
Abstract
Kagami–Ogata syndrome is a rare imprinting disorder and its phenotypic overlap with multiple different etiologies hampers diagnosis. Genetic etiologies include paternal uniparental isodisomy (upd(14)pat), maternal allele deletions of differentially methylated regions (DMR) in 14q32.2 or pure primary epimutations. We report a patient with Kagami–Ogata syndrome and an atypical diagnostic odyssey with several negative standard-of-care genetic tests followed by epigenetic testing using methylation microarray and a targeted analysis of whole-genome sequencing to reveal a 203 bp deletion involving the MEG3 transcript and MEG3:TSS-DMR. Long-read sequencing enabled the simultaneous detection of the deletion, phasing, and biallelic hypermethylation of the MEG3:TSS-DMR region in a single assay. This case highlights the challenges in the sequential genetic testing paradigm, the utility of long-read sequencing as a single comprehensive diagnostic assay, and the smallest reported deletion causing Kagami–Ogata syndrome allowing important insights into the mechanism of imprinting effects at this locus.
Similar content being viewed by others


Introduction
Kagami–Ogata syndrome (KOS) is a rare imprinting disorder of 14q32.2 with an estimated incidence of less than 1 in 1,000,000 liveborn individuals. Over 80 cases of KOS have been reported in the literature with a characteristic phenotype including full cheeks with a protruding philtrum, increased angulation of the ribs, and developmental delay/intellectual disability1. Imprinting disorders are due to changes in the epigenetic phenomena that allow gene expression predominantly from a single parental allele2. Imprinted genes typically form a cluster with differentially methylated regions (DMRs) which regulate expression of the imprinted genes in a parent-of-origin-dependent manner. There are more than 70 imprinting-associated DMRs and each locus has unique transcriptional regulation impacted by imprinting. The human 14q32 imprinting region consists of maternally expressed non-protein-coding genes and paternally expressed protein-coding genes3,4. The maternally expressed genes (MEGs) include two long noncoding RNAs: MEG3 and MEG8. The paternally expressed genes include DLK1 and RTL1. There are two DMRs in the 14q32 imprinted region: intergenic DMR (MEG3/DLK1:IG-DMR) and MEG3:TSS-DMR in the promoter region of MEG3 (Supplemental figure).
Inherited and presumed non-inherited mechanisms can cause imprinting diseases at this locus. MEG3/DLK1:IG-DMR maintains the parent-of-origin-dependent methylation patterns in the body and placenta, while MEG3:TSS-DMR does so only in the body3,4. KOS can be caused by paternal uniparental disomy of chromosome 14, maternal deletion of the 14q32.3 imprinted region, or an epimutation with hypermethylation of the DMRs5. In contrast, maternal uniparental disomy of chromosome 14, paternal deletion of the 14q32.2 imprinted region, or hypomethylation of DMRs cause Temple syndrome which has a distinct phenotype of growth failure, hypotonia, small hands and feet, broad forehead, high arched palate, and premature puberty thus resembling Prader–Willi syndrome and Silver–Russell Syndrome6. The mechanism of disease has important considerations for recurrence risk in future pregnancies.
Here, we report a patient with KOS who was initially diagnosed as having Jeune syndrome and later enrolled in the Undiagnosed Disease Network (UDN). We describe her long diagnostic odyssey including a series of targeted and genome-wide genetic tests, including a negative whole-exome sequencing (WES) and whole-genome sequencing (WGS). A targeted analysis of WGS revealed a de novo 203-bp deletion of the MEG3 transcript and promoter region. Long-read sequencing of the proband enabled the simultaneous detection of the deletion, phasing of the de novo variant, and the methylation status of the MEG3:TSS-DMR locus. The deletion impacts MEG3 expression and demonstrates that loss of MEG3 transcription from the maternal allele leads to hypermethylation of MEG3/DLK1:IG-DMR and MEG3:TSS-DMR. This small deletion refines our understanding of the imprinting mechanism in Kagami–Ogata syndrome.
Results
Case report
A 9-month-old girl presented to the UDN with features felt to be due to Jeune syndrome. She was conceived via in vitro fertilization (IVF) and was born at 31 weeks and 4 days gestation due to premature rupture of the membranes to a 31-year-old gravida 2, para 1 mother. The delivery was vaginal, vertex, and uncomplicated. Polyhydramnios occurred during pregnancy and a 20-week prenatal ultrasound showed fetal hepatosplenomegaly, ventriculomegaly, total body edema, thickened nuchal fold and echogenic kidneys. Amniocentesis was performed for microarray, karyotype, and fluorescence in situ hybridization (FISH) for trisomy 13, 18, and 21 which were all normal.
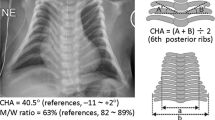
The Apgar scores were 6, 1, and 8 at 1, 5, and 10 minutes of life respectively. The infant required positive-pressure ventilation for cyanosis and apnea. She was noted to have extensive lymphedema, dysmorphic features, and hypotonia on initial exam. Chest X-rays showed a severe bell-shaped chest, handle-bar clavicles, thin ribs, and left lung atelectasis. Abdominal X-ray was notable for hepatomegaly, bilateral acetabular dysplasia, and flaring of iliac wings (Fig. 1). Initial cranial ultrasound was unremarkable except for extensive scalp edema and a spinal ultrasound was concerning for a tethered cord. Cardiac echocardiogram showed patent foramen ovale (PFO), patent ductus arteriosus (PDA), mild right atrial enlargement and a mildly dilated right ventricle with normal systolic wall motion. She passed her newborn hearing test at 3 months old.

A Shortly after birth, the “coat hanger” rib deformities are seen. B Supine view demonstrating the impact of her rib cage and scoliosis on her abdominal content. C A seated view of the impact of the rib cage and scoliosis on her abdominal contents. D The acetabular dysplasia is seen on this view and the progression of the scoliosis. E Toddler and F infant views of the child. Her full cheeks and small chin can be observed. The patient’s family consented to publication of these photographs.
She was admitted to the NICU for 3 months. On the 45th day of life she underwent surgery for placement of a tracheostomy for respiratory failure and gastrostomy tube for dysphagia. Her neonatal course was further complicated by non-ketotic hypoglycemia, hyperinsulinemia, nephrotic syndrome, and MRSA bacteremia and tracheitis. An immune evaluation was performed due to her infections and was normal. After discharge, she was ventilator-dependent for another 6 months. Her neonatal problems resolved, and she started receiving Early Intervention Services: physical therapy, speech therapy, occupational therapy and feeding therapy; with good progress.
Physical exam findings were significant for small chin, low set/posteriorly rotated ears, narrow and bell-shaped chest, rhizomelic appearing limbs, ulnar deviation at wrists, bent 3rd finger on left hand, hepatomegaly, contractures of extensor tendons on dorsum of feet, mild scaphocephaly, horizontal nystagmus, intermittent R > L esotropia, hypertelorism and shallow coccygeal dimple (Fig. 1). There was concern for increasing head circumference and a brain MRI showed ventriculomegaly involving lateral and third ventricles. She also was found to have collapsed long thoracolumbar scoliosis measuring over 50 degrees when she was sitting. Our patient had some features consistent with KOS1 (Supplemental Table) although her rib features were more severe than most published cases.
She is now 46 months old and was decannulated at 27 months of age. Her bell-shaped chest and respiratory status have improved. Her scoliosis continues to progress and is in the moderate range. She is developmentally delayed in gross and fine motor skills but has been walking independently since 20 months of age and said her first words at 12 months of age. Her receptive language is thought to be normal. She is making progress with oral feeds even though she is still dependent on G-tube feeds. After KOS was diagnosed, her medical regimen has been simplified. She recently had a normal abdominal ultrasound and serum α-fetoprotein level of 3 (<20 ng/mL) and will be regularly screened for hepatoblastoma, an unquantified risk in KOS.
Diagnostic evaluations
The patient was seen in the UDN at 24 months of age. Previous post-natal genetic testing included a non-diagnostic skeletal dysplasia panel (Invitae Skeletal Dysplasia Panel) with multiple variants of uncertain significance in 5 different genes (none felt to be candidates), a negative trio WES (GeneDx XomeDxPlus), negative mitochondrial DNA analysis (GeneDx), and a negative deletion/duplication analysis of skeletal dysplasia genes (Custom testing at Division of Medical Genetics, UPMC Children’s Hospital of Pittsburgh). The reanalysis of WES data by the UDN team was non-diagnostic. Upon review by the UDN team, including an ad hoc member specializing in thoracic dystrophy, we concluded that thoracic dystrophy was unlikely, and we expanded our testing to WGS and DNA methylation. The WGS official report was negative but commercial testing of DNA methylation (Greenwood Genetic Center) was abnormal suggesting a methylation defect. Reanalysis of WGS on site revealed a 203 bp de novo deletion (hg38 chr14: 100825924-100826126/NC_000014.9:g.100825924_100826126del) overlapping the MEG3 transcript and the promoter region which was later confirmed by long-read sequencing (Figs. 2 and 3). MEG3 is a noncoding RNA and is typically not covered in WES. The small deletion was not reported on the WGS report as it was below the resolution of the reportable deletions (typically about 3 kb). The short-read sequencing could not establish the parent-of-origin of the 203 bp deletion, therefore long-read sequencing was performed. The MEG3 region was phased using informative heterozygous SNVs (read-based phasing using whatshap) and the parental origin of the two haplotypes were determined using the nearest paternally inherited SNV revealing the deletion on the maternal allele. Methylation profiling using long-read sequencing also revealed that the DNA methylation of the maternal allele was also aberrant. With this information, we established the diagnosis of KOS in this patient, determined that the deletion was apparently de novo, and based on the etiology were able to provide the family with important recurrence risk information.

A A schematic diagram of the deletion revealed though long-read sequencing. The paternal variant is shown in the right-hand section which allowed phasing of the de novo deletion, which is shown in the left block of reads on the maternal allele. Long-reads were phased using heterozygous variants in the genome and the maternal/paternal haplotypes were differentiated using the previously existing parental WGS data. B The methylation profiles from Illumina Epic 850k array. The proband is the red tracing and six controls from a publicly available dataset (GSE153211) are shown in gray. C The phased methylation probabilities identified from PacBio data. The solid lines show the patient data while an internal control is shown as dashed lines. The patient data shows the hypermethylation of maternal allele confirming the diagnosis of Kagami–Ogata syndrome. D Visualization of methylation probabilities along the reads in the MEG3:TSS-DMR region. Each line represents a read and regions shaded in red are methylated while blue are unmethylated. Unshaded regions denote the lack of methylation data. CpG islands in the region are displayed as green horizontal bars.

A comparison of the 203 bp de novo deletion in our patient is shown in red (hg38 chr14: 100825921-100826123/NC_000014.9:g.100825921_100826123del) and previously reported deletions causing Kagami–Ogata syndrome. Paternally expressed genes are labeled in blue. The length of each deletion is specified on the right.
Discussion
The patient reported here exemplifies many of the pitfalls in establishing a correct diagnosis. Physicians anchored on her chest phenotype, leading to a tentative diagnosis of Jeune syndrome and delaying more genome-wide approaches. Standard-of-care WES and WGS analyses failed to identify the 203 bp deletion involving the promoter region which extended to the exon 1 of MEG3 highlighting the analytic challenges. The MEG3 gene is a noncoding RNA and typical exome sequencing assays may not include baits targeting noncoding RNAs. Single exon copy-number variants are challenging to detect using exome sequencing and first exons are even more challenging due to the high GC content. Similarly, the size of this deletion (203 bp) makes it very challenging to detect with WGS as well. Most of the algorithms to detect copy number variants from WGS use mean depth of coverage across evenly divided bins of a certain size (typically 1 to 5 kb) and a 203 bp deletion is under the reporting limits of most of the read-depth based copy number algorithms. The structural variant callers that use split-read alignments can potentially detect a deletion of this size, but they are known to be enriched for false positives making the prioritization of this variant extremely challenging. Long-read sequencing has several advantages compared to the short-read sequencing assays including WES and WGS. Long-read sequencing can detect all major classes of variants in a more comprehensive manner and methylation with phasing in a single assay. In this patient, long-read sequencing established that the deletion was on the maternal allele, found the aberrant methylation at this locus and confirmed the diagnosis in a single experiment. While long-read sequencing has the potential to become the single most comprehensive assay, some challenges remain. It is challenging to perform a hypothesis free, genome-wide analysis using long-read sequencing data as it has some of the same analytical challenges as WGS short-read sequencing data analysis and lacks resources such as large population scale databases which may aid in variant prioritization (e.g., gnomAD). While it is possible to phase and differentiate two haplotypes using informative heterozygous SNVs, determining the parent-of-origin requires informative parental variants SNVs or known imprinting signatures.
The prognosis of KOS reflects the respiratory status primarily and most reported deaths occur early in life1. Several patients have had hepatoblastoma and one died of hemophagocytic syndrome while under treatment for hepatoblastoma, however, the true frequency of hepatoblastoma in KOS is not known. Thus, current recommendations are to screen with alpha fetoprotein levels every three months until 4 years of age1. The child is currently doing well but faces surgery for scoliosis.
Maternal allele deletions have been recognized as causal for KOS since 20104 with the smallest previous reported size of 5.8 kb for disease3. Mapping of a nested deletion series led to the hypothesis of a hierarchy of effects where the MEG3/DLK1:IG-DMR was presumed to regulate the MEG3:TSS-DMR methylation on the maternal allele3. Expression of MEG3 is downregulated by EZH2, HDAC1, and DNMT1 in cancer suggesting that MEG3 expression is downstream of the DMRs post-embryologically7. In stem cells, MEG3 regulates EZH2-mediated repression of DLK1 on the maternal allele8. There are additional miRNAs and snoRNAs downstream of MEG3 which have been hypothesized to contribute overall to imprinting and this deletion does not encompass them, further isolating the role of MEG39 (Supplemental Figure). The structure of the MEG3 transcript is important and the GC content drives the highly complex structure felt to be important in protein binding10. Many of the effects are presumed to be driven during early development where promoters marked with H3K27me3 gain DNA methylation during embryologic differentiation11,12,13. Our patient’s small deletion impacting the MEG3 promoter and transcript demonstrates that MEG3 transcripts are required to prevent methylation of both the MEG3/DLK1:IG-DMR and MEG3:TSS-DMR and suggests that the snoRNAs and miRNAs at this locus have a much lesser role in DMR methylation and in the clinical phenotype.
This case was instructive due to the very small deletion, essentially comprising the promoter and proximal exon of MEG3. Imprinting disorders are due to changes in the epigenome either secondary to DNA mutations in a cis or trans arrangement or primary epimutations in the absence of recognized DNA sequence changes2. Although epimutations are heritable in subsequent cell divisions, they are not heritable across generations and thus have a distinct inheritance pattern. The distinction between the possible etiologies of imprinting disorders has important implications for the families. In spite of great effort to establish a diagnosis using clinically available testing, this child remained without a definitive diagnosis, hindering assessments of prognosis, and impairing medical decision making regarding best treatments. Our finding of a small deletion impacting MEG3 transcription is important because lack of identification would have led to improper recurrence risk assessment. This also raises the concern that some patients felt to have epimutations might have very small or even single nucleotide variants hypothetically that could be heritable as causes of KOS.
Methods
Ethics
The patient’s family self-enrolled in the UDN and consented to evaluation and sharing of results. This program is NIH-funded and was reviewed by the NIH Institutional Review Board. The child’s parents provided written informed consent for her participation and their own participation. The authors affirm that human research participants provided informed consent, for publication of the images in Fig. 1 and provided consent to publish.
Molecular genetic analysis
DNA on the proband and parents was extracted from whole blood, as per UDN protocol (Promega blood DNA kit AS1400 on the Promega Maxwell instrument, Promega, Madison, WI). Aliquots were used for unbiased DNA methylation analysis (EpiSign, Greenwood Genetic Center), WGS, and long-read sequencing. The WGS was performed at Baylor Human Genome Sequencing Center using a PCR-free library preparation with 500 bp insert size according to the KAPA Hyper Prep kit (Roche, Indianapolis, IN). Sequencing was performed on Illumina Novaseq 6000 platform for 150 bp paired end reads to achieve a minimum average coverage of 60x. Short-read sequencing data was aligned using BWA and GATK best practices were used to produce variant calls14.
PacBio long-read sequencing was performed at the Genomics Core Facility at Icahn School of Medicine using Sequel IIe platform using three SMRTcells to achieve 20x mean coverage across the genome. Raw PacBio sequence reads were aligned to the reference genome hg38 using pbmm215, and the DeepVariant16 variant caller was used to detect single nucleotide variants and small insertion deletions. The pbsv software tool17 was used to call structural variants and whatshap18 was used to phase variants using informative genotypes. Methylation probabilities were computed using ccsmeth/CpG-tools19,20,21 and visualized using modbamtools and IGV22,23. A targeted analysis of MEG3 locus was performed in both short-read and long-read sequencing data. Parental short-read sequencing data was used to determine the parent-of-origin of the apparently de novo variant in the proband.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data is available upon request to the corresponding author and has been deposited in dbGaP (see below). Accession numbers for sequence data are: Phenome Central: P0013787 (https://www.phenomecentral.org/). dbGaP: phs001232.V5.p2 (https://www.ncbi.nlm.nih.gov/gap/).
References
Sakaria, R. P. et al. Kagami-Ogata syndrome: case series and review of literature. AJP Rep. 11, e65–e75 (2021).
Eggermann, T. et al. Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin. Epigenetics 7, 123 (2015).
Beygo, J. et al. Novel deletions affecting the MEG3-DMR provide further evidence for a hierarchical regulation of imprinting in 14q32. Eur. J. Hum. Genet. 23, 180–188 (2015).
Kagami, M. et al. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 6, e1000992 (2010).
Kagami, M. et al. Genome-wide multilocus imprinting disturbance analysis in Temple syndrome and Kagami-Ogata syndrome. Genet. Med. 19, 476–482 (2017).
Kagami, M. et al. Temple syndrome: comprehensive molecular and clinical findings in 32 Japanese patients. Genet. Med. 19, 1356–1366 (2017).
Ye, M. et al. Downregulation of MEG3 and upregulation of EZH2 cooperatively promote neuroblastoma progression. J. Cell. Mol. Med. 26, 2377–2391 (2022).
Sanli, I. et al. Meg3 non-coding RNA expression controls imprinting by preventing transcriptional upregulation in cis. Cell Rep. 23, 337–348 (2018).
Budkova, Z. et al. Expression of ncRNAs on the DLK1-DIO3 locus is associated with basal and mesenchymal phenotype in breast epithelial progenitor cells. Front Cell Dev. Biol. 8, 461 (2020).
Uroda, T. et al. Conserved pseudoknots in lncRNA MEG3 are essential for stimulation of the p53 pathway. Mol. Cell 75, 982–995.e989 (2019).
Ohm, J. E. et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 39, 237–242 (2007).
Schlesinger, Y. et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 39, 232–236 (2007).
Mohn, F. et al. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 30, 755–766 (2008).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11 10 11–11 10 33 (2013).
Biosciences, P. pbmm2, https://github.com/PacificBiosciences/pbmm2. (2023).
Poplin, R. et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983–987 (2018).
Biosciences, P. pbsv, https://github.com/PacificBiosciences/pbsv. (2023).
Martin, M. et al. WhatsHap: Fast and Accurate Long Read-based Phasing (2023).
Tse, O. Y. O. et al. Genome-wide detection of cytosine methylation by single molecule real-time sequencing. Proc. Natl. Acad. Sci. USA. https://doi.org/10.1073/pnas.2019768118 (2021).
Akbari, V. et al. Megabase-scale methylation phasing using nanopore long reads and NanoMethPhase. Genome Biol. 22, 68 (2021).
Biosciences, P. pb-CpG-tools, https://github.com/PacificBiosciences/pb-CpG-tools. (2023).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Razaghi, R. et al. Modbamtools: Analysis of Single-molecule Epigenetic Data for Long-range Profiling, Heterogeneity, and Clustering (2023).
Acknowledgements
The authors gratefully acknowledge the patient and her family, Matthew Deardorff for review of imaging, and the many staff who contribute to the functioning of the Undiagnosed Diseases Network. These studies were funded in part by Children’s Hospital of Philadelphia and in part through the Undiagnosed Diseases Network grant U01 HG010219.
Author information
Authors and Affiliations
Consortia
Contributions
K.I., K.E.S., R.R., Z.Z., and L.C. collected and analyzed the sequencing data. K.H., E.M.B., M.F., A.V., D.R., P.J.C., A.R., D.W., and T.J. analyzed additional data. G.K. wrote the initial draft and R.R. and K.E.S. revised the final draft with approval from all authors. The Undiagnosed Disease Network, listed as collaborators in the Supplemental Data, provided the infrastructure for this research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A list of members and their affiliations appears in the Supplementary Information.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kilich, G., Hassey, K., Behrens, E.M. et al. Kagami Ogata syndrome: a small deletion refines critical region for imprinting. npj Genom. Med. 9, 5 (2024). https://doi.org/10.1038/s41525-023-00389-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-023-00389-2
