
Abstract
Coexisting density-wave and superconducting states along with the large anomalous Hall effect in the absence of local magnetism remain intriguing and enigmatic features of the AV3Sb5 kagome metals (A = K, Rb, Cs). Here, we demonstrate via optical spectroscopy and density-functional calculations that low-energy dynamics of KV3Sb5 is characterized by unconventional localized carriers, which are strongly renormalized across the density-wave transition and indicative of electronic correlations. Strong phonon anomalies are prominent not only below the density-wave transition, but also at high temperatures, suggesting an intricate interplay of phonons with the underlying electronic structure. We further propose the star-of-David and tri-hexagon (inverse star-of-David) configurations for the density-wave order in KV3Sb5. These configurations are strongly reminiscent of p-wave states expected in the Hubbard model on the kagome lattice at the filling level of the van Hove singularity. The proximity to this regime should have intriguing and far-reaching implications for the physics of KV3Sb5 and related materials.
Similar content being viewed by others


Introduction
Kagome geometry of corner-sharing triangles plays a special role in condensed-matter physics. In magnetic insulators, it may cause the highly entangled quantum spin liquid state with exotic fractionalized excitations1. Kagome metals exploit another aspect of this peculiar geometry, the simultaneous presence of linear and flat energy bands. The former cross at the Dirac point that gives rise to Weyl nodes when time-reversal symmetry is broken. These interesting features of the electronic structure were indeed detected in several magnetic kagome metals2,3,4,5,6, where spin texture—the type of magnetic order and its individual spin directions—was shown to have large impact on the energy bands and transport7,8,9.
The discovery of AV3Sb5 (A = K, Rb, Cs)10 opens the way to studying physics of kagome framework in the absence of local magnetic moments. Here, the topological nature of the kagome bands can be intricately intertwined with electronic instabilities, such as superconductivity and charge order. Under ambient conditions, the AV3Sb5 family crystallizes in the hexagonal P6/mmm space group. The V-atoms create the kagome network, while the Sb1 atoms fill the centers of the kagome triangles. These V-Sb1 layers are sandwiched between graphite-like Sb2 layers that are, in turn, separated by the alkali-metal ions (Fig. 1a), creating easily exfoliable quasi-2D structures.

a Crystal structure of KV3Sb5 consists of V-Sb kagome layers and graphite-like Sb layers separated by the potassium atoms. b Representative band structure and corresponding density-of-states (DOS) for the nearest-neighbor tight-binding model on the kagome lattice. Fermi energy is set to the van Hove singularity (VHS) point at the filling factor of 5/12. c Calculated DOS of KV3Sb5 in its normal (undistorted) structure and in the DW state (star structure, see Fig. 3) demonstrates a depression in the energy spectrum at the Fermi level (EF = 0) below the transition. d The dc resistivity of the sample used in the optical study is shown together with the resistivity values obtained by the Hagen–Rubens fits of the optical data. The insets display the 78 K anomaly in the first derivative of the resistivity and in the magnetic susceptibility. e The calculated band structure of KV3Sb5 (normal state) demonstrates the linearly dispersing bands around the Fermi energy. Relevant bands for the optical transitions have been labeled bottom up. f Fermi surface constructed from the calculated band structure and the cut along kz = 0 for KV3Sb5. The high-symmetry points and the k-path are shown in orange.
The AV3Sb5 compounds show abrupt changes in the magnetic susceptibility and electrical resistivity around 78 K (A = K10), 102 K (A = Rb11), and 94 K (A = Cs12). They further become superconducting below 0.9 K (A = K13 and Rb11), and 2.5 K (A = Cs10). On one hand, robust nature of the former anomaly under magnetic field and absence of local magnetism witnessed by muon spin spectroscopy14 suggest that mostly charge degrees of freedom should be involved. On the other hand, the anomalous Hall effect is strongly enhanced in KV3Sb515 and CsV3Sb516 below the 78 K (94 K) transition and has been ascribed to scattering from spin clusters. Furthermore, the recent STM results on KV3Sb5 demonstrate chiral response under reversed magnetic field17 and also suggest that the low-temperature electronic state may not be a conventional charge order. This raises the question what this electronic state is, and in which form spin degrees of freedom can be embedded in it.
Theoretical studies of the kagome Hubbard model predict the formation of unusual spin states in this setting. When the Fermi level is tuned to the van Hove singularity that arises from the band saddle point at M (Fig. 1b), different forms of density-wave order are expected18. Their p-wave symmetry19 implies that no spatial modulation of spin density takes place, and any magnetic order should ensue from spin currents. Doping such states away from the van Hove singularity gives rise to an unconventional superconductivity with different gap symmetries18,20,21. This reveals several interesting parallels to AV3Sb5 compounds where both the electronic transition and superconductivity have been reported.
Another aspect, which is hitherto absent in models of kagome metals but inevitably present in real materials, are phonons that may couple to the electronic background. Phonons are instrumental in driving charge-density-wave instabilities via strong momentum dependence of the electron–phonon coupling22,23. Although density-wave instabilities of a model kagome metal may be purely electronic in nature and do not require phonons per se, their manifestations in real AV3Sb5 materials seem to involve atomic displacements12. This renders phonons and electron–phonon coupling potentially important ingredients of the AV3Sb5 physics.
With these questions in mind, here we use broadband optical spectroscopy and density-functional calculations to examine the electronic structure of and the 78 K anomaly in one of the AV3Sb5 members, namely, KV3Sb5. We demonstrate that the anomaly manifests itself by a density-wave-like behavior in our optical probe. Using strong sensitivity of the infrared spectroscopy to charge degree of freedom, we identify bulk nature of the density-wave order and gauge the associated energy scale. The crossover to this density-wave state is accompanied by a strong renormalization of the unconventional carrier response that appears as a low-energy localization peak and reflects the importance of electronic correlations in this system. Furthermore, phonon anomalies observed above and below the transition indicate a strong coupling of phonons to the underlying electronic structure. Aided by ab initio band-structure calculations, we propose possible structural models of the density-wave (DW) state and identify their similarity to phases predicted for the nearest-neighbor Hubbard model on the kagome lattice18. The respective filling factor puts the Fermi level at the van Hove singularity, in strong resemblance to KV3Sb5 with its high density-of-states at the Fermi energy. We argue that similarities between the proposed DW structures and the predicted phases of the kagome Hubbard model can be the key to understanding the peculiar physics of KV3Sb5.
Results
Normal state
Above 0.3 eV, optical conductivity of KV3Sb5 shows a rather frequency-independent behavior with several interband transitions, which get sharper upon cooling (see Supplementary Fig. 2 for details). This frequency-independent behavior is interrupted with a very pronounced absorption in the low-energy range below 0.3 eV, as shown in Fig. 2a.

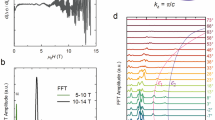
a Temperature-dependent optical conductivity. Spectra shifted by 5000 cm−1 for clarity. Phonon modes are highlighted and the localization peak is demonstrated with the arrow. Optical conductivity in the whole energy range is given in Supplementary Note 2. b Decomposition of the room-temperature optical conductivity: the low-energy Drude-contribution, low-energy localization peak, phonon modes, and interband transitions are visible. c Band-resolved optical conductivity for the undistorted crystal structure with the Fermi level shifted by + 64 meV (see Supplementary Note 6 for details). d Difference optical conductivity in the DW state. A spectral weight transfer from low to high energies is observed. Arrows indicate the zero-crossing and the maximum of the transferred peak. e Temperature evolution of the spectral weight transfer, where the peak position is normalized to the 10 K point. A clear gap opening below 80 K is visible and shows deviations from the mean-field behavior (solid line). f Temperature evolution of the localization peak with the visible redshift upon cooling. The inset shows the peak position as a function of temperature, where a saturation at low temperatures is seen. g SW of the localization peak showing an abrupt drop across TDW. h Overall SW as a function of temperature and frequency. The SW is conserved within the measured energy range. Details of the SW analysis are given in Supplementary Note 4.
A closer look reveals the details. The low-energy part of the spectra can be decomposed into several contributions as demonstrated in Fig. 2b. A very sharp Drude component is present at low energies extrapolating to the dc resistivity values. Narrow peaks that do not shift in frequency upon cooling represent two phonon modes. A broader peak around 0.1 eV does not move either and can be associated with the low-energy interband transition, discussed in detail in the following paragraph. Finally, an even broader peak is observed around 0.06 eV at room temperature and displays a redshift upon cooling. We interpret this feature as a localization peak and defer its discussion to a later part.
Band-structure calculations show that in KV3Sb5 and its sibling compounds the crossings of the linear Dirac bands lie below the Fermi level (see, e.g., Fig. 2e). This should lead to a spectral weight transfer from the interband to the intraband transitions below the Pauli-blocked edge and result in the sharp Drude component24, which is indeed observed in our spectra. The small scattering rate of around 60–70 cm−1 extracted from the fits of the optical conductivity is also in line with the high mobility of the carriers residing in these topologically nontrivial bands9.
The interband transitions are readily identified with the help of DFT calculations for the undistorted P6/mmm crystal structure of KV3Sb5 (Fig. 1e). Linear bands appear in the vicinity of the Fermi level and arise from the underlying kagome geometry of the V-atoms (Fig. 1). Band-resolved calculation (Fig. 2c) reveals that in the 0.25−1.0 eV range the optical conductivity is dominated by two nearly frequency-independent contributions that are indeed expected for transitions between linear bands in 2D Dirac systems24,25. We assign these contributions to the transitions between the linear parts of bands A–C and B–D, respectively.
The sharp absorption peak around 0.1 eV has a different origin and can be traced back to the transitions between bands B and C containing parallel segments along the A − L reciprocal direction. We note in passing that the size of this peak is quite sensitive to the exact position of the Fermi level and may be also influenced by adding electronic correlations within DFT+U. The best agreement with the experimental spectrum was achieved upon shifting the Fermi level by + 64 meV in DFT, or upon adding U = 2 eV. These changes control the occupation of bands B and C around the M-point, see Supplementary Note 6 for details.
Notwithstanding the complex nature of the real KV3Sb5 band structure, several similarities to the idealized bands of a nearest-neighbor kagome metal can be identified (Fig. 1b). First, crossing points of the linear bands are located below the Fermi level, suggesting the effective filling factor above \(\frac{1}{3}\). Second, the Fermi level sits at the energy where bands A–C cross in the vicinity of the M-point. Their saddle points are remarkably similar to the band shape expected in the idealized kagome metal near its van Hove singularity at the filling factor of 5/12. Indeed, in its normal state KV3Sb5 also shows a high density-of-states at the Fermi level (Fig. 1c). This suggests that the 78 K instability in KV3Sb5 may be associated with an instability of a correlated kagome metal at its van Hove singularity18.
Density-wave state
Having identified general trends in the optical conductivity, we inspect the low-temperature behavior in more detail. A transport and magnetic anomaly is observed at TDW = 78 K (Fig. 1d). Its insensitivity to the magnetic field10 suggests an electronic rather than magnetic nature of this instability. Indeed, in the optical spectra, we observed clear signatures of this transition. Although no bandgap opens below TDW, the decrease in the density-of-states at the Fermi energy is witnessed by the reduction in the low-energy optical conductivity, which is transferred toward higher energies as dictated by the optical sum rules. These observations prove the formation of a bulk DW state in KV3Sb526. The phonon modes persist below TDW, albeit with significant anomalies.
Difference spectra illustrate the transfer of spectral weight (Fig. 2d). The low-energy part of σ1(ω) suppressed below ~0.1 eV is recovered at higher energies with a maximum of around ~0.17 eV. The energy scale of the spectral weight redistribution is gauged by ΔDW that can be taken as an order parameter. We employed the zero-crossing of the difference spectra to determine 2ΔDW. Its temperature dependence is given in Fig. 2e. Alternatively, one can choose the maximum of the SW transfer as an analogy to ARPES measurements, resulting in different absolute values of ΔDW but essentially the same temperature dependence when scaled to the 10 K value. This temperature dependence deviates from the mean-field behavior, \({{{\Delta }}}_{{{{\rm{mf}}}}}\approx {{\Delta }}(0)\sqrt{1-\frac{T}{{T}_{c}}}\) (close to TDW), where we assume TDW = 78 K and Δ(0) = 59 meV. The obtained energy scale is of the same order of ~0.03 eV as determined recently by STM17. Note however that here we probe the bulk response, while STM is sensitive to the surface states only.
To identify possible DW states in KV3Sb5, we rely on the experimental constraint of the 2 × 2 in-plane superstructure detected by scanning tunneling microscopy17,27 and single-crystal X-ray diffraction on CsV3Sb512. Different low-symmetry configurations constructed within the 2 × 2 supercell were allowed to relax, and two stable solutions were found (Fig. 3). One of them is star-of-David-type charge order reminiscent of 1T-TaS228,29,30 and denoted as star in the following. The other one involves two types of vanadium clusters, triangles and hexagons, and is labeled simply as hexagon for brevity. In both cases, the V–V distances inside the clusters are below 2.71 Å, to be compared with 2.74 Å in the undistorted structure, whereas V–V distances between the clusters exceed 2.80 Å.

a Energy diagram of the possible DW structures in KV3Sb5. The energies are given per formula unit, and the bonds show V–V distances shorter than 2.71 Å in the DW structures. b, c p-wave states predicted for the kagome Hubbard model at the van Hove singularity18. Charge bond order in b features spatial modulation of charge, whereas spin bond order in (c) features spatial modulation of spin current. d Difference DOS of the undistorted and DW structures demonstrates the redistribution of states across TDW on the 0.1 eV energy scale, which is compatible with ΔDW determined experimentally. e Comparison of the experimental interband transitions with the DFT calculations in the normal and DW states.
The hexagon and star structures lie lower in energy than the parent, undistorted structure by 2.3 meV/f.u. and 1.1 meV/f.u., respectively. These energy differences are only marginally affected by the choice of the exchange-correlation functional or adding the spin–orbit coupling, and all three structures are non-magnetic in DFT. Further on, both hexagon and star types of charge order are compatible with transport properties of KV3Sb5. The metallic behavior observed experimentally even below TDW suggests that a sizable Fermi surface persists below the transition, and the DOS at the Fermi level does not vanish. Indeed, calculated band structures of the DW states are metallic. The changes in DOS (Fig. 3d) between the undistorted and DW structures span the energy range of about 0.1 eV in good agreement with ΔDW determined from optics (see also Fig. 1c).
One peculiar aspect of this density-wave state is its persistent metallic nature. No bandgap opens below TDW, although the DOS at the Fermi level is reduced. This illustrates the multiband nature of KV3Sb5 (Fig. 1e). The vanadium kagome bands with the saddle points around M are prone to the formation of the DW state, whereas other bands crossing the Fermi level in the vicinity of K and Γ are dominated by Sb states and thus less affected by the transition.
Optical conductivity allows a further verification of the DW structures. Experimentally, we observe that the peak due to interband absorption shifts from 0.1 eV above TDW to about 0.15 eV below TDW. Such a blue shift is well reproduced by DFT calculations for the DW states, as demonstrated in Fig. 3e. Therefore, both hexagon and star structures are likely candidates for the DW state of KV3Sb5. The main changes in the electronic structure below TDW are captured by the atomic displacements in the DW states.
An important observation at this juncture is that both hexagon and star structures are also close analogs of the DW states predicted theoretically for the Hubbard model at the filling level of the van Hove singularity18. These states are shown in Fig. 3 and classified as charge bond order and spin bond order or, respectively, as singlet and triplet p-wave states according to the symmetry of their order parameter19. The star structure is immediately recognized as charge bond order, whereas the hexagon structure may be tentatively associated with spin bond order, albeit with one caveat. Spin bond order is not a conventional spin-density wave where spatial modulation of spin density creates local magnetic moments on individual atoms. Instead, it entails a spatial modulation of spin current that can not be captured by DFT. The hexagon structure itself, as obtained by the DFT atomic relaxation, is of charge bond order type, too, and can be understood as an inverse star-of-David, because the short and long V–V bonds are merely inverted compared to the star structure. However, the resemblance of this structure to the spin bond order is far from accidental. The hexagon structure does not support spin-density waves and appears to be immune to conventional magnetism, whereas the star structure readily turns into a spin-density wave when magnetism is introduced on the DFT+U level (see Supplementary Note 6 for details).
The analogy to the p-wave states of the kagome Hubbard model is reinforced by similar filling factors. The p-wave instabilities appear in the model near its van Hove singularity18, whereas real band structure of KV3Sb5 lies close to this regime and shows a peak in the density-of-states around the Fermi level. On the other hand, atomic reconstruction in the DW state should involve phonons, in contrast to the Hubbard model where only electronic instabilities are at play. This dichotomy—competing roles of Fermi surface nesting and phonons—has sparked vivid debates for many of the density-wave states reported earlier.
In the 1D case, electronic instabilities and phonons come hand in hand31. Extending this picture to higher dimensions is usually based on the assumption that Fermi surface nesting occurs at quasi-1D portion of the Fermi surface, and the resulting electronic reconstruction is sufficient to drive a metal-insulator transition or cause drastic changes in carrier scattering. However, other effects may be significant too. For instance, strong electron–phonon coupling was discussed as the source of the charge-density wave observed in metallic 2D dichalcogenides, 2H-NbSe222,32, where strong momentum-dependent electron–phonon coupling can cause it even in the absence of nesting. The situation becomes even more complex in high-temperature cuprate superconductors, where neither weak Fermi surface nesting near the antinodal region33 nor the strong electron–phonon coupling23 were found sufficient to explain the density-wave instability. Strong electronic correlations were then proposed as another crucial ingredient34.
The aforementioned effects may also contribute to the DW formation in KV3Sb5. Their relative importance depends on the balance between electronic correlations and electron–phonon coupling. In the following, we discuss possible experimental fingerprints of these two major ingredients using optical data for KV3Sb5.
Phonon anomalies
Experimental optical spectra reveal two narrow peaks at 188 cm−1 and 482 cm−1, where the latter one is a pronounced antiresonance. Calculated frequencies of Γ-point phonons allow the assignment of the lower mode to the IR-active in-plane E1u phonon (black line in Fig. 4a). In contrast, the upper mode could not be reproduced, because the highest optical phonon is at 299 cm−1. On the other hand, DFT calculations reveal two IR-active modes at 237 cm−1 (E1u) and 241 cm−1 (A2u), fairly close to each other (red line in Fig. 4a). This makes the high-energy mode observable either as a combination of these modes or as an overtone of one, possibly the 241 cm−1 mode, as the resonance frequency is exactly doubled. The overtone/combination modes typically appear via anharmonic effects that may simultaneously reduce the intensity of the fundamental modes and even make them invisible35. Below, we also show that the fundamental mode at 188 cm−1 is unusually broad by virtue of its coupling to the electronic background. A similar broadening of the fundamental modes around 240 cm−1 would make them invisible in our spectrum. Alternatively, the 482 cm−1 mode could be of plasmonic origin36 or a non-Γ phonon that appears in the IR spectrum due to phonon–plasmon interactions37. The latter interpretation seems less likely, though, because recent phonon calculations38 put the upper boundary of single-phonon excitations around 300 cm−1. On the other hand, harmonic approximation assumed in this calculation remains to be verified experimentally, especially in the light of the abnormally high atomic displacement parameter of K atoms10 that potentially indicates strong anharmonic effects in KV3Sb5.

a The calculated IR-active modes are given for all structures. Vertical lines represent the phonon modes relevant to the experimental observations. The black line shows the low-energy phonon mode around 188 cm−1. The red line indicates the phonon modes that can create the overtone/combination high-energy mode around 482 cm−1 in the IR spectra. b Fits to the 188 cm−1 E1u mode for the selected temperatures. The spectra are shifted by 2000 cm−1 for clarity. c Fits to the 482 cm−1 mode for the selected temperatures. The spectra are represented as Δσ1(ω) with all other contributions subtracted and the baselines shifted by 2500 cm−1 for clarity. d–f Fit parameters obtained from the curves in (b). The red line in (f) is the expected broadening behavior due to the electron–phonon coupling39. g–i Fit parameters obtained from the curves in (c). The solid lines mark TDW.
To analyze the aforementioned modes, we fit temperature-dependent spectra, as described in Supplementary Note 3. Both modes show striking signatures of the electron–phonon coupling. For the lower 188 cm−1 mode, strong anomalies are observed across the transition. The resonance frequency (ω0), intensity (Δϵ), and damping (linewidth, γ) obtained from the Lorentzian fit are given in Fig. 4d–f. Below TDW, a strong softening is accompanied by a significant increase in the intensity. Furthermore, the line is very broad and by far exceeds the resolution of our measurement (1 cm−1).
Below TDW, this broadening may be explained by the presence of multiple modes that are expected in the DW states based on our phonon calculations (Fig. 4a). On the other hand, the undistorted structure shows only one mode between 100 and 200 cm−1. Therefore, the broadening above TDW must be caused by the coupling to the electronic background. Moreover, the line gets broader upon cooling, while the sharpening is usually observed when thermal fluctuations abate. Such an unexpected broadening of the phonon modes upon cooling has been discussed in terms of an electron–phonon coupling scenario39,40, for instance in the case of graphene. Here, we used the same formalism and fitted temperature dependence of the linewidth (Fig. 4f) using
Here, ℏω0 = 174 cm−1 is the calculated E1u phonon energy, kB is the Boltzmann constant, and f(x) = 1/[ex + 1]. The intrinsic linewidth of γe−ph(0) = 52 cm−1 signals the crucial role of electron–phonon coupling and makes it a plausible reason for the DW instability in KV3Sb523.
The high-energy mode displays a strong Fano-like behavior, indicating that it couples to the electronic background, too. The fitting parameters for this mode are given in Fig. 4g–i. While the resonance frequency is nearly temperature-independent, the intensity (Δϵ) increases toward TDW and decreases below the transition. On the other hand, the broadening (γ) increases continuously upon cooling. The coupling (asymmetry) constant q suggests the stronger Fano-character at higher temperatures. On approaching TDW, the mode becomes more symmetric, while below TDW it can be represented by a completely symmetric antiresonance. Below, we argue that the peculiar evolution of this mode is strongly intertwined with changes in the localization peak that we discuss in the following.
Localization peak and electronic correlations
Another distinct feature of KV3Sb5 is the presence of a localization peak in the low-energy part of the spectrum. This peak, sometimes understood as a displaced Drude peak, is identified by its redshift upon cooling and signals localization of charge carriers resulting in sub-diffusive transport and major deviations of the intraband optical conductivity from the simple Drude model41,42,43,44. The redshift is accompanied by a sudden reduction in the spectral weight across TDW (Fig. 2g). While the total spectral weight is conserved according to the sum rules (Fig. 2h), the weight of the localization peak drops at TDW, suggesting a redistribution of the intensity upon the DW formation.
The evolution of the localization peak gives one plausible explanation for the behavior of the 482 cm−1 mode as a function of temperature. The strong asymmetry of this mode is probably caused by the coupling to the localization peak located around the same frequency. At lower temperatures, the redshift of the localization peak reduces the coupling, and the 482 cm−1 mode becomes more symmetric. Its intensity decreases (Fig. 4h) because a fraction of the localized carriers is eliminated by the density-wave formation below TDW.
Different microscopic scenarios of the localization peak were discussed in the literature. Whereas mere disorder in the hopping paths42 is excluded by the high quality of our crystals, the electron–phonon45 and electron–electron41 interactions are both likely candidates. Interestingly, even at low temperatures the localization peak does not shift to zero frequency and does not merge with the Drude peak (see the inset of Fig. 2f). This indicates that the slowing down of electron dynamics is caused not only by thermal effects, such as phonons, but also by interactions between the electrons.
Further evidence for such electronic correlations is found upon comparing the experimental optical response of KV3Sb5 with DFT. Experimental optical conductivity could be reproduced, but only with renormalized band energies obtained via shifting the Fermi level by + 64 meV. Moreover, the calculated plasma frequency of 3.48 eV is far above the experimental unscreened one of 2.12 eV. This discrepancy gives rise to a significantly overestimated Drude spectral weight, \({{{{\rm{SW}}}}}_{\exp }/{{{{\rm{SW}}}}}_{{{{\rm{DFT}}}}}={\omega }_{{{{\rm{p,exp}}}}}^{2}/{\omega }_{{{{\rm{p,DFT}}}}}^{2}=0.37\) (see Supplementary Note 5 for the plasma frequency, ωp, estimation), which is a hallmark of electronic correlations in topological semimetals according to ref. 46. Changes in the sample stoichiometry, such as potassium deficiency, can not be responsible for this effect because they lead to a downward shift of the Fermi level, whereas the upward shift is required to reproduce the experimental optical response.
Discussion
The 78 K anomaly was known since the discovery of KV3Sb5, but its nature has remained enigmatic. Our broadband optical spectroscopy experiments witness a strong renormalization of the DOS across TDW and set firm grounds for interpreting this anomaly as a charge-density-wave order. Incidentally, a prominent localization peak in the low-energy optical absorption both above and below TDW signals electronic correlations that coexist with strong phonon anomalies. These findings render electron–phonon and electron–electron interactions two important ingredients of the KV3Sb5 physics. They also suggest that states distinct from a simple charge order may be relevant to the density wave in KV3Sb5. Indeed, candidate states, which we identified for the first time, bear a strong resemblance to the p-wave states of the kagome Hubbard model and allow the interpretation of KV3Sb5 as a correlated kagome metal.
Theoretical results available for the kagome Hubbard model at the van Hove singularity18,21 and in other regimes47,48,49,50,51,52 offer several interesting insights into the KV3Sb5 physics including the large extrinsic anomalous Hall effect15 in KV3Sb5, as well as chiral anisotropy detected in the recent tunneling spectroscopy experiment17. Spin bond order (triplet p-wave) includes spin degrees of freedom in the form of spin currents that can not be detected via conventional probes, such as muon spectroscopy or neutron scattering19, but may have an influence on charge transport, including anomalous Hall effect. Our tri-hexagon structure is the most stable type of a density wave in KV3Sb5 and displays an intriguing similarity to this theoretically predicted spin bond order. On the other hand, doping the kagome Hubbard model away from the van Hove singularity leads to superconducting instabilities20,21,53 that allow further interesting, yet to be explored connections to the recently established superconducting properties of KV3Sb5 and related materials11,12,13,27,38,54,55,56,57.
Note added: After the initial submission of this manuscript, we studied the optical response of the isostructural compound CsV3Sb558. Several important differences from KV3Sb5 are worth noting: (i) different positions of the band saddle points at M lead to a strong change in the interband absorption at low energies in CsV3Sb5 compared to KV3Sb5; (ii) in CsV3Sb5, this interband absorption is perfectly reproduced by DFT without any renormalization of band energies; (iii) at low temperatures, the localization peak in CsV3Sb5 shifts to zero energy and merges with the Drude peak, unlike in KV3Sb5 where the peak position saturates at finite energies, as shown in the inset of Fig. 2f. This indicates that the slowing down of electron dynamics is thermally activated in CsV3Sb5, possibly due to interactions with phonons, while in KV3Sb5 it is intrinsic to electrons and, therefore, persists even at low temperatures. All these observations reinforce our conclusion that electron–electron interactions must play an important role in KV3Sb5, and that KV3Sb5 is significantly different from its Cs sibling. These differences may also be responsible for the complete screening of phonons in CsV3Sb558, as opposed to KV3Sb5.
Methods
Sample characterization
High-quality single crystals were prepared as described elsewhere10,13. Freshly cleaved samples with the dimensions ~ 2 × 3 × 0.2 mm3 were used for optical measurements. In-plane component of the optical conductivity (σxx) was probed in all measurements. dc resistivity was measured with the standard four-point contact method to control the amount of potassium on the exact same piece as used in optical experiments. Magnetic anomaly has also been confirmed via magnetic susceptibility measurements. Here, a field of 1 T has been applied along H ∥ c, and the susceptibility has been measured in field-cooled (FC) configuration with a magnetic properties measurement system (MPMS) from Quantum Design. Comparison to the earlier literature suggests that our sample of KV3Sb5 is nearly stoichiometric, with the possible K deficiency level of well below 8% reported in ref. 10 (see Supplementary Note 1). The optical spectra were well reproducible across measurements in different frequency ranges and on two different crystals, suggesting that no changes in the K stoichiometry could occur during the measurement and that all spectral features revealed by our study are intrinsic to KV3Sb5.
Optical measurements
Broadband reflectivity measurements were performed with two Bruker Fourier Transform Infrared (FTIR) spectrometers. For the high-energy range (0.075−2.25 eV/600−18,000 cm−1), an infrared microscope coupled to a VERTEX80v FTIR spectrometer is utilized, where the infrared light is focused to 200 μm2. Freshly evaporated gold mirrors are used for the reference. For the low-energy measurements (0.01−0.1 eV/70−700 cm−1), an IFS 113v spectrometer coupled with a custom-made cryostat is used. Absolute reflectivity of the sample has been obtained with the gold-overcoating technique59.
Optical conductivity is calculated via Kramers–Kronig (KK) analysis from the measured reflectivity. For the KK analysis, data are extrapolated using Hagen–Rubens relations to the low-energy range, while X-ray scattering functions have been utilized for the high-energy extrapolations60.
Computational details
Density-functional (DFT) band-structure calculations were performed in the Wien2K61,62, FPLO63, and VASP64,65 codes with several cross-checks that ensured the robust nature of our computational results. In all cases, the Perdew–Burke–Ernzerhof flavor of the exchange-correlation potential66 was used, and the k-mesh with L × L × L/2 points adapted to the anisotropic nature of the KV3Sb5 structure was employed (see Supplementary Note 6 for details on the choice of the k-mesh and convergence). Experimental structural parameters from ref. 10 were chosen for the undistorted KV3Sb5 structure, with the Sb1 atom located at \((\frac{2}{3},\frac{1}{3},0.7539)\). Note that zSb1 = 0.7539(1) is the correct value for the powder refinement of the stoichiometric KV3Sb5 sample in ref. 10. The same value is obtained by extrapolating the results of single-crystal refinements of the K-deficient samples10. Band dispersions were independently calculated in Wien2K and FPLO upon converging fully self-consistent calculations on the k-mesh with L = 36. Wien2K was then used to compute optical conductivities with L = 72.
Crystal structures of possible DW states were relaxed in VASP and FPLO in the 2a × 2a × c supercell until residual forces were below 0.002 eV/Å. To facilitate comparison with the optical data and kagome Hubbard model, we focus on the charge order in the ab plane and disregard possible modulation along the c-axis17. Given the doubled a and c parameters, these calculations were performed on the k-mesh with L × L × L points and L = 16. Consequently, optical conductivity was calculated for the fully relaxed structures in Wien2K with L = 24. In addition, phonon calculations were performed in VASP using the built-in procedure with frozen atomic displacements of 0.015 Å.
All calculations were performed with spin–orbit coupling (SOC) included. Whereas the SOC has a minor influence on lattice energies and phonons, it affects band structure and optical transitions in the vicinity of the Fermi level.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request.
References
Broholm, C. et al. Quantum spin liquids. Science 367, eaay0668 (2020).
Lin, Z. et al. Flatbands and emergent ferromagnetic ordering in Fe3Sn2 kagome lattices. Phys. Rev. Lett. 128, 096401 (2018).
Ye, L. et al. Massive Dirac fermions in a ferromagnetic kagome metal. Nature 555, 638–642 (2018).
Liu, D. F. et al. Magnetic Weyl semimetal phase in a kagomé crystal. Science 365, 1282–1285 (2019).
Yin, J.-X. et al. Negative flat band magnetism in a spin-orbit-coupled correlated kagome magnet. Nat. Phys. 15, 443–448 (2019).
Kang, M. et al. Dirac fermions and flat bands in the ideal kagome metal FeSn. Nat. Mater. 19, 163–169 (2020).
Ye, L. et al. de Haas-van Alphen effect of correlated Dirac states in kagome metal Fe3Sn2. Nat. Commun. 10, 4870 (2019).
Guguchia, Z. et al. Tunable anomalous Hall conductivity through volume-wise magnetic competition in a topological kagome magnet. Nat. Comm. 11, 559 (2020).
Biswas, A. et al. Spin-reorientation-induced band gap in Fe3Sn2: optical signatures of Weyl nodes. Phys. Rev. Lett. 125, 076403 (2020).
Ortiz, B. R. et al. New kagome prototype materials: discovery of KV3Sb5, RbV3Sb5, and CsV3Sb5. Phys. Rev. Mater. 3, 094407 (2019).
Yin, Q. et al. Superconductivity and normal-state properties of kagome metal RbV3Sb5 single crystals. Chin. Phys. Lett. 38, 037403 (2021).
Ortiz, B. R. et al. sV3Sb5: A Z2 topological kagome metal with a superconducting ground state. Phys. Rev. Lett. 125, 247002 (2020).
Ortiz, B. R. et al. Superconductivity in the Z2 kagome metal KV3Sb5. Phys. Rev. Mater. 5, 034801 (2021).
Kenney, E. M., Ortiz, B. R., Wang, C., Wilson, S. D. & Graf, M. J. Absence of local moments in the kagome metal KV3Sb5 as determined by muon spin spectroscopy. J. Phys. Condens. Matter 33, 235801 (2021).
Yang, S.-Y. et al. Giant, unconventional anomalous Hall effect in the metallic frustrated magnet candidate, KV3Sb5. Sci. Adv. 6, eabb6003 (2020).
Yu, F. H. et al. Concurrence of anomalous Hall effect and charge density wave in a superconducting topological kagome metal. Phys. Rev. B 104, 1041103 (2021).
Jiang, Y.-X. et al. Unconventional chiral charge order in kagome superconductor KV3Sb5. Nat. Mater. 20, 1353–1357 (2021).
Kiesel, M. L., Platt, C. & Thomale, R. Unconventional fermi surface instabilities in the kagome Hubbard model. Phys. Rev. Lett. 110, 126405 (2013).
Nayak, C. Density-wave states of nonzero angular momentum. Phys. Rev. B 62, 4880 (2000).
Kiesel, M. L. & Thomale, R. Sublattice interference in the kagome Hubbard model. Phys. Rev. B 86, 121105(R) (2012).
Wang, W.-S., Li, Z.-Z., Xiang, Y.-Y. & Wang, Q.-H. Competing electronic orders on kagome lattices at van Hove filling. Phys. Rev. B 87, 115135 (2013).
Johannes, M. D. & Mazin, I. I. Fermi surface nesting and the origin of charge density waves in metals. Phys. Rev. B 77, 165135 (2008).
Zhu, X., Cao, Y., Zhang, J., Plummer, E. W. & Guo, J. Classification of charge density waves based on their nature. Proc. Natl Acad. Sci. USA 112, 2367–2371 (2015).
Gusynin, V. P., Sharapov, S. G. & Carbotte, J. P. Unusual microwave response of Dirac quasiparticles in graphene. Phys. Rev. Lett. 96, 256802 (2006).
Kuzmenko, A. B., van Heumen, E., Carbone, F. & van der Marel, D. Universal optical conductance of graphite. Phys. Rev. Lett. 100, 117401 (2008).
Basov, D. N., Averitt, R. D., van der Marel, D., Dressel, M. & Haule, K. Electrodynamics of correlated electron materials. Rev. Mod. Phys. 83, 471–541 (2011).
Zhao, C. C. et al. Nodal superconductivity and superconducting dome in the topological kagome metal CsV3Sb5. Preprint at https://arxiv.org/abs/2102.08356 (2021).
Wilson, J. A., Di Salvo, F. J. & Mahajan, S. Charge-density waves and superlattices in the metallic layered transition metal dichalcogenides. Adv. Phys. 24, 117–201 (1975).
Fazekas, E. & Tosatti, E. Electrical, structural and magnetic properties of pure and doped 1T-TaS2. Philos. Mag. B 39, 229–244 (1979).
Wang, Y. D. et al. Band insulator to Mott insulator transition in 1T-TaS2. Nat. Commun. 11, 4215 (2020).
Monceau, P. Electronic crystals: an experimental overview. Adv. Phys. 61, 325–581 (2012).
Calandra, M., Mazin, I. I. & Mauri, F. Effect of dimensionality on the charge-density wave in few-layer 2H-NbSe2. Phys. Rev. B 80, 241108 (2009).
Comin, R. et al. Charge order driven by Fermi-arc instability in Bi2Sr2−xLaxCuO6+δ. Science 343, 390–392 (2014).
Fujita, K. et al. Direct phase-sensitive identification of a d-form factor density wave in underdoped cuprates. Proc. Natl Acad. Sci. USA 111, E3026–E3032 (2014).
Du, L., Mackeprang, K. & Kjaergaard, H. G. Fundamental and overtone vibrational spectroscopy, enthalpy of hydrogen bond formation and equilibrium constant determination of the methanol-dimethylamine complex. Phys. Chem. Chem. Phys. 15, 10194–10206 (2013).
Grigorenko, A. N., Polini, M. & Novoselov, K. S. Graphene plasmonics. Nat. Photonics 6, 749–758 (2012).
Lapointe, F. et al. Antiresonances in the mid-infrared vibrational spectrum of functionalized graphene. J. Phys. Chem. C. 121, 9053–9062 (2017).
Tan, H., Liu, Y., Wang, Z. & Binghai, Y. Charge density waves and electronic properties of superconducting kagome metals. Phys. Rev. Lett. 127, 046401 (2021).
Bonini, N., Lazzeri, M., Marzari, N. & Mauri, F. Phonon anharmonicities in graphite and graphene. Phys. Rev. Lett. 99, 176802 (2007).
Lazzeri, M., Piscanec, S., Mauri, F., Ferrari, A. C. & Robertson, J. Phonon linewidths and electron-phonon coupling in graphite and nanotubes. Phys. Rev. B 73, 155426 (2006).
Luca, V. D., Goutéaux, B., Hartnoll, S. A. & Karlsson, A. Bad metals from fluctuating density waves. SciPost Phys. 3, 025 (2017).
Fratini, S., Ciuchi, S. & Mayou, D. Phenomenological model for charge dynamics and optical response of disordered systems: application to organic semiconductors. Phys. Rev. B 89, 235201 (2014).
Fratini, S. & Ciuchi, S. Dynamical localization corrections to band transport. Phys. Rev. Res. 2, 013001 (2020).
Uykur, E., Tanaka, K., Masui, T., Miyasaka, S. & Tajima, S. In-plane optical spectra of Y1−xCaxBa2Cu3O7−δ: overdoping and disorder effects on residual conductivity. Phys. Rev. B 84, 184527 (2011).
Atta-Fynn, R., Biswas, P. & Drabold, D. A. Electron–phonon coupling is large for localized states. Phys. Rev. B 69, 245204 (2004).
Shao, Y. et al. Electronic correlations in nodal-line semimetals. Nat. Phys. 16, 636–641 (2020).
Ohashi, T., Kawakami, N. & Hirokazu, T. Mott transition in kagomé lattice Hubbard model. Phys. Rev. Lett. 97, 066401 (2006).
Guertler, S. & Monien, H. Unveiling the physics of the doped phase of the t − J model on the kagome lattice. Phys. Rev. Lett. 111, 097204 (2013).
Kudo, K., Yoshida, T. & Yasuhiro, H. Higher-order topological Mott insulators. Phys. Rev. Lett. 123, 196402 (2019).
im, H. S., Mishra, A. & Lee, S. Emergent chiral spin ordering and anomalous Hall effect in a kagome lattice at a \(\frac{1}{3}\) filling. Phys. Rev. B 102, 155113 (2020).
Di Sante, D. et al. Turbulent hydrodynamics in strongly correlated kagome metals. Nat. Commun. 11, 3997 (2020).
Kaufmann, J., Steiner, K., Scalettar, R. T., Held, K. & Janson, O. How correlations change the magnetic structure factor of the kagome Hubbard model. Phys. Rev. B 104, 165127 (2021).
Yu, S.-L. & Li, J.-X. Chiral superconducting phase and chiral spin-density-wave phase in a Hubbard model on the kagome lattice. Phys. Rev. B 85, 144402 (2012).
Wang, Y. et al. Proximity-induced spin-triplet superconductivity and edge supercurrent in the topological kagome metal, K1−xV3Sb5. Preprint at https://arxiv.org/abs/2012.05898 (2020).
Du, F. et al. Pressure-induced double superconducting domes and charge instability in the kagome metal KV3Sb5. Phys. Rev. B 103, 1220504 (2021).
Chen, K. Y. et al. Double superconducting dome and triple enhancement of Tc in the kagome superconductor CsV3Sb5 under high pressure. Phys. Rev. Lett. 126, 247001 (2021).
Liang, Z. et al. Three-dimensional charge density wave and surface-dependent vortex-core states in a kagome superconductor CsV3Sb5. Phys. Rev. X 11, 031026 (2021).
Uykur, E. et al. Low-energy optical properties of the nonmagnetic kagome metal CsV3Sb5. Phys. Rev. B 104, 045130 (2021).
Homes, C. C., Reedyk, M., Cradles, D. A. & Timusk, T. Technique for measuring the reflectance of irregular, submillimeter-sized samples. Appl. Opt. 32, 2976–2983 (1993).
Tanner, D. B. Use of x-ray scattering functions in Kramers-Kronig analysis of reflectance. Phys. Rev. B 91, 035123 (2015).
Blaha, P. et al. WIEN2k, An Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties (Karlheinz Schwarz, Techn. Universität Wien, 2018).
Blaha, P. et al. WIEN2k: an APW+lo program for calculating the properties of solids. J. Chem. Phys. 152, 074101 (2020).
Koepernik, K. & Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 59, 1743 (1999).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Acknowledgements
The authors acknowledge the fruitful discussions with Artem Pronin and Sascha Polatkan and technical support by Gabriele Untereiner. We also thank Berina Klis for the dc resistivity measurements. S.D.W. and B.R.O. gratefully acknowledge support via the UC Santa Barbara NSF Quantum Foundry funded via the Q-AMASE-i program under award DMR-1906325. B.R.O. also acknowledges support from the California NanoSystems Institute through the Elings fellowship program. The work has been supported by the Deutsche Forschungsgemeinschaft (DFG) via DR228/51-1 and UY62/2-1. E.U. acknowledges the European Social Fund and the Baden-Württemberg Stiftung for the financial support of this research project by the Elite programme. A.T. was supported by the Federal Ministry for Education and Research via the Sofja Kovalevskaya Award of Alexander von Humboldt Foundation.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
E.U. performed the experiments. A.A.T. performed the DFT calculations. Samples were grown by B.R.O. and S.D.W. The manuscript was written by A.A.T., E.U., and M.D. with suggestions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Uykur, E., Ortiz, B.R., Wilson, S.D. et al. Optical detection of the density-wave instability in the kagome metal KV3Sb5. npj Quantum Mater. 7, 16 (2022). https://doi.org/10.1038/s41535-021-00420-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41535-021-00420-8
This article is cited by
-
Two-dimensional phase diagram of the charge density wave in doped CsV3Sb5
npj Quantum Materials (2024)
-
Coherent phonon and unconventional carriers in the magnetic kagome metal Fe3Sn2
npj Quantum Materials (2024)
-
Anharmonic strong-coupling effects at the origin of the charge density wave in CsV3Sb5
Nature Communications (2024)
-
Interaction of in-plane Drude carrier with c-axis phonon in PdCoO2
npj Quantum Materials (2023)
-
Observation of anomalous Hall resonance of massive Dirac fermions in topological kagome-lattice magnet
npj Quantum Materials (2023)
