
Abstract
We present a protocol that detects molecular conformational changes with two nitroxide electron-spin labels and a nitrogen-vacancy (NV) center in diamond. More specifically, we demonstrate that the NV can detect energy shifts induced by the coupling between electron-spin labels. The protocol relies on the judicious application of microwave and radiofrequency pulses in a range of parameters that ensures stable nitroxide resonances. Furthermore, we demonstrate that our scheme is optimized by using nitroxides with distinct nitrogen isotopes. We develop a simple theoretical model that we combine with Bayesian inference techniques to demonstrate that our method enables the detection of conformational changes in ambient conditions including strong NV dephasing rates as a consequence of the diamond surface proximity and nitroxide thermalization mechanisms. Finally, we counter-intuitively show that with our method the small residual effect of random molecular tumbling becomes a resource that can be exploited to extract inter-label distances.
Similar content being viewed by others

Introduction
A precise knowledge of molecular structure and dynamics would shed new light on the mechanisms by which, e.g., proteins interact with their environment1. This finds important applications in distinct fields such as biochemistry and biology, since macromolecular conformational changes underlie numerous biological processes. In this regard, different techniques have been proposed to study molecular structure and dynamics2,3,4. In particular, methods based on nuclear magnetic resonance (NMR)5,6 and on electron-spin resonance (ESR)7,8 stand out as they are central to investigate a wide variety of molecules, with ESR being more sensitive due to the much larger gyromagnetic ratio of electrons9. One commonly used ESR method is the Double Electron-Electron Resonance (DEER) in which synchronized and/or delayed pulses are delivered to, e.g., estimate average distances among free radicals in molecular ensembles10. However, traditional ESR based on inductive detection with macroscopic coils requires macroscopic samples with a high chemical purity to produce a detectable response. This rules out ESR investigations of static and dynamic properties of individual molecules.
Detectors based on NV centers in diamond11,12 are promising candidates to overcome the limitations of inductive detection. Indeed, such detectors enable ESR and NMR detection of molecular samples down to the micron- and nanoscale and even down to the level of single molecules. The potential of NV centers in diamond originates from a number of appealing properties and early demonstrations. They can be initialized and read out using laser light in the visible spectrum, thereby removing thermal fluctuations from the measurement process13,14,15. Moreover, NV hyperfine states can be manipulated with microwave radiation in such a way that the NV couples coherently to nearby nuclear and electron spins while at the same time being decoupled from environmental fluctuations16,17,18,19,20. These concepts have enabled shallowly implanted NV centers to detect electron spins21 and even nuclear spins 22,23 above the diamond surface with single-spin sensitivity. Detection protocols have also been developed allowing for nanoscale NMR24,25,26,27 and ESR detection28,29,30 with spectral resolution approaching that of macroscopic ESR and NMR based on inductive detection. Fluctuating electron spins of biological samples31,32,33,34 and small groups of statistically polarized nuclear spins in a target protein35 have been detected. Finally and particularly relevant to the present work, an NV center has also been used to detect a single nitroxide electron-spin label36 attached to a protein. The use of nitroxide spin labels is particularly attractive because it enables the study of non-paramagnetic molecules37. In particular, ref. 38 reported the detection of an effective inter-label coupling in molecular ensembles using advanced multi-frequency spectroscopy techniques. Attaching several nitroxides to a single target protein could enable the study of its internal dynamics at room temperature with an NV center. However, this approach is challenging because spin labels have resonance frequencies and interaction strengths that strongly depend on their orientation with respect to the external magnetic field39. Therefore, the identification of these parameters is complicated by the unavoidable molecular motion and environmental noise affecting the NV and the electron-spin labels.
In this work, we show how even a simple pulse sequence applied to a single pair of electron-spin labels and a shallow NV [see Fig. 1(a)] can detect single-molecule conformational transitions at room temperature. Our carefully engineered DEER sequence asymmetrically delivers microwave (radiofrequency) pulses to electron spins of the NV center (nitroxide spin labels) to resolve small energy shifts in the NV spectrum owing to the coupling between labels. Crucially, we find an energy-transition branch of the labels that presents an excellent robustness against unavoidable tumbling motion in ambient conditions. In addition, we show that this branch is especially robust when each nitroxide label contains a distinct nitrogen isotope. Finally, we develop a simple but accurate model which enables us to estimate not only the coupling but also the distance between labels using Bayesian inference. In particular, we counter-intuitively show that the small residual effect of tumbling, instead of being detrimental, enables the extraction of the distance between labels.

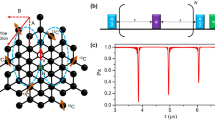
a A protein with two attached labels is near a diamond surface that contains a shallow NV. b Nitroxide molecule including the electron spin (yellow). The principal axes are shown in red, while the laboratory frame is shown in black. The azimuth θ (the angle between the z principal axis and the z laboratory axis) determines the energy-transition branches of the label. c MW and RF driving scheme. This includes initialization and read out with laser and microwave pulses, two free evolution stages of duration τfree, 2N + 1 MW π-pulses on the NV, and a simultaneous RF π-pulse on the labels.
Results
The system
The Hamiltonian of the NV, the two nitroxide electron-spin labels, and the microwave (MW) and radiofrequency (RF) driving fields reads
Here, σz is the Pauli z operator for the NV two-level system and Ji are spin-1/2 operators for each label (i = 1, 2). The Hamiltonian of the ith nitroxide label is \({H}_{{n}_{i}}\) and the coupling of each label to the NV is mediated by the vectors ai. Moreover, \({J}_{i}^{\pm }={J}_{i}^{x}\pm i{J}_{i}^{y}\) are electron-spin ladder operators and g12 is the coupling constant between labels. The last line in Eq. (1) describes a MW field resonant with the NV, leading to the term \(\frac{{{{\Omega }}}_{{{{\rm{MW}}}}}}{2}{\sigma }^{x}\) in a suitable interaction picture. In addition, an RF field of frequency ωRF and Rabi frequency ΩRF excites the electronic resonances of the nitroxide labels. Equation (1) is derived in the Supplementary Note 1 and a sketch of the system is presented in Fig. 1(a).
The term \({H}_{{n}_{i}}\) models each nitroxide label as39,40
where μB is the Bohr magneton, Bz is a magnetic field applied along the z axis, γN is the nuclear gyromagnetic ratio equal to 2π × 3.077 kHz mT−1 for 14N and to 2π × − 4.316 kHz mT−1 for 15N, Ii is the nuclear spin operator for the ith nitroxide label, and \({{\mathbb{G}}}_{i}\), \({{\mathbb{Q}}}_{i}\), and \({{\mathbb{A}}}_{i}\) are respectively the Landé, quadrupolar, and electron-nucleus interaction tensors40. We note that Ii is a spin-1 (spin-1/2) operator when describing a 14N (15N) nuclear spin. The components of the \({{\mathbb{G}}}_{i}\), \({{\mathbb{Q}}}_{i}\), and \({{\mathbb{A}}}_{i}\) tensors in a general reference frame depend on the frame’s relative orientation with respect to the principal axes of the nitroxide [see Fig. 1(b)]. In the principal frame, \({{\mathbb{G}}}_{i}\), \({{\mathbb{Q}}}_{i}\), and \({{\mathbb{A}}}_{i}\) are diagonal40. In particular, the Landé tensor in the principal frame is \({{\mathbb{G}}}_{i}^{(P)}={{{\rm{diag}}}}\left({G}^{x},{G}^{y},{G}^{z}\right)\), with Gx ≈ Gy ≈ 2.007 ≡ G⊥ and Gz ≈ 2.002 ≡ G∥. The quadrupolar tensor for 14N is \({{\mathbb{Q}}}_{i}^{(P)}={{{\rm{diag}}}}\left({Q}^{x},{Q}^{y},{Q}^{z}\right)\), with Qx ≈ 2π × 1.26 MHz, Qy ≈ 2π × 0.53 MHz, and Qz ≈ 2π × − 1.79 MHz. The quadrupolar tensor vanishes for 15N. Finally, the tensor that mediates electron-nucleus interactions in each nitroxide is \({{\mathbb{A}}}_{i}^{(P)}={{{\rm{diag}}}}\left({A}^{x},{A}^{y},{A}^{z}\right)\), where Ax ≈ Ay ≈ 2π × 14.7 MHz ≡ A⊥ and Az ≈ 2π × 101.4 MHz ≡ A∥ for 14N, while Ax ≈ Ay ≈ 2π × 27MHz ≡ A⊥ and Az ≈ 2π × 141 MHz ≡ A∥ for 15N40.
We obtain approximate nitroxide transition frequencies via a perturbative treatment of the nuclear degrees of freedom in Eq. (2), see Supplementary Note 2. For a nitroxide hosting 14N, we find
The three energy-transition branches exhibit transition energies given by \({E}_{1,-1}^{i}={\mu }_{{{{\rm{B}}}}}{B}^{z}G({\theta }_{i})\pm \frac{1}{\sqrt{2}}\sqrt{\left[{({A}^{\parallel })}^{2}-{({A}^{\perp })}^{2}\right]\cos (2{\theta }_{i})+{({A}^{\perp })}^{2}+{({A}^{\parallel })}^{2}}\) and \({E}_{0}^{i}={\mu }_{{{{\rm{B}}}}}{B}^{z}G({\theta }_{i})+\frac{1}{2{\mu }_{{{{\rm{B}}}}}{B}^{z}G({\theta }_{i})}\frac{2{({A}^{\perp }{A}^{\parallel })}^{2}+\left[{({A}^{\perp })}^{4}-{({A}^{\perp }{A}^{\parallel })}^{2}\right]{\sin }^{2}({\theta }_{i})}{{({A}^{\perp })}^{2}{\sin }^{2}({\theta }_{i})+{({A}^{\parallel })}^{2}{\cos }^{2}({\theta }_{i})}\). Here, θi is the azimuth angle between the applied magnetic field and the principal z axis of the nitroxide [see Fig. 1(b)], \(G({\theta }_{i})=\frac{1}{2}[({G}^{\parallel }-{G}^{\perp })\cos (2{\theta }_{i})+{G}^{\perp }+{G}^{\parallel }]\), and \({\left\vert \widetilde{1}\right\rangle }_{i}\), \({\left\vert \widetilde{0}\right\rangle }_{i}\), and \({\left\vert -\widetilde{1}\right\rangle }_{i}\) are the states of the ith nitroxide nucleus dressed by the hyperfine interaction with the nitroxide electron, see Supplementary Note 2. Similarly, for a nitroxide hosting 15N, we find
There are now two energy-transition branches with transition energies given by \({E}_{1/2,-1/2}^{i}={\mu }_{{{{\rm{B}}}}}{B}^{z}G({\theta }_{i})\pm \frac{1}{2\sqrt{2}}\sqrt{{({A}^{\perp })}^{2}+{({A}^{\parallel })}^{2}+\left[{({A}^{\parallel })}^{2}-{({A}^{\perp })}^{2}\right]\cos (2{\theta }_{i})}\). Figure 2a compares the energy-transition branches in Eqs. (3) and (4) (dashed lines) with numerical diagonalization of Eq. (2) (solid lines). Note that since we will target the \({E}_{0}^{i}\) branch for label detection, we have further developed its functional form to include second-order corrections in the electron-nitrogen coupling, see Supplementary Note 2. Figure 2(a) focuses on a single nitroxide (the index i = 1 is removed for clarity) and shows that our expression for E0 is in excellent agreement with numerical diagonalization.

a Energy-transition branches of an electron spin label at Bz = 30 mT for 14N (15N). Solid black (gray) lines show the transition energies as a function of the azimuth θ obtained via numerical diagonalization of Eq. (2). Dashed black (gray) lines correspond to E1,0,−1 in Eq. (3) [E1/2,−1/2 in Eq. (4)]. The vertical line highlights the case θi = 30°, with red dots indicating the transition energies relevant in the following plots. b NV spectrum \({\langle {\sigma }^{x}\rangle }_{{{{\rm{NV}}}}}\) as a function of the RF frequency applied near \({E}_{0}^{1}\). The spectrum was obtained by unitary propagation of Eq. (1) with \({H}_{{n}_{i}}\) given by Eq. (2) (black line) and by \({E}_{0}^{i}{J}_{i}^{z}\) (gray circles). The first nitroxide has azimuth θ1 = 30° while the second nitroxide has azimuth θ2 = 91.7°. The splitting of the peaks is induced by the inter-label coupling g12 ≈ 2π × 1 MHz. The positions of the resonances are indicated by the dashed red lines. In (c, d), shaded areas show the average NV spectra \({\overline{\langle {\sigma }^{x}\rangle }}_{{{{\rm{NV}}}}}\) in the presence of tumbling of the first (near-resonant) nitroxide. Tumbling was mimicked by averaging over a random azimuth following a Gaussian distribution centered at the equilibrium value θ1,eq = 30°. The orientation of the other nitroxide was kept fixed for simplicity. The standard deviation was chosen to be σθ = 6.25°. In (c), we observe that tumbling leads to a loss of contrast and to signal broadening near \({E}_{0}^{1}\). Nevertheless, the spectrum still shows two clearly separated peaks. For comparison, the case without tumbling in (b) is superimposed (black line). d Average NV spectrum \({\overline{\langle {\sigma }^{x}\rangle }}_{{{{\rm{NV}}}}}\) near \({E}_{1}^{1}\). Here, the energy splitting cannot be resolved due to nitroxide tumbling. Also note that tumbling severely reduces contrast. For that reason, the spectrum in the absence of tumbling has much higher contrast and is not shown.
The inter-label coupling leads to an additional splitting ∝ g12 of the energy-transition branches in Eqs. (3), (4). This term typically simplifies as \({g}_{12}\left[{J}_{1}^{z}{J}_{2}^{z}-\frac{1}{4}\left({J}_{1}^{+}{J}_{2}^{-}+{J}_{1}^{-}{J}_{2}^{+}\right)\right]\longrightarrow {g}_{12}{J}_{1}^{z}{J}_{2}^{z}\), since labels with different orientations have different resonance frequencies, thus eliminating the flip-flop contribution. Note that while this simplification is frequently (but not always) valid when using two 14N, it is guaranteed to be valid when using nitroxides hosting different nitrogen isotopes since E0 (for 14N) differs from E1/2,−1/2 (for 15N) for any value of the azimuth [see Fig. 2(a)]. For 14N, the available resonances of, e.g., the first label are then determined by the modified nitroxide Hamiltonian \(H^{\prime}_{{n}_{1}}={H}_{{n}_{1}}+{g}_{12}{J}_{1}^{z}{J}_{2}^{z}\)
That is, any energy-transition branch \({E}_{q}^{1}\) (corresponding to the \(\left\vert \widetilde{q}\right\rangle\) nuclear state of the first nitroxide) splits as
depending on the electronic state \(\left\vert e\right\rangle\), \(\left\vert g\right\rangle\) of the second nitroxide. Supplementary Note 2 includes a complete energy diagram of an electron spin in a nitroxide.
The protocol
Our DEER protocol simultaneously delivers MW and RF pulses to detect energy shifts in the NV spectrum due to inter-label coupling. The sequence contains two free-evolution stages of duration τfree separated by a driving stage [see Fig. 1(c)]. The NV is prepared in the + 1 eigenstate of σx and the nitroxides are assumed to be in a fully thermalized state. During free evolution, the NV accumulates a phase due to the term \({\sigma }^{z}\left({{{{\bf{a}}}}}_{1}\cdot {{{{\bf{J}}}}}_{1}+{{{{\bf{a}}}}}_{2}\cdot {{{{\bf{J}}}}}_{2}\right)\) in Eq. (1). This term can be approximated as \({\sigma }^{z}\left({a}_{1}^{z}{J}_{1}^{z}+{a}_{2}^{z}{J}_{2}^{z}\right)\) in the rotating-wave approximation (RWA) since the \({J}_{i}^{x,y}\) contributions are suppressed by the large electronic precession frequencies of the nitroxides. Indeed, the energies \({E}_{1,0,-1}^{i}\) reach several hundreds of MHz even for moderate values of Bz (we use Bz = 30 mT in our simulations). By contrast, the NV-label coupling takes values of hundreds of kHz for typical NV-label distances of several nanometers. During MW/RF irradiation, the NV undergoes an overall flip σz → − σz via 2N + 1 contiguous π-pulses arising from the same continuous MW drive. At the same time, a single weaker RF π-pulse on the labels induces \({J}_{i}^{z}\to -{J}_{i}^{z}\) if it is near-resonant with an electronic transition (i.e., with \({E}_{1,0,-1}^{i}\) for 14N or with \({E}_{1/2,-1/2}^{i}\) for 15N). The simultaneous flipping of the NV and spin label ensures a constructive and coherent accumulation of the NV phase during free evolution. Meanwhile, the use of 2N + 1π-pulses on the NV ensures that the Rabi frequencies of the two drives are well separated, which effectively averages out spurious NV-label interactions during irradiation. Consequently, scanning the RF frequency near nitroxide transitions yields clean resonance peaks in the NV response. As long as ΩRF < g12, the resonance peaks are split by g12. Figure 2(b) shows the NV spectrum (the expectation value of σx) associated with the transitions \(\left\vert 0g\right\rangle \left\vert g\right\rangle \leftrightarrow \left\vert 0e\right\rangle \left\vert g\right\rangle\) and \(\left\vert 0g\right\rangle \left\vert e\right\rangle \leftrightarrow \left\vert 0e\right\rangle \left\vert e\right\rangle\). The resonances are split by ~ g12 as expected. The solid black line in Fig. 2(b) was obtained by numerically propagating Eq. (1) with \({H}_{{n}_{i}}\) given by Eq. (2), while gray circles were obtained assuming the simplified \({H}_{{n}_{i}}\) in Eq. (3). The two spectra are in excellent agreement. Simulations in Fig. 2(b) assume Bz = 30 mT, which is in the range of values that ensure the stability of the \({E}_{0}^{i}\) energy-transition branch discussed below, see Supplementary Note 2. Moreover, the parameters of the pulse sequence are τfree = 1.3 μs, ΩRF = 2π × 250 kHz, and ΩMW = 31 × ΩRF, for a total sequence time of 4.6 μs. In addition, the labels are separated from the NV by the distances d1 ≈ 6.9 nm and d2 ≈ 7.3 nm, leading to \({{{{\bf{a}}}}}_{1}\approx 2\pi \,\times \left(128,-132,-223\right)\) kHz and \({{{{\bf{a}}}}}_{2}\approx 2\pi \,\times \left(-22,-16,-264\right)\) kHz, while the distance between labels is d12 ≈ 3.297 nm.
The expressions for the energy-transition branches in Eqs. (3) and (4) reveal a dependence on the azimuth angle, i.e., \({E}_{q}^{i}\equiv {E}_{q}^{i}({\theta }_{i})\). Consequently, unavoidable protein motion during the irradiation stage leads to a distortion in the spectrum and to a difficult interpretation of the signal. However, it is important to note the distinct nature of the \({E}_{0}^{i}\) branch, which shows a much weaker dependence on θi than \({E}_{1,-1}^{i}\). This makes the \({E}_{0}^{i}\) branch particularly well suited for robust detection of the energy splitting [see Fig. 2(c, d)]. To maximize the robustness, we chose the magnetic field to be large enough to energetically suppress the effect of the anisotropic hyperfine interaction, but small enough that the anisotropy of the Landé tensor does not become significant, see Supplementary Note 2.
Numerical simulations
We now illustrate our method in realistic ambient conditions including decoherence leading to NV dephasing with decoherence time T2 = 20 μs (for a 4 nm NV depth36), electron-spin label relaxation with T1 = 4 μs36, and molecular tumbling. The dissipative model used in this section is described in the Supplementary Note 3 and captures the main decoherence mechanisms identified in ref. 36. Note that for the specific protocol considered here, decoherence mainly manifests as a reduced line contrast and not as an increased linewidth. This is because we keep the sequence duration fixed and sweep ωRF to find the resonances, as opposed to varying the sequence length at fixed ωRF to observe an echo signal. The molecular tumbling is modelled as a random rigid rotation of both nitroxides around an axis parallel to the laboratory x axis, see Supplementary Note 3. Our simulations use a rotation angle δ that is Gaussian-distributed with standard deviation σδ = 6.25°. This is somewhat smaller but comparable to the fluctuations observed in ref. 36. Note that immobilizing proteins in more rigid matrices41,42 or attaching them to the diamond surface35 through multiple rigid covalent bonds could further reduce tumbling. The resulting tumbling-averaged spectra \({\overline{\langle {\sigma }^{x}\rangle }}_{{{{\rm{NV}}}}}\) are shown in Fig. 3. In Fig. 3(a, b, c), the parameters τfree, ΩMW, ΩRF, and ai are the same as in Fig. 2. In Fig. 3(a), the labels are separated by d12 ≈ 3.297 nm and the inter-label coupling is g12 ≈ 2π × 1 MHz. When the frequency of the RF field is set close to \({E}_{0}^{1}\), we clearly identify two resonance peaks in spite of tumbling. We have verified that the two resonances are still visible even if we choose σδ to be four times larger. Since \({g}_{12}\propto {d}_{12}^{-3}\), a change in the relative position of the labels significantly modifies the observed spectrum. This is shown in Fig. 3(b) where the second label was displaced such that d12 ≈ 4.03 nm, leading to the disappearance of the splitting. This change in the spectrum certifies a change in the relative position between labels and, by extension, a conformational change in the host molecule.

a Simulated average NV spectrum \({\overline{\langle {\sigma }^{x}\rangle }}_{{{{\rm{NV}}}}}\). Two resonance peaks due to g12 are observed. b Similar to (a) but with lower g12. c Simulated average spectrum \({\overline{\langle {\sigma }^{x}\rangle }}_{{{{\rm{NV}}}}}\) for overlapping label resonances with two 14N isotopes (blue shaded area) and with distinct 14N and 15N isotopes (gray shaded area). d Marginal distribution L(d12∣X) of the inter-label distance for simulated data acquisition using the spectrum shown in (a). The true value of the distance and its posterior expectation, 3.297 nm and 3.54(25) nm, are indicated with vertical lines. The error is the standard deviation of the marginal posterior.
So far, all simulations were performed for nitroxides hosting 14N and having distinct transition energies. If both nitroxides have similar transition energies (e.g., due to similar azimuths), the spectra of the two nitroxides overlap and the inter-label flip-flop terms cannot be neglected. This complicates the interpretation of the spectrum. As shown in Fig. 3(c), this problem is avoided by using distinct 14N and 15N isotopes in each nitroxide. In this case, the E0 branch of the 14N nitroxide never overlaps with the branches of the 15N nitroxide and flip-flop terms can be safely neglected. As a result, the interpretation of the spectrum is much simpler. We emphasize that the purpose of using such orthogonal labels is not merely to resolve their distinct spectral signatures43: rather, it is to simplify the form of the inter-label interaction itself for all label orientations.
Bayesian inference
Finally, we show how to infer the inter-label distance d12 under realistic ambient conditions. We first simulate the experimental acquisition of \({\overline{\langle {\sigma }^{x}\rangle }}_{{{{\rm{NV}}}}}\). The simulated dataset has the form X = {X1, …, XM}, where Xj is an experimental estimate of the probability of measuring σx = + 1 after Nm measurements at RF frequency ωRF,j. Second, we use a simplified model to efficiently extract information from X. The model captures the relevant features of the tumbling-averaged spectrum \(\overline{{{{\mathcal{S}}}}}({\omega }_{{{{\rm{RF}}}}})\). More precisely, we derive the approximate expression \({{{\mathcal{S}}}}({\omega }_{{{{\rm{RF}}}}},\theta ,\beta )={{{{\mathcal{S}}}}}_{0}-{\sum }_{s = +,-}{{{{\mathcal{C}}}}}_{s}{\left[\frac{{{{\Omega }}}_{{{{\rm{RF}}}}}}{{{{\Omega }}}_{s}({\omega }_{{{{\rm{RF}}}}},\theta ,\beta )}\right]}^{2}{\sin }^{2}\left[\frac{\pi }{2}\frac{{{{\Omega }}}_{s}({\omega }_{{{{\rm{RF}}}}},\theta ,\beta )}{{{{\Omega }}}_{{{{\rm{RF}}}}}}\right]\) for a specific nitroxide azimuth θ and a specific angle β between the magnetic field and the line joining the nitroxides. Here, \({{{\Omega }}}_{\pm }^{2}({\omega }_{{{{\rm{RF}}}}},\theta ,\beta )={{{\Omega }}}_{{{{\rm{RF}}}}}^{2}+{\left[{\omega }_{{{{\rm{RF}}}}}-{E}_{0}(\theta )\pm {g}_{12}(\beta )/2\right]}^{2}\) and \({{{{\mathcal{S}}}}}_{0}\) and \({{{{\mathcal{C}}}}}_{\pm }\) are parameters that adjust the baseline and contrast, respectively (see Supplementary Note 3). Moreover, E0(θ) is the 14N energy-transition branch, while \({g}_{12}(\beta )\propto {d}_{12}^{-3}(1-3{\cos }^{2}\beta )\) is the inter-label coupling. Both θ and β depend on the tumbling angle δ. Averaging \({{{\mathcal{S}}}}[{\omega }_{{{{\rm{RF}}}}},\theta (\delta ),\beta (\delta )]\) over a Gaussian distribution for δ gives the tumbling-averaged spectrum \(\overline{{{{\mathcal{S}}}}}({\omega }_{{{{\rm{RF}}}}})\). Assuming that the baseline \({{{{\mathcal{S}}}}}_{0}\) is known, our model contains eight free parameters denoted by V. These include d12 and the unknown standard deviation σδ of the tumbling. It must be emphasized that the light tumbling dependence of the E0 branch enables the determination of the inter-label distance. Indeed, a simple measurement of the line splitting \({g}_{12}\propto {d}_{12}^{-3}(1-3{\cos }^{2}\beta )\) cannot give independent access to the distance d12 and the angle β. This is because β is unknown a priori. However, light tumbling fluctuations allow the NV to probe different geometric configurations of the nitroxides. This results in a small distortion of the line shape that yields information beyond that contained in the splitting g12. This in turn enables the independent extraction of the distance d12 and of the angle β. With the help of our model, we can therefore obtain the posterior probability of the parameters V using Markov Chain Monte Carlo sampling44. Assuming a uniform prior for V, the posterior probability for V is \(L({{{\bf{V}}}}| {{{\bf{X}}}})={\prod }_{j = 1,M}{{{{\rm{e}}}}}^{-{[{X}_{j}-(\overline{{{{\mathcal{S}}}}}({\omega }_{{{{\rm{RF}}}},j})+1)/2]}^{2}/2{\sigma }_{m}^{2}}/\sqrt{2\pi {\sigma }_{m}^{2}}\). Here, the noise variance \({\sigma }_{m}^{2}\approx \left(1-{\overline{\langle {\sigma }^{x}\rangle }}_{{{{\rm{NV}}}}}^{2}\right)/4{N}_{m}\) is assumed to be approximately constant and known. In Fig. 3(d) we show the resulting marginal L(d12∣X) of d12 for a dataset X simulated from the spectrum shown in Fig. 3(a). Here, X was obtained by taking Nm = 2 × 104 ideal measurements for each of M = 25 frequencies ranging from ωRF/2π = 839–843 MHz (we estimate that the same accuracy is achieved with Nm ≈ 5 × 105 for imperfect NV detection efficiency45). The expectation of the marginal posterior is d12 = 3.54(25) nm, close to the real value 3.297 nm [see Fig. 3(d)].
Discussion
Our results open many interesting avenues for future investigation. In particular, it would be of great interest to extend our scheme to molecules with more than two attached spin labels. This would yield a stronger NV response, leading to a more detailed observation of conformational changes as well as to an enhancement in the range of inter-label distances to be estimated. The scheme could also be improved through the use of multiple NVs to better triangulate the label positions. In addition, our analytical understanding of the NV response could enable fast Bayesian inference of molecular dynamical properties. Our results pave the way for the observation of conformational dynamics of individual proteins using tools from magnetic resonance.
Data availability
All the data relevant to this work are available upon request to the authors.
Code availability
All the codes employed are available upon request to the authors.
References
Stetz, M. A. et al. Characterization of internal protein dynamics and conformational entropy by nmr relaxation. Meth. Enzymol. 615, 237–284 (2019).
Rhodes, G. Crystallography Made Crystal Clear (Elsevier, 2006).
Duddeck, H., Dietrich, W. & Tóth, G. Structure Elucidation by Modern NMR. (Steinkopff, Heidelberg, 1998).
Alonso, J. L. & López, J. C. Microwave spectroscopy of biomolecular building blocks. In Top. Curr. Chem., 335–401 (Springer, Cham, 2014).
Abragam, A. The Principles of Nuclear Magnetism. (Oxford University Press, London, 1961).
Levitt, M. H. Spin Dynamics: Basics of Nuclear Magnetic Resonance (Wiley, West Sussex, 2008), 2 edn.
Prisner, T., Rohrer, M. & MacMillan, F. Pulsed EPR spectroscopy: biological applications. Annu. Rev. Phys. Chem. 52, 279–313 (2001).
Schweiger, A. & Jeschke, G. Principles of pulsed electron paramagnetic resonance. (Oxford University Press, New York, 2001).
Borbat, P. P., Costa-Filho, A. J., Earle, K. A., Moscicki, J. K. & Freed, J. H. Electron spin resonance in studies of membranes and proteins. Science 291, 266–269 (2001).
Jeschke, G. DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 63, 419–446 (2012).
Doherty, M. W. et al. The nitrogen-vacancy colour centre in diamond. Phys. Rep. 528, 1–45 (2013).
Wu, Y., Jelezko, F., Plenio, M. B. & Weil, T. Diamond quantum devices in biology. Angew. Chem. Int. Ed. 55, 6586–6598 (2016).
Manson, N. B., Harrison, J. P. & Sellars, M. J. Nitrogen-vacancy center in diamond: Model of the electronic structure and associated dynamics. Phys. Rev. B 74, 104303 (2006).
Steiner, M., Neumann, P., Beck, J., Jelezko, F. & Wrachtrup, J. Universal enhancement of the optical readout fidelity of single electron spins at nitrogen-vacancy centers in diamond. Phys. Rev. B 81, 035205 (2010).
Waldherr, G. et al. Dark states of single nitrogen-vacancy centers in diamond unraveled by single shot NMR. Phys. Rev. Lett. 106, 157601 (2011).
Cai, J., Jelezko, F., Plenio, M. B. & Retzker, A. Diamond-based single-molecule magnetic resonance spectroscopy. New J. Phys. 15, 013020 (2013).
Cai, J. et al. Robust dynamical decoupling with concatenated continuous driving. New J. Phys. 14, 113023 (2012).
London, P. et al. Detecting and polarizing nuclear spins with double resonance on a single electron spin. Phys. Rev. Lett. 111, 067601 (2013).
Wang, Z.-Y. et al. Randomization of pulse phases for unambiguous and robust quantum sensing. Phys. Rev. Lett. 122, 200403 (2019).
Munuera-Javaloy, C., Puebla, R. & Casanova, J. Dynamical decoupling methods in nanoscale NMR. Europhys. Lett. 134, 30001 (2021).
Grotz, B. et al. Sensing external spins with nitrogen-vacancy diamond. New J. Phys. 13, 055004 (2011).
Müller, C. et al. Nuclear magnetic resonance spectroscopy with single spin sensitivity. Nat. Commun. 5, 4703 (2014).
Sushkov, A. O. et al. Magnetic resonance detection of individual proton spins using quantum reporters. Phys. Rev. Lett. 113, 197601 (2014).
Schmitt, S. et al. Submillihertz magnetic spectroscopy performed with a nanoscale quantum sensor. Science 356, 832–837 (2017).
Glenn, D. R. et al. High-resolution magnetic resonance spectroscopy using a solid-state spin sensor. Nature 555, 351–354 (2018).
Bucher, D. B., Glenn, D. R., Park, H., Lukin, M. D. & Walsworth, R. L. Hyperpolarization-enhanced NMR spectroscopy with femtomole sensitivity using quantum defects in diamond. Phys. Rev. X 10, 021053 (2020).
Arunkumar, N. et al. Micron-scale NV-NMR spectroscopy with signal amplification by reversible exchange. PRX Quant. 2, 010305 (2021).
Chu, Y. et al. Precise spectroscopy of high-frequency oscillating fields with a single-qubit sensor. Phys. Rev. Appl. 15, 014031 (2021).
Meinel, J. et al. Heterodyne sensing of microwaves with a quantum sensor. Nat. Commun. 12, 2737 (2021).
Staudenmaier, N., Schmitt, S., McGuinness, L. P. & Jelezko, F. Phase sensitive quantum spectroscopy with high frequency resolution. Phys. Rev. A 104, L020602 (2021).
Ermakova, A. et al. Detection of a few metallo-protein molecules using color centers in nanodiamonds. Nano Lett. 13, 3305–3309 (2013).
Ziem, F. C., Götz, N. S., Zappe, A., Steinert, S. & Wrachtrup, J. Highly sensitive detection of physiological spins in a microfluidic device. Nano Lett. 13, 4093–4098 (2013).
Rendler, T. et al. Optical imaging of localized chemical events using programmable diamond quantum nanosensors. Nat. Commun. 8, 14701 (2017).
Barton, J. et al. Nanoscale dynamic readout of a chemical redox process using radicals coupled with nitrogen-vacancy centers in nanodiamonds. ACS Nano 14, 12938–12950 (2020).
Lovchinsky, I. et al. Nuclear magnetic resonance detection and spectroscopy of single proteins using quantum logic. Science 351, 836–841 (2016).
Shi, F. et al. Single-protein spin resonance spectroscopy under ambient conditions. Science 347, 1135–1138 (2015).
Haugland, M. M., Anderson, E. A. & Lovett, J. E. Tuning the properties of nitroxide spin labels for use in electron paramagnetic resonance spectroscopy through chemical modification of the nitroxide framework. In Electron Paramagnetic Resonance, vol. 25 of SPR - Electron Paramagnetic Resonance, 1–34 (Royal Society of Chemistry, 2016).
Schlipf, L. et al. A molecular quantum spin network controlled by a single qubit. Sci. Adv. 3, 1701116 (2017).
Marsh, D. Bonding in nitroxide spin labels from 14n electric-quadrupole interactions. J. Phys. Chem. A 119, 919–921 (2015).
Marsh, D. Spin-Label Electron Paramagnetic Resonance Spectroscopy. (CRC Press, Boca Raton FL, 2019).
Meyer, V. et al. Room-temperature distance measurements of immobilized spin-labeled protein by DEER/PELDOR. Biophys. J. 108, 1213–1219 (2015).
Lira, R. B., Steinkühler, J., Knorr, R. L., Dimova, R. & Riske, K. A. Posing for a picture: vesicle immobilization in agarose gel. Sci. Rep. 6, 25254 (2016).
Galazzo, L., Teucher, M. & Bordignon, E. Orthogonal spin labeling and pulsed dipolar spectroscopy for protein studies. In Meth. Enzymol., 79–119 (Elsevier, 2022).
Gilks, W. R., Richardson, S. & Spiegelhalter, D. J. Markov Chain Monte Carlo in Practice. (Chapman & Hall/CRC, New York, 1996).
Wan, N. H. et al. Efficient extraction of light from a nitrogen-vacancy center in a diamond parabolic reflector. Nano Lett. 18, 2787–2793 (2018).
Acknowledgements
The authors acknowledge financial support from Spanish Government via PGC2018-095113-B-I00 (MCIU/AEI/FEDER, UE) and, from Basque Government via IT986-16. C.M.-J. acknowledges the predoctoral MICINN grant PRE2019-088519. R.P. acknowledges support from European Union’s Horizon 2020 FET-Open project SuperQuLAN (899354). M.B.P. and B.D. acknowledge the ERC Synergy Grants HyperQ (856432), as well as the BMBF project QSens (03ZU1110FF), and Asteriqs (820394). The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster). J.C. acknowledges the Ramón y Cajal (RYC2018-025197-I) research fellowship, the financial support from Spanish Government via EUR2020-112117 and Nanoscale NMR and complex systems (PID2021-126694NB-C21) projects, the EU FET Open Grant Quromorphic (828826), the ELKARTEK project Dispositivos en Tecnologías Cuánticas (KK-2022/00062), and the Basque Government grant IT1470-22.
Author information
Authors and Affiliations
Contributions
All authors contributed to the main ideas and to the paper writing; C.M.-J. developed the analytical study; C.M.-J., R.P., and B.D. produced the numerical simulations; M.B.P. and J.C. led the work jointly.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Munuera-Javaloy, C., Puebla, R., D’Anjou, B. et al. Detection of molecular transitions with nitrogen-vacancy centers and electron-spin labels. npj Quantum Inf 8, 140 (2022). https://doi.org/10.1038/s41534-022-00653-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41534-022-00653-w
This article is cited by
-
Amplified nanoscale detection of labeled molecules via surface electrons on diamond
Communications Physics (2023)
