
Abstract
The onset of colorectal cancer (CRC) is often attributed to gut bacterial dysbiosis, and thus gut microbiota are highly relevant in devising treatment strategies. Certain gut microbes, like Enterococcus spp., exhibit remarkable anti-neoplastic and probiotic properties, which can aid in silver nanoparticle (AgNPs) induced reactive oxygen species (ROS)-based CRC treatment. However, the effects of AgNPs on gut microbial metabolism have not been reported thus far. In this study, a detailed systems-level understanding of ROS metabolism in Enterococcus durans (E. durans), a representative gut microbe, was gained using constraint-based modeling, wherein, the critical association between ROS and folate metabolism was established. Experimental studies involving low AgNP concentration treatment of E. durans cultures confirmed these modeling predictions (an increased extracellular folate concentration by 52%, at the 9th h of microbial growth, was observed). Besides, the computational studies established various metabolic pathways involving amino acids, energy metabolites, nucleotides, and SCFAs as the key players in elevating folate levels on ROS exposure. The anti-cancer potential of E. durans was also studied through MTT analysis of HCT 116 cells treated with microbial culture (AgNP treated) supernatant. A decrease in cell viability by 19% implicated the role of microbial metabolites (primarily folate) in causing cell death. The genome-scale modeling approach was then extended to extensively model CRC metabolism, as well as CRC–E. durans interactions in the context of CRC treatment, using tissue-specific metabolic models of CRC and healthy colon. These findings on further validation can facilitate the development of robust and effective cancer therapy.
Similar content being viewed by others


Introduction
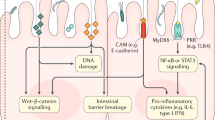
The onset of colorectal cancer (CRC) has been associated with various extrinsic factors such as infection, unhealthy diet, and lifestyle1,2,3, which often result in gut microbiota dysbiosis—a clinical disorder characterized by perturbation in the composition and function of the healthy gut microbiota4,5. In the state of homeostasis, the gut bacteria-derived metabolites synthesize essential nutrients and compounds6,7, some of which have demonstrated remarkable anti-neoplastic properties8. For instance, Enterococcal peptides obtained from clinical strains of Enterococcus genus (a representative gut microbe) exhibit anti-proliferative effects against colorectal adenocarcinoma and other cancer types9. Furthermore, Enterococcus durans was shown to produce butyrate, which is anti-inflammatory in nature and is required to maintain the integrity of intestinal epithelium10. Such studies have highlighted the relevance of gut microbes in devising effective, novel cancer treatment therapies, as in the case of nanoparticle-based CRC treatment (Fig. 1).

AgNPs are known to mediate their direct cytotoxic effects through increased generation of reactive species inside the target cells. Besides, the AgNP-generated oxidative stress can also affect the cellular metabolism and certain pathways of interest (in this case, the anti-cancer metabolites) thereby enhancing indirect cytotoxicity. Gut microbe-CRC interactions can further assist in understanding the role of gut bacterial metabolites (indirect effects of ROS) on CRC metabolism.
The transformation of a healthy colon cell into a cancerous cell type is a complex event, which disrupts cell characteristics at metabolic, signaling, and regulatory levels11,12, that have been captured through various in vitro and in vivo experiments. Moreover, with the advent of constraint-based modeling (CBM), genome-scale metabolic models (GSMMs) too have aimed at investigating the re-programmed metabolism for devising novel therapeutic strategies13,14. To illustrate, context-specific metabolic models of colon and CRC cell types have been developed to understand the aberrant metabolism in CRC, as well as deciphering drug targets and biomarkers for cancer diagnostics. For instance, one such experimental and computational study discovered the FUT9 gene as a crucial promoter of advanced stage colon cancer15. Despite these computational advances, there have been limited computational studies to understand the contribution of reactive species generation (known to exert cytotoxic effects in cancer cells) and gut microbiome secreted metabolites towards CRC metabolism and treatment. Therefore, it would be interesting to investigate and model the role of gut bacteria secreted metabolites in cancer cell killing.
Here we present a computational and experimental study that reveals the effects of AgNP-mediated reactive species generation on gut bacterial metabolism. The effects of reactive oxygen species (ROS) were extensively modeled using the GSMM of E. durans. These computational findings were supported through experiments. Furthermore, the complexity and systems-level understanding of the integral metabolic interactions between gut microbe and host (i.e., colon and CRC cells) was gained using tissue-specific metabolic models. These models were developed from proteomics and literature-based data sources, representing healthy colon and CRC conditions. In addition, we have reported the differences between the CRC vs. healthy colon models in terms of their biomass values and attempted at elucidating the metabolic alterations behind aberrant CRC metabolism using genome-scale metabolic modeling.
Results
AgNP-mediated oxidative stress (ROS) modulated gut microbial metabolism
The AgNPs used in this study were characterized for their physicochemical properties (Supplementary Fig. 1). To study the effects of AgNPs on the growth and cell viability of E. durans, the bacterial cultures were exposed to different AgNP concentrations (Fig. 2). The specific growth rate (µ) for the cultures treated with the lowest AgNP concentration (25 ppm) was 0.198 ± 0.03 h−1, which was 8% lower compared to the control. Further, the highest AgNP concentration (250 ppm) reduced µ to 0.08 ± 0.016 h−1, thereby indicating the deleterious effects of nanoparticles at higher concentrations. Thus, the lowest concentration of AgNPs (25 ppm) exhibited no major detrimental effects on microbial viability. Further, the effects of AgNPs at such low concentrations were quantified experimentally on ROS generation (superoxide and hydroxyl radicals) (Fig. 3a, b). At 6th h (mid-log phase) of AgNP-treated bacterial culture growth, the specific intracellular superoxide concentration increased by 13% (0.273 ± 0.01 nmol/g-cell) compared to control. Similarly, specific intracellular hydroxyl radical levels showed a 48% increase (1.057 ± 0.02 nmol/g-cell, at 9th h) in the late-log phase, compared to control.

Exposure of bacterial cultures to higher concentrations of the nanoparticles results in a) excessive cell death and b) reduced specific growth rates (p-value = 0.0201). Values are expressed as mean ± SD, n = 4.

( indicates control culture;
indicates control culture;  indicates culture treated with 25 ppm AgNP). a The superoxide radical levels increased during the mid-log phase (6th h) of growth (on exposure to AgNPs), and then dropped, when compared to control (p-value = 0.0419). b The hydroxyl radical concentration in the presence of a lower concentration of AgNP, showed increased levels at 9th h when compared to the control (p-value = 0.0357). Values are expressed as mean ± SD, n = 3.
indicates culture treated with 25 ppm AgNP). a The superoxide radical levels increased during the mid-log phase (6th h) of growth (on exposure to AgNPs), and then dropped, when compared to control (p-value = 0.0419). b The hydroxyl radical concentration in the presence of a lower concentration of AgNP, showed increased levels at 9th h when compared to the control (p-value = 0.0357). Values are expressed as mean ± SD, n = 3.
Nevertheless, the AgNP-induced intracellular ROS generation could have altered microbial metabolism. This altered state of metabolism was captured using the CBM approach, which is a preferred computational approach for studying overall metabolic characteristics and metabolite systems profiling16, as experimental studies cannot capture all the metabolic complexities simultaneously. The metabolic effects of ROS (oxidative stress) on E. durans were computationally captured for the first time in this study, wherein CBM of E. durans was used to identify metabolic consequences associated with increased generation of reactive species (ROS/RNS) within the microbial system. Genome-scale metabolic model (GSMM) of E. durans (E. durans ATCC 6056) was downloaded from virtual human metabolic (VMH)17, and an extensive manual curation of the model was performed using scientific literature to incorporate relevant reactive species reactions to generate expanded E. durans model with ROS reactions (Supplementary Data 1 and Supplementary Table 1). The ROS expanded metabolic model was then constrained as per the MRS medium composition (Supplementary Data 1).
Flux variability analysis (FVA)18 was then implemented to capture the relative changes in the network fluxes before and after the addition of ROS reactions. Upon calculating flux span ratio (FSr) for the original and ROS expanded models (detail in the Methods section), seven reactions- most of them associated with folate metabolism, showed heightened fluxes in the expanded ROS model. These reactions were catalyzed by: dihydrofolate reductase (EC:1.5.1.3), methenyltetrahydrofolate cyclohydrolase (EC:3.5.4.9), formate-tetrahydrofolate ligase (EC:6.3.4.3). In addition, minNorm analysis indicated that these seven reactions, in turn, influenced major metabolic pathways, i.e., amino acid/peptide metabolism, nucleotide metabolism, carbohydrate metabolism (pyruvate metabolism, glycolysis/gluconeogenesis), and energy metabolism, as elucidated in Fig. 4 (details in Discussion and Supplementary Table 2). One of the most important and novel CBM predictions highlighted the association between folic acid derivatives (cellular folate pool) and SCFA (propionate) metabolism through the enzymes-malonyl CoA pyruvate carboxytransferase (EC:2.1.3.1) and 2-oxobutanoate formate lyase, as mentioned in Supplementary Table 2.

The metabolic pathways (viz., amino acid, energy and nucleotide metabolism), were influenced by folate metabolism as predicted by the model. The link between SCFA metabolism and folic acid metabolic pathways is a novel model prediction that needs to be confirmed experimentally.
Based on the modeling predictions, the effects of ROS on folate levels were examined experimentally. Interestingly, the findings showed AgNP-induced oxidative stress also elevated both intracellular (Supplementary Fig. 2) and extracellular (Fig. 5a) microbial folate levels, as revealed by HPLC analysis. At 9th h of AgNP-treated microbial growth, maximum specific extracellular folate was 52% higher compared to control (128.84 ± 0.16 nmol/g-cell).

( indicates control culture;
indicates control culture;  indicates culture treated with 25 ppm AgNP). a The extracellular folic acid concentrations increased on exposure to AgNPs. This increase could be an outcome of the oxidative stress induced in the organism, which in turn impact folate related metabolic pathways. b MTT analysis of HCT116 treated with bacterial supernatants from cultures exposed to silver nanoparticles and control. Reduction in viability was observed for cells treated with 9th h supernatants (p-value = 0.0031), the time point corresponding to maximum concentration. Values are expressed as mean ± SD, n = 3.
indicates culture treated with 25 ppm AgNP). a The extracellular folic acid concentrations increased on exposure to AgNPs. This increase could be an outcome of the oxidative stress induced in the organism, which in turn impact folate related metabolic pathways. b MTT analysis of HCT116 treated with bacterial supernatants from cultures exposed to silver nanoparticles and control. Reduction in viability was observed for cells treated with 9th h supernatants (p-value = 0.0031), the time point corresponding to maximum concentration. Values are expressed as mean ± SD, n = 3.
E. durans-secreted metabolites, specifically folate, affected CRC cell viability
Previous studies have reported that many microbe-secreted metabolites exhibit anticancer properties8,19. Folic acid is one such metabolite, which is involved in CRC carcinogenesis20,21. However, for the very first time, we were able to identify the cytotoxic effects of folic acid (at a critical concentration of 0.5 µM) on HCT 116 cells (Supplementary Fig. 3). Moreover, the role of various metabolites produced by E. durans in the context of nanoparticles-based targeting of CRC was studied. To do so, HCT 116 cells were treated with crude bacterial supernatant obtained from AgNP-treated bacterial culture and an MTT viability assay was performed. It was found that AgNP (25 ppm) treated bacterial culture supernatant exerted cytotoxic effects on cancer cells. The viability of the cancer cells treated with 9th h supernatant from nanoparticle treated cultures was reduced by 19% as compared to control (Fig. 5b). Interestingly, this was also the time point corresponding to the release of the optimal cytotoxic concentration of extracellular folate secreted in the culture (Fig. 5a). As microbe secreted metabolites have the potential to affect cancer cell viability, it was crucial to understand the metabolic interrelationships between the E. durans and the host cell (i.e., healthy colon and cancer colon cell types) in detail.
Context-specific metabolic models of CRC and CRC-microbe metabolic interactions facilitated understanding metabolic complexities of CRC
Tissue-specific metabolic models of healthy colon and CRC cells were developed from Recon 3D22. The generic human metabolic network was constrained using proteomic data obtained from the Human Protein Atlas (HPA; Version: 19.1)23, as well as gene regulation data (genes specifically upregulated in colon cancer) from the literature24. The multi-omics data was then integrated using iMAT algorithm25. The data-constrained models were further conditioned to grow on a defined nutrient medium for mammalian cells, i.e., DMEM growth medium as per the experimental studies (Supplementary Data 2). These nutrient-constrained colons and CRC models were then independently integrated with ROS expanded E. durans (Supplementary Data 3 and Supplementary Table 1) using the Microbiome Modeling Toolbox26 to simulate host–microbe interactions.
The first step in investigating the metabolic attributes differentiating healthy colon and CRC was to model the growth of the models, which is a function of the flux obtained through their biomass reactions, respectively using flux balance analysis (FBA). The flux through the biomass reaction of the CRC model was found to be higher (0.0223 mmol/g-DW/h), compared to that of the healthy colon model (0.0003 mmol/g-DW/h), thus, indicating that transformed cells grow and proliferate at relatively higher rates. This defining characteristic of CRC cells is attributed to their ability to cater to the increasing biosynthetic demands for increased proliferation, which was further explored by computing variation in fluxes of the entire metabolic network. To do so, FVA and subsequently, flux distribution ratio (FSr, considering values in the range 0.8 > FSr > 2) analysis of colon and CRC metabolic models, as well as colon-microbe and CRC–microbe models was performed. A surge in biochemical flux was observed through different major metabolic pathways in CRC compared to the colon (Fig. 6a) and CRC-microbe to colon-microbe models (Fig. 6b). Most of these pathways were common to both sets of models and included:
-
i.
Fatty acid oxidation: The reactions participating in fatty acid oxidation catalyzed by Carnitine O-Palmitoyltransferase (CPT; EC:2.3.1.21), Enoyl Coenzyme A Hydratase (ECH, EC:1.3.8.7), and Enoyl Coenzyme A Reductase (EC:1.3.1.38) showed increased fluxes, indicating beta-oxidation of fatty acids was a metabolically pronounced phenomenon in both the CRC and CRC- E. durans integrated models.
-
ii.
Fatty acid synthesis: The pathways involving reactions catalyzed by Very-Long-Chain 3-Oxoacyl Coenzyme A Synthase (EC:2.3.1.199) and Very-Long-Chain 3-Oxoacyl Coenzyme A Reductase (EC:1.1.1.330) also had increased flux values, indicative of fulfilling the metabolic requirements of cancer cells.
-
iii.
Amino acid metabolism: Tyrosine metabolic pathway displayed enhanced flux through the reaction catalyzed by 3, 4-Dihydroxymandelaldehyde:NADP + Oxidoreductase (EC:1.2.1.5).
-
iv.
Squalene and cholesterol metabolic pathways were also affected in these metabolic networks. For instance, Isopentenyl-Diphosphate D-Isomerase (IDI, EC:5.3.3.2) which is important in squalene synthesis showed increased flux value in both models.
-
v.
The reaction catalyzed by the enzyme Deoxyuridine Phosphorylase (DURIPP; EC:2.4.2.23) in nucleotide interconversion also showed increased flux.

a Colon vs. CRC and b colon-microbe vs. CRC–microbe models.
The relevance of these enzymes and related pathways in cancer cells have been discussed at great length in the Supplementary Discussion. Interestingly, these computational predictions have been concordant with the existing literature findings (summarized in Table 1), thereby supporting the reliability of these models in evaluating metabolic aspects of CRC as a disease. However, certain reactions catalyzed by Prostaglandin I2 synthase (E.C:5.3.99.4; eicosanoid metabolism) and Palmitoyl Coenzyme A Hydrolase (E.C:3.1.2.2; fatty acid oxidation showed reduced fluxes. This contrasts with the findings reported in the literature (Supplementary Discussion).
E. durans-driven changes in CRC metabolism highlight the relevance of gut microbe
The FSr analysis between the host (CRC and colon)–microbe model sets emphasized changes brought about in the fluxes of the following CRC biochemical reactions in presence of E. durans (ROS exposed) model when compared to colon–E. durans:
-
i.
Steroid metabolism: In the CRC–E. durans integrated metabolic model, steryl sulfatase (STS, EC:3.1.6.2) and Hydroxysteroid (17-Beta) Dehydrogenase 4 (17βHSDs, EC: 1.1.1.35) showed increased flux values.
-
ii.
Oxidative phosphorylation (OXPHOS): One of the crucial highlights of the CRC–microbe integrated metabolic model was increased flux through Sn-Glycerol-3-Phosphate: (Acceptor) 2-Oxidoreductase (EC:1.1.5.3) catalyzed reaction of the OXPHOS pathway. This established the importance of OXPHOS in rewired CRC metabolism when exposed to E. durans integration.
-
iii.
Arachidonic acid metabolism: Arachidonate 5-Lipoxygenase (EC:1.13.11.34) catalyzed reaction showed increased flux value in the CRC–microbe model.
Moreover, we also compared fluxes between CRC and CRC–microbe models to identify metabolic changes specifically driven by E. durans. These included the following:
-
i.
Phosphatidylinositol (PI) and inositol phosphate metabolism: The flux through various enzyme-catalyzed reactions involved in the PI pathway exhibited increased fluxes in the CRC-microbe integrated model compared to the CRC model alone. These enzymes included 1-Phosphatidylinositol-4-Phosphate 5-Kinase (PIP kinase, EC:2.7.1.68) and Phosphatidylinositol N-acetylglucosaminyltransferase (EC:2.4.1.198).
-
ii.
Glycerophospholipid metabolism: The reaction catalyzing the generation of 1-Palmitoylglycerophosphoinositol and 1-Arachidonoylglycerophosphoinositol showed increased fluxes in the CRC-E. durans integrated model. These reactions are catalyzed by phospholipase A2 (EC:3.1.1.4).
-
iii.
Propanoate metabolism: 2-Hydroxybutyrate dehydrogenase (2HBDH; EC:1.1.1.30), catalyzed reaction showed increased flux in the CRC–E. durans integrated model.
Metabolome analysis of the host–microbe models aid in predicting novel therapeutic drug targets in CRC
Another important aspect of our computational studies involving FSr analysis for different sets of metabolic models, viz. CRC and colon; CRC–E. durans and colon–E. durans, was to identify any affected reactions/enzymes that behaved differentially in CRC (or CRC–E. durans) models and could serve as promising new drug targets. These included:
-
1.
Sn-Glycerol-3-Phosphate: (Acceptor) 2-Oxidoreductase (E.C.1.1.3.21) catalyzed reaction of the OXPHOS pathway which displayed increased flux in CRC-microbe integrated model (16.67% increment) indicating that the CRC cells favor oxidative phosphorylation (OXPHOS) over glycolysis.
-
2.
3, 4-Dihydroxymandelaldehyde Dehydrogenase (E.C.1.2.1.5) catalyzed reactions carried increased flux in CRC and CRC–microbe metabolic models (increase by 62.5%).
-
3.
Catechol O-Methyltransferase (COMT; E.C.2.1.1.6) catalyzed reaction from phenylalanine metabolism in CRC–microbe model showed increased flux by 41.67%.
These novel predictions have been summarized in Table 2 and their significance in CRC has been discussed in detail in the ensuing discussion.
The secreted metabolite profiles (secretome) of the host and host-microbe models were also analyzed to better understand the metabolic changes associated with CRC metabolism (in the presence and absence of gut microbe). Based upon the FSr analysis of the colon v/s CRC and colon–microbe vs. CRC–microbe models, the exchanges with FSr values falling in the defined range (0.8 > FSr > 2) were further analyzed to establish the secretome of the CRC model. The maximum fluxes for these exchanges were subsequently observed to identify the metabolites with increased secretion in cancer cells (Fig. 7). In case of colon v/s CRC models, the metabolites pertaining to fatty acid metabolism (EX_lnlncg (gamma-linolenic acid), EX_ocdca (stearic acid), EX_hdca (palmitic acid) etc.), sphingolipid and glycolipid metabolism (EX_dolichol_L, EX_acnam (N-acetylneuraminic acid), EX_sphgn (sphinganine)), glucose metabolism (EX_glyc (glycerate), EX_glcn (gluconic acid)), amino acid metabolism (EX_Nacasp (N-acetyl-L-aspartic acid), EX_glu_L, EX_asn_L) showed increased flux in CRC model compared to healthy colon (Fig. 7a). Similarly, a comparison of colon-microbe and CRC–microbe in terms of fluxes through host exchange reactions captured increased fluxes for certain metabolites of importance in cancer (Fig. 7b). These included pyrimidine metabolism (EX_cytd[e]b (cytidine), EX_dcyt[e]b (deoxycytidine), EX_orot[e]b (orotic acid)), steroid metabolism (EX_dheas[e]b (Dehydroepiandrosterone sulfate), EX_eandrstrn[e]b (16a-hydroxydehydroisoandrosterone), EX_prgnlone (pregnenolone)), catechol metabolism (EX_34dhoxpeg[e]b (3,4-dihydroxyphenylglycol)). Further, E. durans secreted metabolites can also affect CRC metabolism. In this study, gut bacteria secreted thymidine (intercellular exchange) showed increased flux in CRC–microbe model, thus acting as the point of interaction between the two models. The secretome analysis of cancer cells and its relevance in promoting/controlling the disorder has been discussed in the next section.

a Colon vs. CRC and b colon–microbe vs. CRC–microbe. Exchange reactions with increased fluxes are depicted in red, with the same fluxes in white and reduced fluxes in black.
Discussion
The primary aim of this study was to uncover the role of gut microbes in nanoparticle-aided CRC management using guided computational and experimental approaches. Specifically, genome-scale metabolic modeling was utilized to substantiate the metabolic interactions between gut microbe and CRC metabolisms, which were identified through experimental analysis. Firstly, the response of E. durans to nanoparticle exposure was examined, as AgNPs have been reported to generate intracellular oxidative stress27,28. in biological systems. Despite the notable antioxidant properties displayed by E. durans29, a lower concentration (25 ppm) of AgNPs resulted in an increased generation of intracellular reactive species (Fig. 3). Furthermore, the hydroxyl levels increased noticeably during the late log phase in nanoparticle-treated cultures, indicating that AgNPs are potent inducers of ROS. In turn, the nanoparticle-generated oxidative stress has been reported to affect the cellular metabolism in different biological systems, thereby increasing the productivity of certain metabolites (for instance, xanthan gum and pyocyanin in Xanthomonas campestris)30,31. Based on these findings, it was hypothesized that increased ROS levels may impact the gut microbial metabolism and secretion profiles of metabolites, such as folate, while the latter being of major relevance in key metabolic pathways.
To test this hypothesis, we used the systems biology modeling approach (COBRA toolbox)32 to analyze the systems-level effects of ROS on the metabolism of E. durans. The modeling framework assisted in establishing the critical link between folic acid metabolism and other important intermediary metabolic pathways (i.e., amino acid, energy, lipids, as well as SCFA metabolism) in the gut microbe exposed to ROS (Supplementary Table 2). For instance, the minNorm analysis indicated that the addition of ROS to the model system affected glycine hydroxymethyl transferase (EC:2.1.2.1)33 catalyzed reaction, wherein the 3-carbon serine serves as one of the major sources in transferring one-carbon moiety to tetrahydrofolate (THF) to form 5, 10-methylene THF (MLTHF). A positive flux was generated in this reaction, implying an increased release of folate derivatives, thereby affecting folate metabolism. Similarly, an increased flux was observed in methylenetetrahydrofolate dehydrogenase (EC:1.5.1.5) catalyzed reaction that synthesizes METHF (methenyl THF) from MLTHF (derived from serine metabolism), thus associating energy (NADPH) and nucleic acid metabolism with folate metabolism34.
Interestingly, the novel link between folic acid derivatives and SCFA (propionate) metabolism was predicted by the model (Fig. 4). The two enzymes linking these essential pathways were malonyl CoA pyruvate carboxytransferase (EC:2.1.3.1) and 2-oxobutanoate formate lyase. Both these enzymes are required for synthesizing propionate CoA, which further participates in propionate metabolism. The role of ROS in increasing the folate levels was further validated through experiments, wherein an increase in extracellular folate levels was observed in AgNP treated bacterial culture (Fig. 5a). Thus, this is the first study of its kind that establishes the interplay between gut bacterial metabolism and AgNP-generated intracellular oxidative stress.
Moreover, the interactions between E. durans and CRC were extensively modeled to assess the role of gut microbes in disease etiology. To do so, multi-omics (proteomics and transcriptomics) data-driven metabolic models of CRC and colon were generated (refer methods section). These models were used to analyze distinct metabolic features of CRC cells often represented through rapid growth and division in tumor cells when compared to their healthy counterparts (colonocytes)35. A higher rate of cell proliferation was observed in the CRC model in terms of increased biomass flux (i.e., approximately 98%), as compared to the healthy colon model, thus corroborating the existing literature reports36. Moreover, these tissue-specific models were also able to capture various metabolic pathways (Fig. 6a), responsible for providing various biosynthetic components (ATP, NADPH, NAD+, acetyl-CoA, and amino acids) to the transformed cell for sustaining its growth and proliferation.
Further, to understand the effect of gut microbes on CRC metabolism, the host–microbe interactions were simulated by constraining the host-microbe integrated model as per the experimental conditions. These metabolic interactions can aid in deciphering the interplay between gut microbes and tumor cells, and hence be advantageous in devising novel therapeutic targets in the context of AgNP-based CRC treatment. The metabolites secreted by gut microbes also affected CRC cell viability, as a significant reduction in viability was observed in HCT 116 cells treated with supernatants obtained at the 9th h of bacterial culture growth—both control and AgNP treated cultures (Fig. 5b). Moreover, the extracellular folate concentrations measured in AgNP treated cultures at the 9th h of bacterial growth were equivalent to the optimal-cytotoxic concentration of synthetic folic acid that resulted in increased cell death (compared to control). These experimental findings observations found that oxidative stress heightened the production of folate to optimal-cytotoxic levels, affecting cancer cell viability.
Contrary to experimental findings, the CRC–microbe model was unable to capture the anti-cancer effects of gut bacterial metabolite(s), as no decrease in flux was observed in the flux through the CRC-biomass reaction. Typically, an oncogenic transformation operates at multiple levels of signaling, metabolic, and regulatory variations, all culminating in the re-wiring of cellular metabolic pathways12,37. However, owing to the limitations of GSMs, these models were unable to capture other significant cellular aspects (i.e., signaling and regulatory) associated with metabolism. Nonetheless, the tissue-specific models (CRC and CRC–E. durans) effectively captured various metabolic pathways that contribute to aberrant CRC metabolism (Fig. 6b).
One of the major outcomes of this study was the agreement observed between the computational predictions and literature findings for the affected pathways in CRC and CRC–microbe models. A detailed review of the same has been discussed in the Supplementary Information (Supplementary Figs. 4–6) along with additional analysis such as computing shadow prices, flux enrichment analysis, etc. (details in Supplementary Discussion). Furthermore, these metabolic models were able to predict enzymatic reactions that could serve as novel drug targets while managing CRC (Table 2). For instance, the constraint-based CRC-microbe integrated metabolic model showed increased flux through Sn-Glycerol-3-Phosphate: (Acceptor) 2-Oxidoreductase (EC:1.1.3.21) catalyzed reaction of the OXPHOS pathway when compared with colon–microbe model. Experimental studies have shown that the cancer cells derive their energy mainly through aerobic glycolysis (Warburg effect)38. In such cases, the cells prefer to metabolize carbohydrates through glycolysis, despite having completely functional mitochondria, thus not resorting to a more efficient oxidative phosphorylation (OXPHOS) pathway. However, recent studies have shown that certain cancers (prostate and ovarian) exhibited a switch from glycolysis to OXPHOS for energy generation39. This change in energy metabolism is another prominent characteristic feature of cancer metabolism, which makes the OXPHOS pathway an effective target for cancer therapeutics. This phenomenon has not been reported in CRC-related experimental studies this far. In addition to linking carbohydrate and lipid metabolism, this enzyme also catalyzes the generation of hydrogen peroxide (H2O2), the latter resulting in increased intracellular oxidative stress40. This chronic oxidative stress in turn is conducive to the pro-inflammatory micro-environment of CRC tissues. Thus, given its significance in the OXPHOS pathway, this enzyme can serve as a novel chemotherapeutic drug target.
Another enzyme, 3, 4-dihydroxymandelaldehyde: NADP + oxidoreductase (EC:1.2.1.5) facilitates the conversion of 3,4-dihydroxymandelaldehyde to 3,4-dihydroxymandelic acid (DHMA) in tyrosine metabolism. DHMA, a catechol, is a norepinephrine metabolite known to possess strong antioxidative potential41. Certain in vitro studies have shown that catechol inhibits the growth of lung cancer, and therefore possesses anticancer attributes42. However, its role in CRC metabolism and treatment has not been investigated. Interestingly, 3, 4-dihydroxymandelaldehyde: NADP + oxidoreductase catalyzed reactions showed increased flux in CRC and CRC–microbe metabolic models, thus, favoring its importance as a therapeutic target while designing novel cancer treatment drugs.
Catechol O-methyltransferase (COMT; EC:2.1.1.6) catalyzes the synthesis of S-Adenosyl-homocysteine (SAH) from S-Adenosyl-methionine (SAM), the latter being a major methyl donor in transmethylation reactions that can downregulate the expression of oncogenic promoters by reversing DNA hypomethylation, thereby inhibiting tumor growth43. Furthermore, this tumor-inhibiting attribute of SAM is well established in gastric and colon cancers44. In CRC–microbe model, an increase in flux through this reaction was observed (41.67%). On the other hand, high SAH serum levels inhibit SAM-dependent methyltransferases (that catalyzes methionine to SAM conversion). Elevated serum concentrations of SAH have been reported in chronic diseases like cardiovascular and chronic kidney diseases45. However, the role of SAH has not been established in colon cancer yet. The reduced levels of SAM (on account of increased flux through COMT catalyzed reaction in the CRC–microbe model) can result in increased cancer cell proliferation and growth making COMT a promising drug target, an aspect that can motivate future experimental studies.
To better understand the anomalous behavior of CRC, the secretome profiles obtained from medium constrained models focused on the metabolites accountable for deviant cellular characteristics observed in CRC by altering the tumor micro-environment. The models were constrained as per the DMEM composition, wherein various macro- and micro-nutrients were provided as substrate inputs. Several metabolite secretions were affected in diseased conditions compared to a healthy colon. For instance, increased secretion of alpha-ketoglutarate (EX_akg), an intermediate of the tricarboxylic acid cycle, reinstated its relevance in the oxidation of fatty acids, amino acids, and glucose46. Other metabolites partaking in fatty acid metabolism, glycolysis, sphingolipid, and amino acid metabolism also showed increased exchange fluxes in the CRC model, compared to the colon (Fig. 7a). An essential metabolite, N-acetyl-l-aspartic acid (EX_Nacasp) which has a prominent role in promoting tumor growth and can act as a potential target for anticancer therapy47 also showed increased secretion in the CRC model.
Similarly, CRC-microbe secretome analysis also featured elevated fluxes in various metabolites secretions (Fig. 7b) like orotic acid (EX_orot[e]b) and its derivative (EX_orot5p[e]b), which is a powerful tumor promoter in hepatocellular carcinoma48. Pregnenolone (EX_prgnlone[e]b) and sulfites (EX_so3[e]b) which promote prostate cancer49 and ulcerative colitis, potentially leading to colon cancer50 respectively, also showed increased secretion in the integrated model. In addition to the tumor-promoting metabolite secretions, dehydroepiandrosterone sulfate (EX_dheas[e]b) a key adrenal steroid that is known to have some protective role in CRC51 also showed elevated levels in the host–microbe integrated model. Moreover, it was thymidine secreted by E. durans that acted as an important metabolite connecting the microbe metabolism with that of the host. Increased extracellular transport of thymidine from microbe to CRC could have possibly boosted the latter’s pyrimidine metabolism compared to the healthy colon, as captured and supported by the FSr analysis. Thus, all the computational predictions cumulatively emphasized the applicability of GSMMs in comprehending CRC metabolism and potential drug targets.
To summarize, CBM effectively predicted the association between ROS and folate metabolism, which was also verified through experiments. Moreover, context-specific models of CRC and colon could accurately capture the fundamental biochemical differences between a healthy colon- and CRC-cell, further demonstrating the metabolic adaptations essential for CRC survival. The integration of CRC and gut microbe metabolic models to simulate host-microbe interactions was beneficial in understanding CRC metabolism in presence of a gut microbe. These metabolic interactions can help mimic the interplay between gut microbes and tumor cells under in vivo conditions and can be advantageous while devising novel therapeutic targets in the context of AgNP-based CRC treatment.
Methods
Characterization of silver nanoparticles (AgNPs)
Silver nanoparticles (AgNPs) were obtained from Sigma Aldrich (catalog No. 7440-22-4). Size distribution analysis was carried out using dynamic light scattering and zeta potential was measured using Horiba Scientific Nanopartica nanoparticle analyzer (SZ-100). A scanning electron microscope was used to study NP–bacteria interaction.
Bacterial culture and treatment with AgNPs
E. durans, a facultative aerobe, procured from MTCC (MTCC No. 3031) was used as the model organism. The bacterial culture was grown in shake flasks containing MRS broth at 37 °C, 180 rpm, in a shaker (Scigenics Orbitek). The total cell concentrations at different time points were measured through optical density (cell scatter) at 600 nm (JASCO V-630 Spectrophotometer), and a comparison with a standard plot of OD vs. cell concentration was done.
The AgNPs (25–250 ppm) were dispersed in the medium using a water bath sonicator. The medium was then inoculated with the appropriate volume of the subculture (inoculum), such that the OD value at the 0th h was 0.1.
Quantifying intracellular ROS concentrations
Intracellular ROS were measured by following the procedures from literature52. 3′-(p-amino-phenyl) fluorescein, APF (Invitrogen, USA) and dihydroethidium, DHE (Sigma-Aldrich, India) were used to detect hydroxyl and superoxide radicals, respectively. The samples (microbial cell cultures at different time points) were incubated with the dyes (2 mM DHE at 37 °C and 5 mM APF at room temperature) for 30 min. Upon detection through a fluorescent spectrophotometer, the hydroxyl and superoxide radical concentrations were evaluated using the standards plots of hydrogen peroxide and potassium superoxide, respectively. The concentration of superoxide and hydroxyl radicals was reported in nmol/(g-cell).
Sample preparation for folate estimation and HPLC analysis
The bacterial culture was harvested every 3 h and culture volume corresponding to 10 OD was used for sample extraction. The extracted culture volume was centrifuged, and the bacterial pellet obtained was suspended in 1 ml of milli Q water. It was then sonicated (Q Sonica sonicator), at an amplitude of 70%, for a processing time of 4 min (pulse on and off time being 2 s). The sonicated sample was then placed in a water bath at 100 °C and subjected to heat for 5 min to release any folate bound to the folate binding proteins. The cell-free extract was obtained by centrifuging the sample. The supernatant was collected, filtered, and used further for folate estimation. For quantifying extracellular folate, released by the bacterial cells into the growth medium, 1 ml of culture was collected every 3 h and the filtered supernatant was used for HPLC using UFLC Shimadzu HPLC setup. C18 Hypersil column (25 cm*4.6 mm, 5-micron spherical packing)53. Carbinol in 0.05 M KH2PO4 which was used as the mobile phase was filtered through 0.46-micron filters before use and then sonicated in a bath sonicator for 10 min for degassing the solution. The flow rate was maintained at 0.4 mL/min.
Folic acid (Himedia, catalog No. CMS175) was used as standard (0–125 μM) in the estimation of folate in samples (cell extracts and bacterial supernatant).
Treatment of HCT 116 with bacterial supernatant
HCT 116 (colon cancer cell line) was obtained from Dr. Bert Vogelstein, John Hopkins University, Baltimore, USA. The tumor cells were grown in Dulbecco’s Modified Eagle Media (DMEM), with 5% serum. The cells were seeded and grown in a 96-wells plate and were treated with bacterial supernatant (control and 25 ppm AgNPs), and different concentrations of synthetic folic acid. MTT cell proliferation assay was then performed to quantify cell viability on exposure to the drug after 48 h of treatment54.
Modeling gut bacteria—ROS interplay: constraint-based model formulation
In our study, the unconstrained E. durans GSMM was downloaded from the VMH database (https://vmh.uni.lu/) and expanded using ROS reactions curated from the literature.
rBioNet was used to add the different reactions (ROS reactions; missing transport and exchange reactions; sink and demand reactions) to the model. rBioNet enables the user to add these reactions in a quality-controlled manner, by exempting sources of manual error55. A total of eight ROS reactions and 11 metabolites were added to the model. These ROS reactions focused on the biochemical interactions between reactive species (predominantly superoxide, hydroxyl radical, and nitric oxide), and amino acids56, and nucleic acids57. The resulting reconstruction (with ROS reactions) was then merged with the downloaded metabolic model, thus, formulating the ROS expanded model (Supplementary Data 1). FBA was carried out on the newly added reactions individually to check if the reactions were blocked (carrying zero net flux). Blocked reactions might result from incomplete reaction information about the consumption of substrates or the generation of products. These blocked reactions were then resolved by adding complete metabolic reactions and pathways from the literature). In case there was no literature evidence supporting reactions pertaining to the blocked reaction, the reaction was un-blocked (by adding demand or sink reactions for products and reactants, respectively). For E. durans model, seven demand reactions and one sink reaction were added. Upon un-blocking the newly added reactions, dead-end metabolites were then identified. Dead-end metabolites are the ones that are either produced or consumed but never both. These dead-end metabolites were also resolved using literature information for adding the required reactions to the model. The debugged model was medium constrained as per MRS medium composition.
FVA was then performed that provided the flux values (minimum and maximum) FSr, which is the ratio between absolute flux values of reactions for healthy model (original E. durans) to absolute flux values of reactions for disease model (ROS-expanded E. durans) was calculated. The range for FSr is user-defined. The reactions with FSr values in the range 0.8 > FSr > 2 were identified. These were the reactions that were considered as affected due to the addition of ROS reactions to the model. minNorm analysis was then carried out on these reactions. minNorm function in MATLAB is a tool for frequency estimation of the data vector, for a given function.
Context-specific model building
Recon 3D, a human metabolic reconstruction, consisting of metabolic reactions and their corresponding enzymes/genes was used to generate the colon and CRC condition-specific tissue models. The ‘omics’ data (obtained from HPA)23 for healthy and CRC colon was mapped onto Recon 3D, and the resultant model was derived using iMAT algorithm25 to provide a better understanding of the molecular phenomena occurring in cancer and normal cell types. iMAT is an integrative metabolic analysis tool, that allows the integration of transcriptomic and proteomic data with genome-scale metabolic networks to calculate fluxes associated with enzyme metabolized reactions. It works best for capturing reactions/pathways associated with the given data input. This is advantageous in cases where a single objective is not obeyed/maintained, for example, mammalian cells. The reactions are grouped in three categories based on the expression levels of proteins as high, medium, and low (as per HPA). A data matrix is constructed, wherein the primary reaction set represents reactions with a high and medium confidence level to be present in the cell type, and this data matrix is fed in the iMAT algorithm. The gap-filling was then done using MILP (mixed-integer linear programming) approach, maintaining the defined stoichiometric and thermodynamic constraints. The preliminary models once generated are further curated to satisfy the sanity checks as per COBRA toolbox standards. Further, to the healthy colon model, transport reactions were added as reported in literature58, and the biomass reaction was modified to exclude replicating precursors and include dependency on cellular folate59. However, the CRC model was retained with the biomass reaction as in Recon 3D. Additionally, both the healthy and CRC colon models were expanded to include exchange reactions for all the metabolites appearing in the extracellular compartment.
Microbiome modeling toolbox
The Microbiome Modeling Toolbox26 provided by the COBRA toolbox was used to integrate host and microbe models. This toolbox is primarily used for modeling microbe-microbe/host-microbe interactions, as well as personalized microbial communities.
Statistical analysis
All cultures and measurements were carried out in triplicates (each subjected to at least three technical replicates). Values have been reported as mean ± SD (please refer to individual results in the Results section). One-way ANOVA (level of significance, α = 0.05) and Tuckey’s multiple comparison tests were carried out.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The AGORA resource is freely available at the Virtual Metabolic Human website (https://vmh.life). The models used in the study are growth medium constrained and have been provided as excel sheets in Supplementary Data 1–3.
Code availability
MATLAB codes for performing simulations and data analyses can be accessed at https://opencobra.github.io/cobratoolbox/latest/modules/analysis/index.html and Supplementary Note.
References
Tuan, J. & Chen, Y.-X. Dietary and lifestyle factors associated with colorectal cancer risk and interactions with microbiota: fiber, red or processed meat and alcoholic drinks. Gastrointest. Tumors 3, 17–24 (2016).
Bardou, M., Barkun, A. N. & Martel, M. Obesity and colorectal cancer. Gut 62, 933–947 (2013).
Dulal, S. & Keku, T. O. Gut microbiome and colorectal adenomas. Cancer J. 20, 225–231 (2014).
Carding, S., Verbeke, K., Vipond, D. T., Corfe, B. M. & Owen, L. J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 26, 26191 (2015).
Belizário, J. E. & Faintuch, J. Microbiome and gut dysbiosis. Exp. Suppl. 109, 459–476 (2018).
Jandhyala, S. M. et al. Role of the normal gut microbiota. World J. Gastroenterol. 21, 8787–8803 (2015).
Koppel, N., Rekdal, V. M. & Balskus, E. P. Chemical transformation of xenobiotics by the human gut microbiota. Science 356, eaag2770 (2017).
Ríos-Covián, D. et al. Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 7, 185 (2016).
Karpiński, T. M. & Adamczak, A. Anticancer activity of bacterial proteins and peptides. Pharmaceutics 10, E54 (2018).
Avram-Hananel, L., Stock, J., Parlesak, A., Bode, C. & Schwartz, B. E durans strain M4-5 isolated from human colonic flora attenuates intestinal inflammation. Dis. Colon Rectum 53, 1676–1686 (2010).
Zhu, H., Ji, Y., Li, W. & Wu, M. Identification of key pathways and genes in colorectal cancer to predict the prognosis based on mRNA interaction network. Oncol. Lett. 18, 3778–3786 (2019).
Koveitypour, Z. et al. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 9, 97 (2019).
Jerby, L. & Ruppin, E. Predicting drug targets and biomarkers of cancer via genome-scale metabolic modeling. Clin. Cancer Res. 18, 5572–5584 (2012).
Agren, R. et al. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 10, 721 (2014).
Auslander, N. et al. An integrated computational and experimental study uncovers FUT9 as a metabolic driver of colorectal cancer. Mol. Syst. Biol. 13, 956 (2017).
Becker, S. A. & Palsson, B. O. Context-specific metabolic networks are consistent with experiments. PLOS Comput. Biol. 4, e1000082 (2008).
Noronha, A. et al. The Virtual Metabolic Human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 47, D614–D624 (2019).
Mahadevan, R. & Schilling, C. H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 5, 264–276 (2003).
Hinnebusch, B. F., Meng, S., Wu, J. T., Archer, S. Y. & Hodin, R. A. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J. Nutr. 132, 1012–1017 (2002).
Keum, N. & Giovannucci, E. L. Folic acid fortification and colorectal cancer risk. Am. J. Prevent. Med. 46, S65–S72 (2014).
Petersen, L. F. et al. Elevated physiological levels of folic acid can increase in vitro growth and invasiveness of prostate cancer cells. BJU Int. 109, 788–795 (2012).
Brunk, E. et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 36, 272–281 (2018).
Thul, P. J. & Lindskog, C. The human protein atlas: a spatial map of the human proteome. Protein Sci. 27, 233–244 (2018).
van der Post, S. & Hansson, G. C. Membrane protein profiling of human colon reveals distinct regional differences. Mol. Cell Proteom. 13, 2277–2287 (2014).
Zur, H., Ruppin, E. & Shlomi, T. iMAT: an integrative metabolic analysis tool. Bioinformatics 26, 3140–3142 (2010).
Baldini, F. et al. The Microbiome Modeling Toolbox: from microbial interactions to personalized microbial communities. Bioinformatics 35, 2332–2334 (2019).
Durán, N. et al. Potential use of silver nanoparticles on pathogenic bacteria, their toxicity and possible mechanisms of action. J. Braz. Chem. Soc. 21, 949–959 (2010).
Khalandi, B. et al. A review on potential role of silver nanoparticles and possible mechanisms of their actions on bacteria. Drug Res. 67, 70–76 (2017).
Pieniz, S., Andreazza, R., Anghinoni, T., Camargo, F. & Brandelli, A. Probiotic potential, antimicrobial and antioxidant activities of Enterococcus durans strain LAB18s. Food Control 37, 251–256 (2014).
Rao, Y. M. & Sureshkumar, G. K. Improvement in bioreactor productivities using free radicals: HOCl-induced overproduction of xanthan gum from Xanthomonas campestris and its mechanism. Biotechnol. Bioeng. 72, 62–68 (2001).
Rao, Y. M. & Sureshkumar, G. K. Oxidative-stress-induced production of pyocyanin by Xanthomonas campestris and its effect on the indicator target organism Escherichia coli. J. Ind. Microbiol. Biotech. 25, 266–272 (2000).
Heirendt, L. et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protocols 14, 639–702 (2019).
Piper, M. D., Hong, S. P., Ball, G. E. & Dawes, I. W. Regulation of the balance of one-carbon metabolism in Saccharomyces cerevisiae. J. Biol. Chem. 275, 30987–30995 (2000).
Fan, J. et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302 (2014).
Susnow, N. J. & Hockenbery, D. M. Bak to basics of colonocyte renewal. Gastroenterology 136, 763–766 (2009).
Keibler, M. A. et al. Metabolic requirements for cancer cell proliferation. Cancer Metab. 4, 16 (2016).
Fritz, V. & Fajas, L. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene 29, 4369–4377 (2010).
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Porporato, P. E., Filigheddu, N., Pedro, J. M. B.-S., Kroemer, G. & Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 28, 265–280 (2018).
Singh, G. Mitochondrial FAD-linked glycerol-3-phosphate dehydrogenase: a target for cancer therapeutics. Pharmaceuticals 7, 192–206 (2014).
Ley, J. P., Engelhart, K., Bernhardt, J. & Bertram, H.-J. 3,4-Dihydroxymandelic acid, a noradrenalin metabolite with powerful antioxidative potential. J. Agric. Food Chem. 50, 5897–5902 (2002).
Lim, D. Y. et al. A natural small molecule, catechol, induces c-Myc degradation by directly targeting ERK2 in lung cancer. Oncotarget 7, 35001–35014 (2016).
Gao, J. et al. S-adenosyl methionine and transmethylation pathways in neuropsychiatric diseases throughout life. Neurotherapeutics 15, 156–175 (2018).
Wu, W. et al. Catechol-O-methyltransferase inhibits colorectal cancer cell proliferation and invasion. Arch. Med. Res. 46, 17–23 (2015).
Valli, A. et al. Elevated serum levels of S-adenosylhomocysteine, but not homocysteine, are associated with cardiovascular disease in stage 5 chronic kidney disease patients. Clin. Chim. Acta 395, 106–110 (2008).
Abla, H., Sollazzo, M., Gasparre, G., Iommarini, L. & Porcelli, A. M. The multifaceted contribution of α-ketoglutarate to tumor progression: an opportunity to exploit? Semin. Cell Dev. Biol. 98, 26–33 (2020).
Zand, B. et al. Role of Increased n-acetylaspartate Levels in Cancer. J. Natl Cancer Inst. 108, djv426 (2016).
Jung, E.-J., Lee, K.-Y. & Lee, B.-H. Proliferating effect of orotic acid through mTORC1 activation mediated by negative regulation of AMPK in SK-Hep1 hepatocellular carcinoma cells. J. Toxicol. Sci. 37, 813–821 (2012).
Grigoryev, D. N., Long, B. J., Njar, V. C. & Brodie, A. H. Pregnenolone stimulates LNCaP prostate cancer cell growth via the mutated androgen receptor. J. Steroid Biochem. Mol. Biol. 75, 1–10 (2000).
Teigen, L. M. et al. Dietary factors in sulfur metabolism and pathogenesis of ulcerative colitis. Nutrients 11, 931 (2019).
Alberg, A. J., Gordon, G. B., Hoffman, S. C., Comstock, G. W. & Helzlsouer, K. J. Serum dehydroepiandrosterone and dehydroepiandrosterone sulfate and the subsequent risk of developing colon cancer. Cancer Epidemiol. Biomark. Prev. 9, 517–521 (2000).
Menon, K. R., Balan, R. & Suraishkumar, G. K. Stress induced lipid production in Chlorella vulgaris: relationship with specific intracellular reactive species levels. Biotechnol. Bioeng. 110, 1627–1636 (2013).
Jose, S., Bhalla, P. & Suraishkumar, G. K. Oxidative stress decreases the redox ratio and folate content in the gut microbe, Enterococcus durans (MTCC 3031). Sci. Rep. 8, 12138 (2018).
Riss, T. L. et al. Cell viability assays. in Assay Guidance Manual (eds Sittampalam, G. S. et al.) (Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2004).
Thorleifsson, S. G. & Thiele, I. rBioNet: A COBRA toolbox extension for reconstructing high-quality biochemical networks. Bioinformatics 27, 2009–2010 (2011).
Hawkins, C. L. & Davies, M. J. Generation and propagation of radical reactions on proteins. Biochim. Biophys. Acta 1504, 196–219 (2001).
Gates, K. S. An overview of chemical processes that damage cellular DNA: spontaneous hydrolysis, alkylation, and reactions with radicals. Chem. Res. Toxicol. 22, 1747–1760 (2009).
Sahoo, S., Aurich, M. K., Jonsson, J. J. & Thiele, I. Membrane transporters in a human genome-scale metabolic knowledgebase and their implications for disease. Front. Physiol. 5, 91 (2014).
Sahoo, S. & Thiele, I. Predicting the impact of diet and enzymopathies on human small intestinal epithelial cells. Hum. Mol. Genet. 22, 2705–2722 (2013).
Shi, L. & Tu, B. P. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr. Opin. Cell Biol. 33, 125–131 (2015).
Zaugg, K. et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 25, 1041–1051 (2011).
Nakajima, Y. et al. Three-dimensional structure of the flavoenzyme acyl-CoA oxidase-II from rat liver, the peroxisomal counterpart of mitochondrial acyl-CoA dehydrogenase. J. Biochem. 131, 365–374 (2002).
Swanson, K. M. & Hohl, R. J. Anti-cancer therapy: targeting the mevalonate pathway. Curr. Cancer Drug Targets 6, 15–37 (2006).
Ducker, G. S. & Rabinowitz, J. D. One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017).
Adeva-Andany, M. M., López-Maside, L., Donapetry-García, C., Fernández-Fernández, C. & Sixto-Leal, C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids 49, 1005–1028 (2017).
Gwaltney-Brant, S. M. Chapter 7—nutraceuticals in hepatic diseases. in Nutraceuticals (ed Gupta, R. C.) 87–99 (Academic Press, 2016). https://doi.org/10.1016/B978-0-12-802147-7.00007-3.
Rawłuszko-Wieczorek, A. A. et al. Significance of intratissue estrogen concentration coupled with estrogen receptors levels in colorectal cancer prognosis. Oncotarget 8, 115546–115560 (2017).
Anwar, Y., Sabir, J. S. M., Qureshi, M. I. & Saini, K. S. 5-lipoxygenase: a promising drug target against inflammatory diseases-biochemical and pharmacological regulation. Curr. Drug Targets 15, 410–422 (2014).
Acknowledgements
This work was supported by the Department of Science and Technology, Government of India (grant No. EMR/2016/002069) to G.K.S., R.R. and D.K. DST-INSPIRE Faculty award (DST/INSPIRE/04/2015/000036) to S.S.
Author information
Authors and Affiliations
Contributions
G.K.S., S.S. and P.B. designed the wet lab experiments and computational studies. R.R. provided novel inputs in the computational part, and D.K. in the experimental part. P.B. performed all the experiments and computational analysis. P.B. and S.S. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhalla, P., Rengaswamy, R., Karunagaran, D. et al. Metabolic modeling of host–microbe interactions for therapeutics in colorectal cancer. npj Syst Biol Appl 8, 1 (2022). https://doi.org/10.1038/s41540-021-00210-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-021-00210-9
This article is cited by
-
Gut microbiota modulation: a tool for the management of colorectal cancer
Journal of Translational Medicine (2022)
-
Recent Progress in Mass Spectrometry-based Metabolomics for Colorectal Cancer
Chemical Research in Chinese Universities (2022)
