM” and received a PhD fellowship funding from CONACYT (CVU659273) and the German Academic Exchange Service, DAAD (ref. 91693321). CTCFL (BORIS) plasmid constructs were kindly donated by Prof. Lobanenkov and Dr. Pugacheva.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
MS-A obtained and analyzed the experimental data, and wrote the manuscript. JS performed the Random Survival Forest and Cox PH analyses and helped in the manuscript writing. RG-B provided critical feedback and helped to improve the manuscript. JB and ES-R were in charge of overall direction, planning, and supervision. ES-R. is the guarantor of this work and, as such, has full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salgado-Albarrán, M., Späth, J., González-Barrios, R. et al. CTCFL regulates the PI3K-Akt pathway and it is a target for personalized ovarian cancer therapy. npj Syst Biol Appl 8, 5 (2022). https://doi.org/10.1038/s41540-022-00214-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-022-00214-z
This article is cited by
-
The function of brother of the regulator of imprinted sites in cancer development
Cancer Gene Therapy (2023)
-
Single-cell and bulk RNA sequencing reveal ligands and receptors associated with worse overall survival in serous ovarian cancer
Cell Communication and Signaling (2022)
