
Abstract
This study proposes a nonvariational scheme for geometry optimization of molecules for the first-quantized eigensolver, which is a recently proposed framework for quantum chemistry using probabilistic imaginary-time evolution (PITE). In this scheme, the nuclei in a molecule are treated as classical point charges while the electrons are treated as quantum mechanical particles. The electronic states and candidate geometries are encoded as a superposition of many-qubit states, for which a histogram created from repeated measurements gives the global minimum of the energy surface. We demonstrate that the circuit depth per step scales as \({{{\mathcal{O}}}}({n}_{{\rm {e}}}^{2}{{{\rm{poly}}}}(\log {n}_{{\rm {e}}}))\) for the electron number ne, which can be reduced to \({{{\mathcal{O}}}}({n}_{{\rm {e}}}{{{\rm{poly}}}}(\log {n}_{{\rm {e}}}))\) if extra \({{{\mathcal{O}}}}({n}_{{\rm {e}}}\log {n}_{{\rm {e}}})\) qubits are available. Moreover, resource estimation implies that the total computational time of our scheme starting from a good initial guess may exhibit overall quantum advantage in molecule size and candidate number. The proposed scheme is corroborated using numerical simulations. Additionally, a scheme adapted to variational calculations is examined that prioritizes saving circuit depths for noisy intermediate-scale quantum (NISQ) devices. A classical system composed only of charged particles is considered as a special case of the scheme. The new efficient scheme will assist in achieving scalability in practical quantum chemistry on quantum computers.
Similar content being viewed by others
Introduction
Modern computational designs for materials1, proteins2, and drug discovery3 often include atomistic simulations instead of coarse-grained models for distinguishing microscopic subtleties. Electronic-structure calculations based on the density functional theory4,5 or wave function theory6 must be performed to optimize the geometries of solids and molecules in their ground states to ensure that simulations are as quantitatively reliable as possible. Although target systems with a diverse number of atoms and elements are found in physics, chemistry, and biochemistry, there are two main approaches for determining the optimal geometry of a molecule using a classical computer: energy- and force-based.
The energy-based approach is based on the calculated total energies of all the candidate geometries. The procedure in a naive form typically begins by determining the discretization of the positions for each nucleus and calculating the total energies of all possible geometries. This approach leads to an exhaustive search for the optimal geometry among all candidates and the search can be easily parallelized for many classical computers. However, the required computational resources grow exponentially with respect to the size of the target molecule. This extensive scaling makes the naive energy-based approach impractical for systems of practical interest.
The force-based approach is based on the forces acting on the nuclei within the Born–Oppenheimer (BO) approximation. This optimization procedure for a target molecule is performed by calculating the total energy and forces acting on the constituent nuclei. More precisely, the procedure typically calculates the Hellmann–Feynman forces7. If necessary, the Pulay forces are calculated to compensate for the incompleteness of the adopted basis set8. These forces can be calculated using only a small amount of additional computational resources for the total energy calculation. The nuclear positions are iteratively updated until convergence according to the forces. The steepest-descent and conjugate-gradient methods are force-based approaches in the simplest forms. However, the updating process used in these methods is not parallelizable in principle. In addition, the search is prone to becoming stuck in a local minimum on the energy surface. Various elaborate force-based approaches have been proposed to achieve the efficient and robust optimization of molecular geometries. For details, refer to ref. 9.
While quantum computation has been regarded as a promising alternative for storing many-electron wave functions living in a huge Hilbert space10 since long before the advent of quantum computers, we find that geometry optimization of electronic systems is still going through the phase of establishing basic techniques, on the contrary to classical computation. Hirai et al.11 proposed recently a method within the first-quantized formalism12,13,14 by finding the lowest-energy geometry based on the imaginary-time evolution (ITE) with variational parameters15,16,17 for nonadiabatically coupled electrons and nuclei. Their approach, which we refer to as the variational ITE (VITE) in what follows, is a kind of variational quantum eigensolver (VQE)18,19. The major difference between our approach described later and their approach exists in how the qubits for nuclear degrees of freedom are used: we use them to encode the nuclear positions as classical data instead of their femtometer-scale wave functions so that we perform an exhaustive search for the optimum among candidates via quantum parallelism. We point out here that a quantum algorithm for force-based geometry optimization has been proposed20.
Since the prevalent paradigm of electronic-structure calculations on classical computers has been developed primarily for computing the total energies of systems built up of electrons and nuclei, we might overlook the important fact, that is, there is no need for knowing the values of the total energies of candidate geometries to find the optimal one. We can find it only by knowing which geometry has the unknown lowest energy. Given this fact and the first-quantized eigensolver (FQE)21, this study presents a quantum algorithm for efficient geometry optimization that outperforms classical algorithms. FQE is a recently proposed framework based on probabilistic ITE (PITE) for nonvariational energy minimization in quantum chemistry21. For a brief review of generic PITE, see Supplementary Note 1. The second-quantized formalism is useful for calculating the dynamical properties related to the excitation processes of a molecule, where the electron number can increase and decrease22,23. However, the first-quantized formalism for finding the ground state offers better scaling of operation numbers21. This characteristic is inherited even when geometry optimization is involved, as will be demonstrated later.
Results
Exhaustive search for optimal geometries
Let us consider a molecular system consisting of ne electrons as quantum mechanical particles and nnucl nuclei as classical point charges fixed at Rν(ν = 0, …, nnucl−1), as depicted in Fig. 1. These two kinds of particles interact with each other via pairwise interactions v dependent only on the distance between two particles. The Hamiltonian is given by
where the nuclear positions appear as parameters. \(\hat{T}\) is the kinetic-energy operator of electrons having the mass me = 1. All the quantities in this paper are in atomic units unless otherwise stated. \({\hat{{{{\boldsymbol{r}}}}}}_{\ell }\) and \({\hat{{{{\boldsymbol{p}}}}}}_{\ell }\) are the position and momentum operators, respectively, of the ℓth electron. Zν is the charge of the νth nucleus, while that of an electron is −1. We can introduce a position-dependent external field vext felt by each electron. Although we have adopted the common interaction v for \({\hat{V}}_{{{{\rm{ee}}}}},{\hat{V}}_{{{{\rm{en}}}}}\), and \({\hat{V}}_{{{{\rm{nn}}}}}\) for simplicity, distinct interactions for them could be introduced with only small modifications to the following discussion. Also, the formulations for one- and two-dimensional spaces will be possible similar to the three-dimensional case.

We treat the ne electrons contained in a target molecule as quantum mechanical particles having the kinetic energies \(\hat{T}\), while the nnucl nuclei as fixed classical point charges. The Hamiltonian of the total system involves the electron–electron interactions \({\hat{V}}_{\mathrm {{ee}}}\), the electron–nucleus interactions \({\hat{V}}_{{{{\rm{en}}}}}\), and the nucleus–nucleus interactions Enn. The electrons can feel an external field \({\hat{V}}_{{{{\rm{ext}}}}}\).
We encode the ne-electron wave function in real space by using nqe qubits for each direction per electron, as usual in the first-quantized formalism12,13,14,21,24,25, or equivalently the grid-based formalism. We refer to the 3nenqe qubits collectively as the electronic register. We generate uniform grid points in a cubic simulation cell of size L to encode the normalized many-electron spatial wave function ψ by using the register as
where kℓ is the three integers specifying the position eigenvalue (kℓxex + kℓyey + kℓzez)Δx for the ℓth electron. Δx ≡ L/Nqe is the spacing of \({N}_{\mathrm {{qe}}}\equiv {2}^{{n}_{\mathrm {{qe}}}}\) grid points for each direction. We introduced the volume element ΔV ≡ Δx3 for the normalization of \(\left\vert \psi \right\rangle .\)
We construct a composite system consisting of the electrons and nuclei and define an appropriate Hamiltonian, for which we perform energy minimization based on PITE to find the optimal combination \({\{\Delta {{{{\boldsymbol{R}}}}}_{\nu }^{({{{\rm{opt}}}})}\}}_{\nu }\) of displacements from the original positions \({\{{{{{\boldsymbol{R}}}}}_{\nu 0}\}}_{\nu }.\) To this end, we first decide upon the largest possible displacement \(\Delta {R}_{\nu \mu \max }\,(\mu =x,y,z)\) in each direction μ for each nucleus ν. We introduce nqn qubits for encoding the displacement in each direction for each nucleus. Specifically, we define the x position operator \({\hat{\boldsymbol{\mathcal{R}}}}_{\nu x}\) of the νth nucleus such that each of the computational basis \({\left\vert {j}_{\nu x}\right\rangle }_{{n}_{{\mathrm {q}}{{{\rm{n}}}}}}\,({j}_{\nu x}=0,\ldots ,{2}^{{n}_{{\mathrm {q}}{{{\rm{n}}}}}}-1)\) is the eigenstate as follows:
where \({N}_{{\rm {q}}{{{\rm{n}}}}}\equiv {2}^{{n}_{{\rm {q}}{{{\rm{n}}}}}}.\) The operators \({\hat{{{{\mathcal{R}}}}}}_{\nu y}\) and \({\hat{{{{\mathcal{R}}}}}}_{\nu z}\) for the y and z positions, respectively, are defined similarly. We refer to the 3nnuclnqn qubits for the nuclear positions as the nuclear register. There exists one-to-one correspondence between the \({N}_{{\mathrm {q}}{{{\rm{n}}}}}^{3{n}_{{{{\rm{nucl}}}}}}\) computational basis vectors and the possible molecular geometries. It is noted that nqn is a parameter that determines the resolution of the search for the optimal geometry and has no direct relation to the physical properties of the nuclei. Also, we emphasize here that we have introduced the nuclear register and the operators \({\{{\hat{\boldsymbol{\mathcal{R}}}}_{\nu}\}}_{\nu}\) not for encoding quantum states of nuclei, but for encoding the data for the nuclei as distinguishable classical particles. Having defined the nuclear position operators, we rewrite the Hamiltonian in Eq. (1) by replacing the nuclear positions as c-numbers with the corresponding operators: \({{{\mathcal{H}}}}({\left\{{{{{\boldsymbol{R}}}}}_{\nu }\right\}}_{\nu })\to {{{\mathcal{H}}}}({\left\{{\hat{\boldsymbol{\mathcal{R}}}}_{\nu }\right\}}_{\nu }),\) leading to the new Hamiltonian for the (3nenqe + 3nnuclnqn)-qubit system. Enn has become an operator \({\hat{V}}_{{{{\rm{nn}}}}}\).
The preparation of an initial state consists of Uguess and Uref gates. Uguess generates the superposition of Ncand possible geometries having nonzero desired weights, as in Fig. 2a. Uref is designed to generate the desired reference electronic state for the indistinguishable electrons26,27 in the specified geometry, as in Fig. 2b. Possible implementation of the initial-state preparation that expedites the convergence of subsequent energy minimization is outlined in Supplementary Note 2. By using these two gates, we construct the circuit \({{{{\mathcal{C}}}}}_{{{{\rm{opt}}}}}\) for the entire optimization procedure within FQE, as shown in Fig. 2c. For details, see Supplementary Note 2. The state of the composite system undergoing this circuit is written in the form
where J is the collective notation of 3nnuclnqn integers specifying one of the candidate geometries. \(\left\vert \psi [{{{\boldsymbol{J}}}}]\right\rangle\) is the normalized trial electronic state for the geometry J, whose weight is wJ. When we perform a measurement on the nuclear register comprising \(\left\vert {{{\Psi }}}_{s}\right\rangle\) of the form in Eq. (4) immediately after the sth step, the probability for observing the molecular geometry corresponding to a specific J is clearly wsJ, which is the weight of geometry contained in \(\left\vert {{{\Psi }}}_{{\mathrm {s}}}\right\rangle .\) The composite state having undergone sufficiently many PITE steps will thus provide the lowest-energy geometry with the highest probability:
from which the optimal displacements \({\{\Delta {{{{\boldsymbol{R}}}}}_{\nu }^{({{{\rm{opt}}}})}\}}_{\nu }\) are calculated from Eq. (3). In practice, J(opt) can be found by drawing a histogram of observed values of J from repeated measurements. Our scheme is also applicable to a geometry optimization problem for point charges as a classical system (see Supplementary Note 2).

a Initial-guess gate Uguess for assigning the weight w0J to each molecular geometry specified by J. b Reference state gate Uref for generating the reference electronic state \(\left\vert {\psi }_{{{{\rm{ref}}}}}[{{{\boldsymbol{J}}}}]\right\rangle\) for the geometry specified by J. c Circuit \({{{{\mathcal{C}}}}}_{{{{\rm{opt}}}}}\) for the entire optimization procedure within FQE. It contains nsteps PITE steps for energy minimization governed by the Hamiltonian \({{{\mathcal{H}}}}\) of the system made up of the electrons and the nuclei. The ancilla qubit is for observing the success or failure state at each PITE step.
Let us consider a plausible case of Ncand candidate geometries for which good reference states are available from sophisticated classical calculations. As considered in Supplementary Note 2, the energy shift technique by Nishi et al.28 leads to the required number of steps for obtaining the optimal state with a tolerance δ estimated to be
where ΔEcand is the energy difference between the optimal and second optimal geometries. Δτ is the amount of each imaginary-time step. For a practical PITE circuit, an upper bound on Δτ needs to be respected in order for the Taylor expansion of the ITE operator to be justified (see Supplementary Note 1).
In the actual optimization procedure for a given molecule, we will be confronted with a dilemma: while a more accurate prediction of the optimal geometry requires finer discretization of nuclear displacements, such discretization inevitably leads to smaller energy differences between “neighboring” candidate geometries, which are more difficult to detect via the finite number of PITE steps. The histogram of observed geometries will thus exhibit a shape formed by multiple maxima, each of which has a finite width around it and corresponds to possibly one of the local minima on the energy surface of the molecule. If we want to predict one of the local-minima geometries more accurately, we should start newly an optimization procedure by restricting the nuclear displacements within the vicinity of the local minimum, only for which the nuclear register is spent.
Kassal et al.14 demonstrated that nonadiabatic treatment of nuclei as quantum mechanical particles in a molecule as well as the electrons, is computationally much more efficient for a chemical-reaction simulation than the BO approximation, except for the smallest molecules. On the other hand, one finds that the classical treatment of nuclei in our approach for geometry optimization is more efficient than the nonadiabatic treatment for the following reasons. If we used the 3nnuclnqn qubits for the nuclei as quantum mechanical particles to encode their wave function, the grid spacing in the simulation cell has to be on the order of femtometer (fm) to detect the finite width of the wave function of each nucleus. The grid spacing Δx for the electronic wave function has also to be on the same order for a reliable simulation, while that may be on the order of Å in our original approach. The required number nqn of qubits for the nonadiabatic treatment is thus larger than that for the classical treatment roughly by \({\log }_{2}\) (Å/fm) ≈ 16.6, which is also the case for nqe. Furthermore, we will then give up the superposition of candidate geometries since the nuclear register has already been reserved for the many-nucleus wave function. Therefore we have to perform the energy minimization starting from some single initial geometry. These considerations indicate that the classical treatment of nuclei is practically more favorable than the nonadiabatic one unless the result of the optimization is affected qualitatively by the nonadiabatic treatment.
Circuit depths
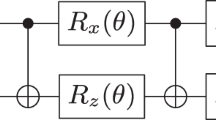
The PITE circuit \({{{{\mathcal{C}}}}}_{{{{\rm{PITE}}}}}\) consists mainly of controlled real-time evolution (RTE) operators.21 We implement the RTE operator \({{\mathrm {e}}}^{-i{{{\mathcal{H}}}}\Delta t}\) for a time step Δt by employing the first-order Suzuki–Trotter as usual to decompose it approximately into the kinetic part \({{\mathrm {e}}}^{-i\hat{T}\Delta t}\) and the position-dependent part \(\exp [-i({\hat{V}}_{{\mathrm {ee}}}+{\hat{V}}_{{\mathrm {e}}{{{\rm{n}}}}}+{\hat{V}}_{{{{\rm{nn}}}}}+{\hat{V}}_{{{{\rm{ext}}}}})\Delta t].\) While the former can be implemented using the quantum Fourier transform (QFT)-based techniques21,29,30 as in the electrons-only cases, the latter is further decomposed exactly into the four parts, as shown in Fig. 3. The evolution \({{\mathrm {e}}}^{-i{\hat{V}}_{\kappa }\Delta t}\,(\kappa ={\mathrm {ee}},{\mathrm {e}}{{{\rm{n}}}},{{{\rm{nn}}}})\) is implemented by applying the pairwise phase gate Uκ(Δt) that acts diagonally as
to every pair of interacting particles. \(\left\vert {{{\boldsymbol{s}}}}\right\rangle\) and \(\left\vert {{{\boldsymbol{s}}}}{\prime} \right\rangle\) are the position eigenstates of the particles with the interaction energy \(v({{{\boldsymbol{s}}}},{{{\boldsymbol{s}}}}{\prime} ).\) On the other hand, \({{\mathrm {e}}}^{-i{\hat{V}}_{{{{\rm{ext}}}}}\Delta t}\) is implemented by applying the phase gate Uext(Δt) that acts diagonally as \({U}_{\kappa }(\Delta t){\left\vert {{{\boldsymbol{k}}}}\right\rangle }_{3{n}_{{\mathrm {qe}}}}=\exp (-i{v}_{{{{\rm{ext}}}}}({{{{\boldsymbol{r}}}}}^{({{{\boldsymbol{k}}}})})\Delta t){\left\vert {{{\boldsymbol{k}}}}\right\rangle }_{3{n}_{{\mathrm {qe}}}}\) to each electron. The details of their implementation and the scaling of circuit depths with respect to the particle numbers are explained in Supplementary Note 3. It is clear from Fig. 3 that the partial circuits for \({e}^{-i{\hat{V}}_{ee}\Delta t}\) and \({{\mathrm {e}}}^{-i{\hat{V}}_{{{{\rm{nn}}}}}\Delta t}\) are deeper than those for \({{\mathrm {e}}}^{-i{\hat{V}}_{{\mathrm {e}}{{{\rm{n}}}}}\Delta t}\) and \({{\mathrm {e}}}^{-i{\hat{V}}_{{{{\rm{ext}}}}}\Delta t}\) from the viewpoint of scaling with respect to ne and nnucl.

The latter is shown in the left part of this figure, which is further decomposed into the evolution operators generated by \({\hat{V}}_{{\mathrm {ee}}},{\hat{V}}_{{\mathrm {e}}{{{\rm{n}}}}},{\hat{V}}_{{{{\rm{nn}}}}},\) and \({\hat{V}}_{{{{\rm{ext}}}}}\) separately, as shown in the right part. The scaling of depths with respect to the numbers of electrons and nuclei is also shown.
While we will be focusing on the first-order Suzuki–Trotter with the fixed Δt below, it is possible instead to employ a generic pth-order product formula with controlling the error ε originating from the noncommutativity between the kinetic and position-dependent parts of the Hamiltonian. Specifically, the depth per PITE step takes on a factor of \({{{\mathcal{O}}}}({\widetilde{\alpha }}_{{{{\rm{comm}}}}}^{1/p}\Delta {t}^{1+1/p}/{\varepsilon }^{1/p}),\) where \({\widetilde{\alpha }}_{{{{\rm{comm}}}}}\) is a function of L and Δx31.
Although our PITE circuit does not assume specific implementation of the pairwise phase gates comprising \({{\mathrm {e}}}^{-i{\hat{V}}_{{\mathrm {ee}}}\Delta t},{{\mathrm {e}}}^{-i{\hat{V}}_{{\mathrm {e}}{{{\rm{n}}}}}\Delta t},\) and \({{\mathrm {e}}}^{-i{\hat{V}}_{{{{\rm{nn}}}}}\Delta t},\) we propose here a plausible alternative by exploiting the fact that the pairwise interaction v is common to these three types of evolution and depends only on the distance between particles. By dividing the task we have to go into the computation of distances between the particles and that of the phases for evolution, we find the systematic construction of the circuits, as explained in Supplementary Note 3. Figure 4 shows the circuit that implements the pairwise e–e phase gate Uee(Δt), defined in Eq. (15) in Supplementary Note 3 as a building block of \({{\mathrm {e}}}^{-i{\hat{V}}_{{\mathrm {ee}}}\Delta t}\) operation. The pairwise phase gates \({U}_{{\mathrm {e}}{{{\rm{n}}}}}^{(\nu )}(\Delta t)\) and \({U}_{{{{\rm{nn}}}}}^{(\nu ,\nu {\prime} )}(\Delta t)\) as building blocks of \({{\mathrm {e}}}^{-i{\hat{V}}_{{\mathrm {e}}{{{\rm{n}}}}}\Delta t}\) and \({{\mathrm {e}}}^{-i{\hat{V}}_{{{{\rm{nn}}}}}\Delta t}\), respectively, can also be implemented similarly. For example, the circuit for computing the distance can be implemented efficiently by combining the addition32,33,34, multiplication35,36,37, and square root38.

\({U}_{{\mathrm {ee}}}^{({{{\rm{d}}}})}\) defined in Eq. (23) in Supplementary Note 3 computes the distance between two electrons at r(k) and \({{{{\boldsymbol{r}}}}}^{({{{\boldsymbol{k}}}}{\prime} )}\), which is then stored into the distance register consisting of \({n}_{\mathrm {{ee}}}^{({{{\rm{d}}}})}\) qubits. The interaction phase gate Uint,ee(Δt) defined in Eq. (26) in Supplementary Note 3 refers to the distance register to generate the phase required for the evolution coming from the electron pair. The inverse of \({U}_{{\mathrm {ee}}}^{({{{\rm{d}}}})}\) performs uncomputation for disentangling the distance register from the electronic register.
If we approximate the functional shape of the interaction v as a simple or a piecewisely defined polynomial, the interaction phase gates Uint,κ(Δt)(κ = ee, en, nn), defined in Eq. (26) in Supplementary Note 3, can be implemented with polynomial depths30,39 in the numbers \({n}_{\kappa }^{({{{\rm{d}}}})}\) of qubits for the distance registers. (See also Supplementary Note 4.) Although \({n}_{\kappa }^{({{{\rm{d}}}})}\) can be set independently of nqe and nqn, it is suitable to set them such that the resolutions induced by the former are on the same order as by the latter: \({n}_{ee}^{({{{\rm{d}}}})}={{{\mathcal{O}}}}({n}_{qe}),{n}_{e{{{\rm{n}}}}}^{({{{\rm{d}}}})}={{{\mathcal{O}}}}(\max ({n}_{qe},{n}_{q{{{\rm{n}}}}})),\) and \({n}_{{{{\rm{nn}}}}}^{({{{\rm{d}}}})}={{{\mathcal{O}}}}({n}_{q{{{\rm{n}}}}}).\) These considerations tell us that the pairwise phase gates \({U}_{ee}(\Delta t),{U}_{e{{{\rm{n}}}}}^{(\nu )}(\Delta t),\) and \({U}_{{{{\rm{nn}}}}}^{(\nu ,\nu {\prime} )}(\Delta t)\) can be implemented with polynomial depths in nqe and nqn.
As discussed in ref. 21, the number of qubits for the electronic wave function with a resolution Δx typically scales as \({n}_{{\mathrm {qe}}}={{{\mathcal{O}}}}(\log ({n}_{{\mathrm {e}}}^{1/3}/\Delta x)).\) On the other hand, that for the nuclear displacements scales as \({n}_{{\mathrm {q}}{{{\rm{n}}}}}={{{\mathcal{O}}}}(\log (\Delta {R}_{\max }/\Delta R))\) for typical values of a resolution ΔR and the maximal displacement \(\Delta {R}_{\max }.\) Recalling the fact that ne is much larger than nnucl despite their common scaling for a generic molecule, we find that \({{\mathrm {e}}}^{-i{\hat{V}}_{{\mathrm {ee}}}\Delta t}\) dominates the scaling of circuit depth of the entire position-dependent evolution when \(\Delta {R}_{\max }\) and ΔR are fixed. In fact, the \({{\mathrm {e}}}^{-i{\hat{V}}_{{{{\rm{nn}}}}}\Delta t}\) circuit does not contribute to the total depth since it and \({{\mathrm {e}}}^{-i{\hat{V}}_{{\mathrm {ee}}}\Delta t}\), which is much deeper than it, can be performed in parallel, as seen in Fig. 3. The scaling coming from the electron–electron interactions, given by Eq. (31) in Supplementary Note 3, is dominant even in the entire RTE circuit:
For details, see Supplementary Note 3. Since the single PITE step contains the controlled RTE operations, its depth exhibits the same scaling: \({{{\rm{depth}}}}({{{{\mathcal{C}}}}}_{{{{\rm{PITE}}}}})={{{\mathcal{O}}}}({{{\rm{depth}}}}({{\mathrm {e}}}^{-i{{{\mathcal{H}}}}\Delta t})).\)
If the same number 3nenqe of extra qubits as in the electronic register are available, the scaling of depth for \({{\mathrm {e}}}^{-i{\hat{V}}_{{\mathrm {ee}}}\Delta t}\) can be reduced. Specifically, \({n}_{\mathrm {{e}}}^{2}\) on the RHS in Eq. (31) in Supplementary Note 3 becomes ne via the technique described therein (see also ref. 24). The scaling of depth for \({{\mathrm {e}}}^{-i{\hat{V}}_{{{{\rm{nn}}}}}\Delta t}\) can be reduced similarly if the same number of qubits as in the nuclear register are available. With these techniques, the depth of the entire RTE circuit is
instead of Eq. (8). It is noted that, if the number of available extra qubits is \({{{\mathcal{O}}}}({n}_{{\mathrm {e}}}^{2}{n}_{{\mathrm {qe}}})\), the technique proposed in ref. 24 leads to a more drastic reduction of the depth: \({n}_{{\mathrm {e}}}^{2}\) on the RHS in Eq. (31) in Supplementary Note 3 becomes 1.
Let us estimate the computational cost for finding the optimal geometry for the case considered above [see Eq. (6)], where the good reference states are available for the Ncand candidates. From the required number of steps for a tolerance δ and the depth for the single step [see Eq. (9)], the total depth scales as
with respect to ne, Ncand, and δ. The RHS of this equation imposes a lower bound on the coherence time of hardware being used. Since the expected number nmeas(δ) of measurements performed until we reach the optimal state (see Supplementary Note 2) is larger than nsteps(δ) due to the probabilistic nature, the scaling of computational time apart from Uref is estimated to be
As for energy-based geometry optimization on a classical computer, Ncand total-energy calculations are needed and each of them involves the construction of Hamiltonian matrix of dimension \({N}_{{\mathrm {qe}}}^{3{n}_{{\mathrm {e}}}}.\) The classical-operation number for finding the optimal geometry is thus at least \({{{\mathcal{O}}}}({({N}_{{\mathrm {qe}}}^{3{n}_{{\mathrm {e}}}})}^{2}{N}_{{{{\rm{cand}}}}})\) whether using the good reference states or not. This should be compared with the quantum scaling in Eq. (11). Specifically, the scaling in ne for classical computational time is exponential, while that for quantum computational time is at most polynomial. The scaling in Ncand for classical computation is linear, while that for quantum computation is \({{{\mathcal{O}}}}({N}_{{{{\rm{cand}}}}}\log {N}_{{{{\rm{cand}}}}}).\) These observations imply that our optimization scheme with a fixed number of candidates exhibits quantum advantage in molecule size (ne and nnucl). When the candidate number also varies independently of molecule size, the quantum scaling is still at most polynomials. Since the quantum scaling in Ncand is worse than the classical one only logarithmically, it may not cause serious disadvantage that would cancel the advantage in ne. In this sense, our scheme may offer overall quantum advantage when molecule size and candidate number vary, as long as we have an implementation of Uguess and Uref that do not spoil this quantum scaling. Although the pursuit of efficient preparation of reference states is a crucial and challenging task not only for our optimization scheme but also for all the first-quantized schemes, we do not go into further details than Supplementary Note 2.
Quantum amplitude amplification (QAA)40,41, known as a generalization of Grover’s search algorithm, can raise the success probability at each PITE step42. This technique is also applicable for multiple steps by delaying the measurements, as demonstrated by Nishi et al.43 recently. If we introduce the QAA technique to our optimization scheme, the total success probability undergoes quadratic speedup, that is, it changes from ~1/Ncand to \(\sim 1/\sqrt{{N}_{{{{\rm{cand}}}}}}.\) The scaling of computational time in terms of the candidate number is then \({{{\mathcal{O}}}}(\sqrt{{N}_{{{{\rm{cand}}}}}}\log {N}_{{{{\rm{cand}}}}})\) instead of Eq. (11). The optimization scheme for this case offers a quantum advantage with respect to Ncand itself, in addition to ne.
It should be noted that for a case where all the possible displacements of all the nuclei are candidates (\({N}_{{{{\rm{cand}}}}}={N}_{{\mathrm {q}}{{{\rm{n}}}}}^{3{n}_{{{{\rm{nucl}}}}}}\)) with uniform initial weights, the quantum scaling of computational time is exponential in nnucl as well as the classical scaling. This comes from the exponential decrease in the initial weight of the optimal geometry following the increase in the molecule size, lowering the success probability at each step. A situation in which such quantum computation is demanded is, however, actually unlikely. It is because the uniform distribution of weights for the \({N}_{{\mathrm {q}}{{{\rm{n}}}}}^{3{n}_{{{{\rm{nucl}}}}}}\) geometries means that we are completely ignorant of the relative stability among them. The modern sophisticated techniques for electronic-structure calculations and molecular dynamics are, as assumed in our resource estimation, able to enumerate a very small number (compared to \({N}_{{\mathrm {q}}{{{\rm{n}}}}}^{3{n}_{{{{\rm{nucl}}}}}}\)) of promising candidates by spending moderate classical resources. Implementation of Uguess that assigns significant weights to those candidates will be a practical strategy.
PITE simulation for a model LiH molecule
We consider here an effective model of a lithium hydride molecule in one-dimensional space used in ref. 44. This model regards the 1s electrons of the Li atom to be frozen so that the system consists of the two valence electrons, the H ion with ZH = 1, and the Li ion with ZLi = 1. The interactions between the particles are modeled basically by the soft-Coulomb interaction \({v}_{{{{\rm{soft}}}}}(r;\lambda )\equiv 1/\sqrt{{\lambda }^{2}+{r}^{2}},\) where r is the distance between two particles and the parameter λ measures the softness of the interaction. This family of potentials is often used for avoiding the singular behavior of the bare-Coulomb potential45. The adopted values for the interactions are as follows: vee(r) = vsoft(r; λee) between the electrons with \({\lambda }_{{\mathrm {ee}}}^{2}=0.6\), veH(r) = vsoft(r; λeH) between each electron and the H ion with \({\lambda }_{{\mathrm {e}}{{{\rm{H}}}}}^{2}=0.7\), veLi(r) = vsoft(r; λeLi) between each electron and the Li ion with \({\lambda }_{{\mathrm {e}}{{{\rm{Li}}}}}^{2}=2.25\), and vLiH(r) = vsoft(r; λLiH) between the ions with \({\lambda }_{{{{\rm{LiH}}}}}^{2}\equiv {\lambda }_{{\mathrm {e}}{{{\rm{H}}}}}^{2}+{\lambda }_{{\mathrm {e}}{{{\rm{Li}}}}}^{2}-{\lambda }_{{\mathrm {ee}}}^{2}=2.35.\) The potential felt by each electron is thus ven(x) = −ZHveH(∣x−XH∣)−ZLiveLi(∣x−XLi∣), where XH and XLi are the positions of the H and Li ions, respectively. The details of the following simulations are described in Supplementary Note 6.
Figure 5a shows the energy eigenvalues of the molecule as functions of the bond length d ≡ ∣XLi−XH∣ obtained by numerical diagonalization of the Hamiltonian matrix. By using nqe = 6 qubits per electron for a simulation cell with L = 15, we obtained the equilibrium bond length deq = 1.55, in reasonable agreement with that in the earlier paper44. Figure 5b shows the electron densities of the energy eigenstates obtained by numerical diagonalization for d = deq, 4. For d = deq, the electrons are localized near the H ion to exhibit the single-peak shape. For d = 4, on the other hand, they are localized at each ion, indicative of dissociation. We found for both bond lengths that the ground state \(\left\vert {\phi }_{{{{\rm{gs}}}}}\right\rangle\) and the second excited state \(\left\vert {\phi }_{{{{\rm{ex2}}}}}\right\rangle\) are symmetric under the exchange of the spatial coordinates x0 and x1 of the two electrons, while the first excited state \(\left\vert {\phi }_{{{{\rm{ex1}}}}}\right\rangle\) is antisymmetric under the exchange. Recalling that our encoding of wave functions does not incorporate explicitly the spin parts [see Eq. (2)], the ground state and the second excited state are spin-singlet states, while the first excited state is a spin-triplet state.

a Energy eigenvalues of the LiH model system as functions of the bond length d obtained by numerical diagonalization. \(\left\vert {\phi }_{{{{\rm{gs}}}}}\right\rangle ,\left\vert {\phi }_{{{{\rm{ex1}}}}}\right\rangle ,\) and \(\left\vert {\phi }_{{{{\rm{ex2}}}}}\right\rangle\) are the ground state, the first excited state, and the second excited state, respectively. The horizontal dashed line indicates the dissociation limit, that is, the sum of total energies for the isolated H and Li atoms. b Left panel shows the electron densities of the energy eigenstates for the equilibrium bond length deq. The vertical lines indicate the positions of the ions. The potential ven felt by each electron due to the nuclei is also shown. The right panel is a similar plot for d = 4. The x coordinates in the figures have been shifted so that the midpoint of the bond is at the origin.
We performed simulations of geometry optimization among eight candidates represented by nqn = 3 qubits. To be specific, we tried the bond lengths dJ = 0.55 + 0.5J(J = 0, …, 7). The amount of imaginary-time step does not need to be constant. For example, we can define it for the kth PITE step as \(\Delta {\tau }_{k}=(1-{{\mathrm {e}}}^{-k/\kappa })(\Delta {\tau }_{\max }-\Delta {\tau }_{\min })+\Delta {\tau }_{\min },\) so that it changes gradually from \(\Delta {\tau }_{\min }\) to \(\Delta {\tau }_{\max }.\,\)κ determines the rate of change. We adopted \(\Delta {\tau }_{\min }=0.2,\Delta {\tau }_{\max }=0.3,\) and κ = 8 for the following simulations.
To find the optimal bond length for the ground state, we assigned a uniform weight distribution to the candidate geometries, for which we generated the initial spatial wave functions
for the geometries. Xm ≡ (XH + XLi)/2 is the midpoint of the bond and w = 3 is the width of the wave function. Since Ψs is symmetric under exchange of the electrons, it is for obtaining a spin singlet state. Figure 6a shows the weight wJ of each geometry J during the steps contained in the state \(\left\vert {{\Psi }}\right\rangle\) for the composite system of the electrons and nuclei. The weight wJ,gs of the ground state \(\left\vert {\psi }_{{{{\rm{gs}}}}}\right\rangle\) for each geometry is also shown in the figure. It is seen that the uniform distribution of weights in the initial state undergoes deformation via the PITE steps. It has the peak around the geometry for J = 2 already after the 9th step, corresponding to the equilibrium bond length deq. This peak structure becomes more prominent after the 19th step. These observations corroborate the validity of our generic scheme.

a Those for eight candidates starting from the symmetric spatial wave function in Eq. (12).
Using the fact that the ground state and the first excited state \(\left\vert {\psi }_{{{{\rm{ex1}}}}}\right\rangle\) of this system have different symmetry, we can perform geometry optimization for the first excited state. To this end, we adopted the initial spatial wave functions
for the geometries. Since Ψa is antisymmetric under exchange of the electrons, it is for obtaining a spin triplet state. The results are shown in Fig. 6b. In contrast to the case of the ground state, the resultant weight distribution does not have a peak between J = 0 and 7, which lets the observer recognize that there exists no equilibrium bond length among the candidate geometries.
Although the non-optimal geometries in Fig. 6a were found to have significant weights even after the 19th step, our scheme worked thanks to the detectable peak in the histogram. This means that a severe tolerance δ for quashing the near-optimal geometries that would lead to more steps threatening the coherence time [see Eq. (6)] is not necessary for this small system. If it is also the case for a generic large molecule whose energy surface possibly has many local minima, one practical strategy is to continue to pile up data points on a histogram using a moderate tolerance until the optimal and near-optimal geometries become detectable via statistical data processing. How practical compromise between the tolerance for PITE steps and the number of data points for a histogram is met and quantum advantage taking it into account should be examined in the future.
VITE simulation for a model H\({}_{2}^{+}\) molecule
Since the essence of our approach is the superposition of nuclear-register states where the candidate geometries are encoded, geometry optimization based on VITE instead of PITE is straightforwardly formulated. To demonstrate that, we consider here an effective model of a hydrogen molecular ion in a one-dimensional space used in Ref. 46. The charge of each ion is ZH = 1. Here, we also use the soft-Coulomb interaction to model the interactions between the particles. We adopt the softness \({\lambda }_{{\mathrm {e}}{{{\rm{H}}}}}^{2}=1\) for the interaction veH(r) between the electron and each ion and \({\lambda }_{{{{\rm{HH}}}}}^{2}=1\) for vHH(r) between the ions. The potential felt by the electron is thus ven(x) = −ZHveH(∣x−XHα∣)−ZHveH(∣x−XHβ∣), where XHα and XHβ are the positions of the H ions.
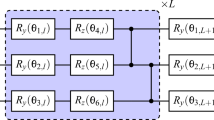
The VITE approach is explained briefly in Supplementary Note 5. Figure 7 shows our ansatz circuit for geometry optimization of the H\({}_{2}^{+}\) model system. We adopted the hardware-efficient connectivity47 for the circuit simulations48, which is desirable for noisy intermediate-scale quantum (NISQ) devices due to shallow circuit depths. In addition, the accuracy of the quantum computation systematically improves by incrementing the repetition d of the layer. Here, we use the full coupling model; CZ gates connect every pair of qubits for entangling all qubits. We allocated nqnucl = 3 qubits for encoding the nuclear positions and nqe = 6 qubits for encoding the single-electron wave function in a simulation cell with L = 15. As demonstrated below, the VITE-based scheme can, despite the absence of Uguess and Uref, find the optimal geometry going through more than a thousand of steps, while the PITE-based scheme finds the optimal one in much fewer steps (see Supplementary Note 6). Such many steps are practically possible since the circuit depth is related not to the number of steps, but to the depth of the ansatz. This feature renders the VITE-based scheme NISQ-friendly, in contrast to the PITE-based one.

The part inside the parentheses is applied to the nuclear and electronic registers d times. The purple boxes stand for single-qubit rotations whose angles are specified by distinct variational parameters. The nuclear register is measured at the end of the circuit to find the optimal bond length.
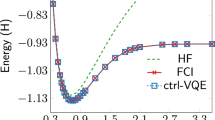
The VITE calculation was performed for candidates whose bond lengths were specified by dJ = 0.5 + (7.5/8)J (J = 0, …, 7). We simulated the updating process of variational parameters with d = 12 for 6000 VITE steps with Δτ = 0.01. All the initial values of the variational parameters were set to random values. The expected energy of the trial state \(\left\vert {{\Psi }}\right\rangle\) at each VITE step measured from the numerically exact ground state energy is shown in Fig. 8a. We recognize the monotonic but slow decrease in the energy difference. Figure 8b shows the weights wJ of candidate geometries contained in the trial wave function at each VITE step. The weight of the most stable geometry labeled by J = 2 monotonically increases and reaches close to unity at the final step. The second most stable structure, J = 3, is amplified once in the first 1500 steps and then turns to decrease. We draw the electronic wave function component contained in the most stable state, J = 2, in Fig. 8c. The ground state \(\left\vert {\phi }_{{{{\rm{gs}}}}}\right\rangle\) for the geometry J = 2 quickly increases, and the excited states decrease to zero within 1000 steps. These results support our ideas of encoding candidate geometries for optimization work and also for the variational scheme. The convergence of nuclear states was rather slow compared to that of the electronic states for the individual geometries, as seen in Fig. 8b and c. This observation reflects the generic fact that the continuous energy of classical nuclei leads to a small energy difference between neighboring candidate geometries, as discussed above.

a Expected energy of the trial state \(\left\vert {{\Psi }}\right\rangle\) at each VITE step measured from the lowest energy eigenvalue of the molecule. b The weights wJ of candidate geometries contained in the trial state at each step. c The weight wJ=2,gs of the ground state \(\left\vert {\phi }_{{{{\rm{gs}}}}}\right\rangle\) for the geometry J = 2 contained in the trial state at each step. Those of the first- and second-excited states, wJ=2,ex1 and wJ=2,ex2, are also shown.
PITE simulation for a classical C6H6–Ar system
As stated in Supplementary Note 2, our scheme is also applicable to a geometry optimization problem for point charges as a classical system. It is known that the improved Lennard-Jones (ILJ)49,50 potentials describe the experimental data well for hydrocarbon molecules interacting with rare-gas atoms. We adopt here these model potentials to consider a classical system consisting of a benzene molecule interacting weakly with an argon atom49, as depicted in Fig. 9a. We perform simulations of geometry optimization for this system by using our PITE scheme.

a Classical target system for geometry optimization based on PITE approach. The C6H6 molecule lies on the xy plane with its center of mass located at the origin. Two of the C-H bonds are along the y-axis. We consider an Ar atom on the xz plane. b The interaction energy as a function of the position of the Ar atom. c The weight of each candidate during the PITE steps contained in the state of nuclear register.
The C–C and C–H bond lengths are fixed at 1.39 and 1.09 Å51, respectively, throughout the simulations. The explicit expressions for the ILJ potentials are provided in Supplementary Note 6. Figure 9b shows the interaction energy between the C6H6 molecule and the Ar atom on the xz plane as a function of the position of the Ar atom. The interaction energy takes a minimum value at z = 3.57 Å with x = y = 0 Å49.
We performed simulations of geometry optimization among 64 candidates represented by nqn = 3 qubits for each of the x and z coordinates of the Ar atom. Each of the candidates is specified by two integers J = (Jx, Jz) with Jx, Jz = 0, …, 7, which generate the coordinates xJ = −2.4 + 0.8Jx Å and zJ = 3.2 + 0.4Jz Å. We used a constant amount Δτ = 0.004 meV−1 of each PITE step in the following simulations.
In each simulation of the circuit shown in Supplementary Fig. 3, we assigned a uniform weight distribution to the candidate geometries for an initial state. Figure 9c shows the weight wJ of each candidate during the steps contained in the state of the nuclear register. It is seen that the uniform distribution of weights in the initial state undergoes the deformation via the steps, as expected. The largest weight is already seen after the 11th step at J = (3, 1), which is closer to the true optimal geometry than any other candidate is. This peak structure becomes more prominent after the 19th step, as seen in the figure.
Discussion
In summary, this study proposed a nonvariational scheme for geometry optimization of a molecule within the framework of FQE, where the electrons and nuclei are treated as quantum mechanical particles and classical point charges, respectively. The scheme encodes their information as a many-qubit state, for which repeated measurements give the global minimum among all the candidate geometries. We demonstrated that the total computational time may exhibit an overall quantum advantage in terms of molecule size and candidate number. The circuit depth of RTE operation, which is the central component of each PITE step, was found to scale as \({{{\mathcal{O}}}}({n}_{{\mathrm {e}}}^{2}{{{\rm{poly}}}}(\log {n}_{{\mathrm {e}}}))\) for the electron number ne. This can be reduced to \({{{\mathcal{O}}}}({n}_{\mathrm {{e}}}{{{\rm{poly}}}}(\log {n}_{{\mathrm {e}}}))\) if the same number of extra qubits as in the original circuit are available. If \({{{\mathcal{O}}}}({n}_{{\mathrm {e}}}^{2}\log {n}_{{\mathrm {e}}})\) extra qubits are available, the depth can be reduced to \({{{\mathcal{O}}}}({{{\rm{poly}}}}(\log {n}_{{\mathrm {e}}})).\) The validity of the new scheme was verified through numerical simulations. The scheme will assist in achieving scalability in practical quantum chemistry on quantum computers. Additionally, this approach will support the realization of geometry optimization using NISQ devices.
There may be room for elaborating the sampling strategy for candidate geometries for this scheme to be more efficient from a practical perspective. That is, adaptively changing the range and resolution of nuclear displacements under the constraint of a fixed total number of measurements may more accurately determine the optimal geometry, which could be examined in the future.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
The code developed for the current study is available from the corresponding author on reasonable request.
References
Axelrod, S. et al. Learning matter: materials design with machine learning and atomistic simulations. Accounts Mater. Res. 3, 343–357 (2022).
Pereira, J. M., Vieira, M. & Santos, S. M. Step-by-step design of proteins for small molecule interaction: a review on recent milestones. Protein Sci. 30, 1502–1520 (2021).
Pandey, M. et al. The transformational role of GPU computing and deep learning in drug discovery. Nat Mach. Intell. 4, 211–221 (2022).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Helgaker, T., Jørgensen, P. & Olsen, J. Molecular Electronic-Structure Theory (Wiley, 2000).
Feynman, R. P. Forces in molecules. Phys. Rev. 56, 340–343 (1939).
Pulay, P. Ab initio calculation of force constants and equilibrium geometries in polyatomic molecules. Mol. Phys. 17, 197–204 (1969).
Schlegel, H. B. Geometry optimization. WIREs Comput. Mol. Sci. 1, 790–809 (2011).
Feynman, R. P. Simulating physics with computers. Int. J. Theor. Phys. 21, 467–488 (1982).
Hirai, H. et al. Molecular structure optimization based on electrons-nuclei quantum dynamics computation. ACS Omega 7, 19784–19793 (2022).
Wiesner, S. Simulations of many-body quantum systems by a quantum computer. arXiv e-prints quant-ph/9603028 (1996).
Zalka, C. Simulating quantum systems on a quantum computer. Proc. R. Soc. Lond. Ser. A 454, 313–322 (1998).
Kassal, I., Jordan, S. P., Love, P. J., Mohseni, M. & Aspuru-Guzik, A. Polynomial-time quantum algorithm for the simulation of chemical dynamics. Proc. Natl Acad. Sci. USA 105, 18681–18686 (2008).
Jones, T., Endo, S., McArdle, S., Yuan, X. & Benjamin, S. C. Variational quantum algorithms for discovering Hamiltonian spectra. Phys. Rev. A 99, 062304 (2019).
McArdle, S. et al. Variational ansatz-based quantum simulation of imaginary time evolution. npj Quantum Inf. 5, 75 (2019).
Yuan, X., Endo, S., Zhao, Q., Li, Y. & Benjamin, S. C. Theory of variational quantum simulation. Quantum 3, 191 (2019).
Peruzzo, A. et al. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun. 5, 4213 EP (2014).
McClean, J. R., Romero, J., Babbush, R. & Aspuru-Guzik, A. The theory of variational hybrid quantum-classical algorithms. N. J. Phys. 18, 023023 (2016).
Kassal, I. & Aspuru-Guzik, A. Quantum algorithm for molecular properties and geometry optimization. J. Chem. Phys. 131, 224102 (2009).
Kosugi, T., Nishiya, Y., Nishi, H. & Matsushita, Y.-i Imaginary-time evolution using forward and backward real-time evolution with a single ancilla: first-quantized eigensolver algorithm for quantum chemistry. Phys. Rev. Res. 4, 033121 (2022).
Kosugi, T. & Matsushita, Y.-i Construction of green’s functions on a quantum computer: Quasiparticle spectra of molecules. Phys. Rev. A 101, 012330 (2020).
Kosugi, T. & Matsushita, Y.-i Linear-response functions of molecules on a quantum computer: charge and spin responses and optical absorption. Phys. Rev. Res. 2, 033043 (2020).
Jones, N. C. et al. Faster quantum chemistry simulation on fault-tolerant quantum computers. N. J. Phys. 14, 115023 (2012).
Chan, H. H. S., Meister, R., Jones, T., Tew, D. P. & Benjamin, S. C. Grid-based methods for chemistry simulations on a quantum computer. Sci. Adv. 9, eabo7484 (2023).
Abrams, D. S. & Lloyd, S. Simulation of many-body fermi systems on a universal quantum computer. Phys. Rev. Lett. 79, 2586–2589 (1997).
Berry, D. W. et al. Improved techniques for preparing eigenstates of fermionic Hamiltonians. npj Quantum Inf. 4, 22 (2018).
Nishi, H., Hamada, K., Nishiya, Y., Kosugi, T. & Ichiro Matsushita, Y. Optimal scheduling in probabilistic imaginary-time evolution on a quantum computer. Phys. Rev. Res. 5, 043048 (2023).
Somma, R. D. Quantum simulations of one dimensional quantum systems. arXiv e-prints arXiv:1503.06319 (2015).
Ollitrault, P. J., Mazzola, G. & Tavernelli, I. Nonadiabatic molecular quantum dynamics with quantum computers. Phys. Rev. Lett. 125, 260511 (2020).
Childs, A. M., Su, Y., Tran, M. C., Wiebe, N. & Zhu, S. Theory of trotter error with commutator scaling. Phys. Rev. X 11, 011020 (2021).
Draper, T. G. Addition on a quantum computer. arXiv e-prints quant–ph/0008033 (2000). quant-ph/0008033.
Draper, T. G., Kutin, S. A., Rains, E. M. & Svore, K. M. A logarithmic-depth quantum carry-lookahead adder. Quant. Inf. Comp. 6, 351–369 (2006).
Cuccaro, S. A., Draper, T. G., Kutin, S. A. & Moulton, D. P. A new quantum ripple-carry addition circuit. Quant-ph. 9, https://doi.org/10.48550/arXiv.quant-ph/0410184. (2004).
Kowada, L. A. B., Portugal, R. & de Figueiredo, C. M. H. Reversible Karatsuba’s algorithm. J. Univers. Comput. Sci. 12, 499–511 (2006).
Parent, A., Roetteler, M. & Mosca, M. Improved reversible and quantum circuits for Karatsuba-based integer multiplication. https://doi.org/10.48550/arXiv.1706.03419 (2017).
Dutta, S., Bhattacharjee, D. & Chattopadhyay, A. Quantum circuits for toom-cook multiplication. Phys. Rev. A 98, 012311 (2018).
Hadfield, S. Quantum algorithms for scientific computing and approximate optimization. arXiv e-prints arXiv:1805.03265 (2018).
Benenti, G. & Strini, G. Quantum simulation of the single-particle schrödinger equation. Am. J. Phys. 76, 657–662 (2008).
Brassard, G. & Hoyer, P. An exact quantum polynomial-time algorithm for Simon’s problem. In Proc. Fifth Israeli Symposium on Theory of Computing and Systems pp. 12–23 (IEEE, Ramat Gan, Israel, 1997).
Brassard, G., Hoyer, P., Mosca, M. & Tapp, A. Quantum amplitude amplification and estimation. arXiv e-prints quant-ph/0005055 (2000).
Nishi, H., Kosugi, T., Nishiya, Y. & Matsushita, Y.-i. Acceleration of probabilistic imaginary-time evolution method combined with quantum amplitude amplification. arXiv e-prints arXiv:2212.13816 (2022).
Nishi, H., Kosugi, T., Nishiya, Y. & Matsushita, Y.-i. Quadratic speedups of multi-step probabilistic algorithms in state preparation. arXiv e-prints arXiv:2308.03605 (2023).
Tempel, D. G., Martínez, T. J. & Maitra, N. T. Revisiting molecular dissociation in density functional theory: a simple model. J. Chem. Theory Comput. 5, 770–780 (2009).
Li, C. Exact analytical solution of the ground-state hydrogenic problem with soft coulomb potential. J. Phys. Chem. A 125, 5146–5151 (2021).
Wagner, L. O., Stoudenmire, E. M., Burke, K. & White, S. R. Reference electronic structure calculations in one dimension. Phys. Chem. Chem. Phys. 14, 8581–8590 (2012).
Kandala, A. et al. Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets. Nature 549, 242–246 (2017).
Suzuki, Y. et al. Qulacs: a fast and versatile quantum circuit simulator for research purpose. Quantum 5, 559 (2021).
Pirani, F., Alberti, M., Castro, A., Teixidor, M. & Cappelletti, D. Atom-bond pairwise additive representation for intermolecular potential energy surfaces. Chem. Phys. Lett. 394, 37–44 (2004).
Pirani, F. et al. Beyond the Lennard-Jones model: a simple and accurate potential function probed by high resolution scattering data useful for molecular dynamics simulations. Phys. Chem. Chem. Phys. 10, 5489–5503 (2008).
Pirani, F., Porrini, M., Cavalli, S., Bartolomei, M. & Cappellettigaga, D. Potential energy surfaces for the benzene-rare gas systems. Chem. Phys. Lett. 367, 405–413 (2003).
Acknowledgements
This work was supported by MEXT as “Program for Promoting Researches on the Supercomputer Fugaku” (JPMXP1020200205) and JSPS KAKENHI as “Grant-in-Aid for Scientific Research(A)” Grant Number 21H04553. The computation in this work has been done using (supercomputer Fugaku provided by the RIKEN Center for Computational Science/Supercomputer Center at the Institute for Solid State Physics at the University of Tokyo).
Author information
Authors and Affiliations
Contributions
T.K. developed the methods and wrote the simulation code. H.N. and Y.-i.M. discussed our approach with T.K. from the viewpoint of quantum chemistry and solid-state physics. All the authors contributed equally to the manuscript preparation and presentation of results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kosugi, T., Nishi, H. & Matsushita, Yi. Exhaustive search for optimal molecular geometries using imaginary-time evolution on a quantum computer. npj Quantum Inf 9, 112 (2023). https://doi.org/10.1038/s41534-023-00778-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41534-023-00778-6
