
Abstract
Exome sequencing is utilized in routine clinical genetic diagnosis. The technical robustness of repurposing large-scale next-generation sequencing data for pharmacogenetics has been demonstrated, supporting the implementation of preemptive pharmacogenetic strategies based on adding clinical pharmacogenetic interpretation to exomes. However, a comprehensive study analyzing all actionable pharmacogenetic alleles contained in international guidelines and applied to diagnostic exome data has not been performed. Here, we carried out a systematic analysis based on 5001 Spanish or Latin American individuals with diagnostic exome data, either Whole Exome Sequencing (80%), or the so-called Clinical Exome Sequencing (20%) (60 Mb and 17 Mb, respectively), to provide with global and gene-specific clinical pharmacogenetic utility data. 788 pharmacogenetic alleles, distributed through 19 genes included in Clinical Pharmacogenetics Implementation Consortium guidelines were analyzed. We established that Whole Exome and Clinical Exome Sequencing performed similarly, and 280 alleles in 11 genes (CACNA1S, CYP2B6, CYP2C9, CYP4F2, DPYD, G6PD, NUDT15, RYR1, SLCO1B1, TPMT, and UGT1A1) could be used to inform of pharmacogenetic phenotypes that change drug prescription. Each individual carried in average 2.2 alleles and overall 95% (n = 4646) of the cohort could be informed of at least one actionable pharmacogenetic phenotype. Differences in variant allele frequency were observed among the populations studied and the corresponding gnomAD population for 7.9% of the variants. In addition, in the 11 selected genes we uncovered 197 novel variants, among which 27 were loss-of-function. In conclusion, we provide with the landscape of actionable pharmacogenetic information contained in diagnostic exomes, that can be used preemptively in the clinics.
Similar content being viewed by others


Introduction
Adverse Reactions to Drugs (ADRs) and therapeutic failure are major public health care problems1,2. Over 200 genes have been associated with therapy response and are recognized as pharmacogenetic genes3 and between 80 and 99% of individuals are estimated to carry genetic variants that modify drug response4,5. Pharmacogenomic information is included in drug labels and several clinical guidelines have been elaborated to adjust drug prescription to the genetic background of the individual (e.g. by the Clinical Pharmacogenetics Implementation Consortium-CPIC or by the Dutch Pharmacogenetics Working Group-DPWG)6,7,8,9. However, the implementation of pharmacogenetics in the clinics is still low.
Genotyping-based technologies are useful strategies for reactive pharmacogenetics, by guiding the use of particular medications or explaining specific ADRs10,11,12,13. They also promote preemptive pharmacogenetics4, but the recent explosion of next-generation sequencing (NGS) techniques creates a unique opportunity to accelerate this field14. Whole exome sequencing (WES) or whole-genome sequencing (WGS) are suitable approximations for repurposing available sequencing data and report pharmacogenetic actionable alleles4,10,14,15,16,17, while they can also uncover novel potentially relevant variants18,19. The accuracy of NGS for detection of pharmacogenetic variants is elevated, showing high concordance with genotyping techniques10,14,17, and customized pharmacogenetic NGS panels and WGS can resolve the full set of actionable pharmacogenetic alleles17,20,21, including copy number variations (CNVs) in important pharmacogenes such as CYP2D614,18,21,22,23,24,25,26. Exome sequencing is limited by design (i.e. lack of non-coding regions, low resolution for CNVs, and incomplete HLA-typing accuracy), but at the moment, the large-scale NGS techniques most commonly used for diagnosis are WES together with the so-called Clinical Exome Sequencing (CES), which are smaller designs targeting disease-causing genes (typically, 12–17 Mb compared to 50–60 Mb of WES)27,28,29. Diagnostic WES and CES provide an extraordinary opportunity to repurpose this data to recover actionable pharmacogenetic information and boost preemptive pharmacogenomic testing.
After some pioneer studies with small sample sizes and/or lacking phenotype assignments14,15,17, two recent large biobank studies used research-oriented data to compare the pharmacogenetic allele resolution of genotyping arrays, WES and WGS10, and to investigate the frequency of pharmacogenetic alleles in the WES of 50,000 individuals in the UK Biobank30. Regarding diagnostic WES, a study in >1500 individuals investigated the feasibility of extracting 42 selected variants in 11 pharmacogenes16, and two additional studies with >1000 individuals explored the secondary use of WES for pharmacogenomics in specific patient groups31,32. However, these studies have missed some clinically relevant pharmacogenes (e.g. CACNA1S10,14,15,16,31, CYP2B6 and UGT1A131,32, CYP4F2 and G6PD15,16, NUDT15 and RYR114,15,16), they are population-specific10,30,32, and none has investigated the potential utility of CES. Therefore, a systematic analysis including all pharmacogenetic variants defined as actionable by international consensus, within a routine clinical diagnostic WES context, is still lacking.
In this study, we performed a comprehensive pharmacogenetic analysis in 5001 individuals that underwent WES or CES for genetic diagnosis in Spain and Latin American countries, by extracting and analyzing the 788 pharmacogenetic alleles defined in any of the 25 Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines currently available. In addition, we investigated differences in allele frequencies among populations and extracted novel loss-of-function (LOF) variants to estimate their contribution to the different pharmacogenetic phenotypes.
Results
Study cohort
We retrospectively collected pharmacogenetic data from 5001 unrelated individuals that underwent routine WES or CES aimed at medical diagnosis of diverse hereditary conditions (NIMGenetics, Madrid, Spain; between July 2017 and May 2019) using two frequently used commercial library kits (n = 4002 WES and n = 999 CES; see Methods section for platform details). 53.8% of the cases were females (n = 2690), 44.6% males (n = 2231), and for 1.6% (n = 80) gender was unknown. Regarding the country of origin, 2862 individuals (57.2%) were from Spain, 2016 (40.3%) were from Latin America (1342 Colombia, 568 Brazil, 92 Mexico, 11 Ecuador, 1 Argentina, 1 Chile and 1 from an unknown location in Latin America), 40 (0.01%) were from other diverse countries and 83 individuals (1.7%) were from an unknown location.
Selected pharmacogenetic alleles with clinical actionability
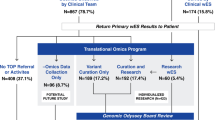
Figure 1 summarizes the workflow that was followed for allele selection. All pharmacogenetic alleles described in any of the 25 available CPIC clinical guidelines (https://cpicpgx.org/guidelines/; accessed in July 2021) were selected for the study (788 alleles distributed in 19 genes). We filtered out those with no application in the clinics (with normal function, with unclear/uncertain/unknown impact in function or with a limited/inadequate strength of evidence, when available; 345 alleles discarded and 443 retained), then we identified alleles defined by intronic, intergenic or upstream regions variants not present in the exome data (5001 exomes analyzed; 33 alleles discarded and 410 retained). At this filtering step, no differences were observed between WES and CES. At this point, we reasoned that 8 genes (CYP2C19, CYP3A5, IFNL3, VKORC1, CYP2D6, HLA-A, HLA-B, and CFTR) could not be fully informed because (i) a major actionable allele was absent in exome data (CYP2C19*17-promoter region, CYP3A5*3-deep intronic, IFNL3 rs12979860-non-coding region and VKORC1 rs9923231-promoter region) and this would lead to an incomplete pharmacogenetic report with no associated clinical recommendation; (ii) an important actionable allele is a Copy Number Variant (CYP2D6) or is a HLA allele (HLA-A and HLA-B) which cannot be accurately resolved by exome data. Partial gene deletions in CYP2B6 (CYP2B6*29 and CYP2B6*30 alleles) cannot be detected by exome, but due to their rarity in tested populations (<1% in African American and Asians individuals)33,34, this gene was not excluded from the analysis13; and (iii) these were disease causative alleles (CFTR) that are out of the objectives of preemptive pharmacogenetics. Therefore, we filtered out all the alleles in these 8 genes, and retained a total of 280 alleles distributed in 11 pharmacogenes (CACNA1S, CYP2B6, CYP2C9, CYP4F2, DPYD, G6PD, NUDT15, RYR1, SLCO1B1, TPMT, and UGT1A1) that were considered suitable to generate a comprehensive exome-based pharmacogenetic report and were kept for the analysis (Fig. 1).

*Evidence: “evidence level” is obtained from CPIC “allele functionality” tables. This information is only available for CYP2C19, CYP2C9, and DPYD genes.
Sequencing depth of coverage data and quality control
We performed a sequencing coverage quality control (QC) of all genetic loci required to resolve the 280 actionable alleles (Supplementary Fig. 1A). All interrogated loci were covered by both exome designs, despite we observed some differences between WES and CES for some genes. In CES there was a lower coverage in CACNA1S caused by the variant rs772226819 (40% of samples covered <40x, although 99.5% had >20x). In WES there was a lower coverage in TPMT at variant rs1142345, defining TPMT*3C and TMPT*3A alleles (80% of samples <40x, although 90% had >20x). The gene with the lowest coverage was UGT1A1, especially in CES, due to the indel variant rs34983651, which defines the alleles UGT1A1*28, *36, and *37 (the percentage of samples covered >20x were 93 and 34% for WES and CES, respectively). Of note, CYP2B6 and CYP2C9, with the lowest level of sequence uniqueness (Supplementary Fig. 1B), did not show lower coverage nor evidence of potential calling errors in our analysis. Overall, an average coverage >20x was observed for all loci (except for rs34983651) in 99.3% of the samples, indicating that repurposing of diagnostic WES and CES data for clinical pharmacogenomics is a suitable approximation for the selected genes.
Pharmacogenetic diplotypes
After variant genotype to allele diplotype conversion, 96.4% (n = 4823) of individuals harbored one or more actionable alleles (Fig. 2a). The average number of actionable alleles per individual was 2.2, with 6 individuals having 7 actionable alleles. Most individuals (79.3%) had 2 or more pharmacogenetic alleles, which illustrates the high pharmacogenetic heterogeneity (Supplementary Fig. 2), and 55% of the cohort carried either one (n = 2735) or two (n = 11) alleles defined by indel variants (Fig. 2a). Regarding the different genes involved, UGT1A1 and CYP2B6 were the two genes that contributed the most to the total alleles called, followed by CYP4F2, CYP2C9, and SLCO1B1, while a low number of alleles were observed for TPMT, DPYD, NUDT15, G6PD, and RYR1, and no individuals were found to carry any CACNA1S actionable allele (Fig. 2b).

a Colored stacked bar plots show the percentage of individuals which carry none, one, or multiple alleles for SNVs and indel variants. b Pie chart showing the percentage of alleles found per gene out of the total number of actionable alleles (n = 12,309) in the whole cohort (n = 5001 individuals). c For each actionable gene, a group of four stacked bar plots depicts alleles distributions within all (n = 5001) individuals, the Spanish, Colombian, and Brazilian subgroups, respectively. Y-axis labels represent the number of carriers in each specific group. For each allele depicted, the fraction of compound heterozygous (CH) and homozygous (Homoz) individuals is represented in gray bars versus remaining individuals in white bars. “Other” indicates low prevalent alleles represented together. The full list of alleles per gene can be found in Supplementary Data 1. Some allele names in G6PD gene (marked with an asterisk) have been shortened: Seattle Lodi Modena FerraraII Athens-like: Seattle*, Mediterranean Dallas Panama‚Sassari Cagliari Birmingham: Mediterranean*, Union Maewo Chinese-2 Kalo: Union*, G6PDA-968C-376G: A-968C-376G.
The most frequent alleles in each gene are shown in Fig. 2c, for the whole cohort and for the largest populations analyzed (Spain, Colombia, and Brazil), together with the frequency in which those alleles are found in homozygosity or compound heterozygosity (gray bars in Fig. 2c). In CYP4F2 and NUDT15 only one or two actionable alleles were detected (CYP4F2*3, NUDT15*2, and NUDT15*3, respectively). In SLCO1B1, UGT1A1, and CYP2B6, one allele was far more frequent than the rest (SLCO1B1*15, UGT1A1*28, and CYP2B6*6, respectively), with CYP2B6 having a significant proportion of less frequent alleles. In TPMT, the most common allele was TPMT*3A, followed by TPMT*2 in Spain or TPMT*3C in Colombia. In CYP2C9, the dominating alleles were CYP2C9*2 and CYP2C9*3, but additional minor alleles were detected, especially in Latin American population (10.6% and 9.9% in Colombia and Brazil versus 4.2% in Spain). In DPYD, the two most frequent alleles were DPYD HapB3 and DPYD c.2846A > T across the populations studied, followed by DPYD*2A in Spain and by c.557A > G in Colombia and Brazil. RYR1 and G6PD genes were the more heterogenous genes, with diverse and low prevalent alleles across populations, in the case of G6PD with -202A-376G being the most common allele. The full list of alleles per individual is provided in Supplementary Data 2.
We compared the frequency of variants found in Spain and Latin America and the corresponding gnomAD population (Supplementary Fig. 3). Among high-frequency variants with differences, SLCO1B1 rs2306283 (present in several SLCO1B1 alleles) had the highest frequency in Brazil, CYP4F2 rs2108622 (*3) in Spain and CYP2B6 rs3745274 and rs2279343 (*6) in Colombians. Among medium and low-frequency variants, several G6PD variants had a large variability among populations, TPMT rs1800462 (*2) had the highest frequency in Spain and DPYD rs115232898 (c.557A > G) had higher frequencies in our populations (i.e. 0.00087 versus 0.000053 for Spain and gnomAD NFE; 0.003 versus 0.00078 for Latin American countries and gnomAD AMR) suggesting a contribution from Sub-Saharan Africa in our cohorts35. Differences with CPIC European and Latino allele frequency data is presented in Supplementary Fig. 4.
Pharmacogenetic phenotype analysis
Allele diplotype to phenotype conversion revealed that 4646 individuals (95.1%) had a clinical actionable pharmacogenetic phenotype that could be reported based on exome data (Supplementary Fig. 5B). The proportion of individuals with an actionable phenotype differed according to the gene, with the highest numbers corresponding to UGT1A1, CYP2B6, and CYP4F2, followed by CYP2C9 and SLCO1B1 (Table 1 and Supplementary Fig. 5B). When only the more extreme phenotypes were considered (those in which an alternative drug is recommended due to an increased risk of severe/fatal toxicity, i.e. CYP2C9 PM (Poor Metabolizer), DPYD PM, G6PD deficient, NUDT15 PM, RYR1 MHS (Malignant Hyperthermia Susceptibility) carriers, SLCO1B1 LF (Low Function), and TPMT PM), these were present in 5.5% of the individuals.
Comparing the phenotypes of Spanish and the Latin American individuals, some statistically significant differences were observed in four genes (Table 1 and Supplementary Fig. 5C). For example, NUDT15 Intermediate Metabolizer (IM) phenotype was higher in Latin America than in Spain (4.5% versus 1%, p < 0.00001; Supplementary Fig. 5C; and Table 1), while CPIC indicates frequencies of 8% and 0.8% for Latino and in European populations, respectively36. The combination of TPMT and NUDT15 phenotypes resulted in 15 individuals being IMs for both genes, the majority being from Latin America (0.5% versus 0.1% in Spain).
Novel Loss-of-Function variants
In the eleven pharmacogenes analyzed in the exomes, we retrieved a total of 1012 variants, 19.5% of which were novel (167 missense, 11 frameshift, 7 canonical splice site, 8 stop gain, and 4 inframe variants) (Fig. 3a, b and Supplementary Data 3). As expected, the average allele frequency of the novel variants was lower than that of the known variants (0.00025 and 0.028, respectively, p value = 0.02) and 99% of the novel variants were found in one single country (either Spain, Colombia or Brazil), compared to 71% for known variants. Differently from the novel missense variants, which have an unknown effect in protein activity, LOF variants are expected to lead to a non-functional protein. The 26 novel LOF variants found were estimated to increase in average 0.54% the total number of actionable alleles, with a greatest impact on G6PD and DPYD and the lowest in UGT1A1 and TPMT (3.4%, 1.1% and 0.04%, 0%, respectively; Fig. 3c). It is important to note that for the calcium release channels RYR1 and CACNA1S the nonsense variants found have unknown significance, as only specific missense variants contribute to MHS. The full list of known and novel variants found in the selected list of pharmacogenes can be found in Supplementary Data 3.

a Two stacked bar plots showing the number of known variants (left, variants reported in gnomAD or dbSNP databases) and novel variants (right, not reported in these databases). Blue, red and green stacked bars represent variants found exclusively in Spanish, Colombian, or Brazilian individuals, respectively, and yellow for variants found in a mixture of individuals from different countries. b Pie chart summarizing the fraction of different novel variants (missense, in-frame, frameshift, splice, or start/stop gained/lost). c Bar plots representing the number of novel LOF variants per gene. The percentages over each bar is an estimation of the contribution of the novel LOF in each gene over the total number of actionable alleles previously found. LOF variants in CACNA1S and RYR1 have not been associated with increased risk of malignant hyperthermia, thus, the contribution of them to this phenotype is not applicable (NA).
Discussion
Repurposing diagnostic exome data for clinical pharmacogenomics has the potential to globally change drug prescription. WGS is taking the lead for genomics medicine37, but WES and CES provide with an invaluable resource38,39,40 and a unique opportunity to implement pharmacogenetics, since they represent a competitive option among the large-scale NGS techniques used in the clinic41,42. In this study, by performing a systematic analysis of all pharmacogenetic alleles included in CPIC guidelines in 5001 exomes, we illustrate the pharmacogenetic actionable landscape contained in routine diagnostic WES and CES data.
Analyzing the 19 pharmacogenes included in CPIC guidelines revealed that 11 of these genes can be comprehensively informed (CACNA1S, CYP2B6, CYP2C9, CYP4F2, DPYD, G6PD, NUDT15, RYR1, SLCO1B1, TPMT, and UGT1A1) using WES or CES data. These genes are included in eight guidelines that describe relevant changes in therapeutic management of patients treated with volatile anesthetic agents and succinylcholine, antiretroviral therapy with efavirenz and atazavir, nonsteroidal anti-inflammatory drugs, fluoropyrimidines and thiopurines, rasburicase, and statins (see Supplementary Data 4). In addition, phenytoin and warfarin guidelines are partially covered: in the first case, informing about the recommended drug doses but missing the identification of patients with increased risk of hypersensitivity reactions43,44 and in the second case, missing VKORC1 genotype information precludes comprehensive estimation of warfarin dose44. Thus, the pharmacogenetic information that is easily retrieved from exome data, is crucial to prevent drug adverse reactions (e.g. life-threatening malignant hyperthermia when treated with inhalation anesthetics or severe/fatal drug toxicity when treated with fluoropyrimidine drugs) and to guide drug dosing adjustments (e.g. to stabilize phenytoin plasma concentration within the targeted therapeutic range). Although data for seven relevant pharmacogenes (CYP2C19, CYP3A5, CYP2D6, HLA-A, HLA-B, IFNL3, VKORC1) could not be retrieved from exome data, the implementation of pharmacogenetics in the clinics is still low and repurposing routinely generated diagnostic WES/CES data to provide, without laboratory testing, the actionable phenotypes of 11 crucial pharmacogenes, is a step forward in preemptive pharmacogenetics.
Repurposing exome data for clinical preemptive pharmacogenomics requires the same QC that applies to any other genetic diagnostic test. To ensure this, first, we performed a depth of coverage analysis for each pharmacogenetic actionable allele and retrieved the genotype information in all loci. This step avoids misleading genotype data derived from low coverage. In the 11 selected genes, 99.3% actionable variants were covered at a standard diagnostic coverage threshold of 20x in both WES and CES data45. The genetic loci of the variant rs34983651, which informs UGT1A1*28, *36, and *37 alleles, was the only exception as it achieved a lower coverage in the CES data, that would result in UGT1A1 gene exclusion in a substantial number of cases (22%). Some previous studies marked UGT1A1*2816 as a low-coverage allele also in WES, but we and others21 have reported them with high quality. Differences are probably due to technical reasons related to the panel design. Furthermore, the performance of other exome platforms for this position located outside the coding region might differ, marking this indel as a complex variant that requires specific analysis. Second, indel variants are an additional bioinformatic challenge and they constitute 21 (7%) of the actionable pharmacogenetic alleles interrogated. Thus, we manually reviewed them from the variant calling output, recovering high frequency (e.g. UGT1A1*28, *37, and *36) and low-frequency indels (e.g. CYP2C9*6 and DPYD*7). This step was necessary to avoid missing indel variants that were called differently due to reasons such as read sequence alignment30 (e.g. UGT1A1*28 can be called as C > CAT or CAT > CATAT). Indel normalization or a robust characterization of indel annotation by the variant callers could be strategies to automate this process. Third, diplotype translation according to CPIC definitions required manual curation for ambiguous calls and haplotype phasing, notably in CYP2B6, TPMT, and G6PD. These three highlighted issues are being addressed by novel bioinformatic approaches30,46 to facilitate automatization. In addition, although WGS and long-read sequencing face their own technical problems (e.g. coverage issues), repurposing their data for preemptive pharmacogenomics may help to overcome some issues derived from exome sequencing7,19,46,47,48,49,50.
Ancestry data is relevant for pharmacogenetic implementation, as several actionable alleles are subjected to important ethnic differences51. International efforts are contributing to expand country-based pharmacogenetics data10,16,30,32. However, in comparison to countries with national genomics medicine initiatives, the access of Latin American countries to NGS is still limited11. In this study, we compared variant frequencies in Spain, Colombia and Brazil and evaluated deviations from their closest gnomAD population, as this database is frequently used as reference in genetic studies35. Relevant differences included the variants defining CYP2B6*6, CYP4F2*3 and DPYD c.557 A > G alleles; which had higher frequencies in our studied populations than in reference populations previously reported35,52,53. Although our study is limited by using the country for the diagnostic test rather than ethnic group data in the analysis, from a healthcare point of view it is the pharmacogenetic landscape in each country, with current population admixture context, what will be most relevant to design genetic testing.
We and others have previously estimated the contribution of NGS to the detection of novel variants in pharmacogenes24,25,30. In this study, we complement these studies analyzing diagnostic exome data, since novel LOF variants detected by NGS will change the pharmacogenetic phenotype classification of the individual. For the 11 pharmacogenes that can be informed using exome data, the novel LOF variants contributed in average 0.54% to the total of known actionable alleles. However, most of the novel variants detected were missense variants (87%; Fig. 3b), part of which will affect protein activity and will contribute to the variability in drug response. In silico protein activity predictions are improving19, however, these evidences are still insufficient for clinical application and, thus, these rare missense variants have to be classified as of unknown significance. Beyond their importance for pharmacogenetics implementation, the incremental use of Electronics Health Records combined with prospective and retrospective genomics data and the development of novel computations tools predicting variant consequences, will help to elucidate the clinical impact of these variants and will reinforce the use of NGS13,40.
In conclusion, the high number of actionable pharmacogene alleles carried by the individuals and the urgent need for safer and more efficient personalized treatments, support the implementation of pharmacogenetics in genomic medicine12. The exponential growth in large-scale NGS diagnostics, with exomes being the most widely used platforms, argue for repurposing these data for clinical pharmacogenetics. The landscape of high-quality pharmacogenetic information that can be extracted from exomes and used to adjust drug treatments according to international guidelines, together with population-specific allele variations and estimations of the contribution of novel variants identified by NGS, all provided in this study, will aid in removing barriers and facilitating the clinical implementation of pharmacogenetics.
Methods
Study cohort and Whole Exome Sequencing/Clinical Exome Sequencing
We retrospectively collected NGS data from 5001 unrelated individuals who had undergone genetic testing aimed at medical diagnosis of diverse hereditary conditions at NIMGenetics´s genetic laboratory (28108 Madrid, Spain; https://www.nimgenetics.com/). While per-sample NGS data and sensitive information had been previously anonymized, cohort information included gender, country of origin for NGS testing, and trio availability. DNA samples were not available. The study was approved by the ethics committee of the ISCIII (IRB nr. CEI PI 105_2020-v2). As the study was performed on a retrospective set of anonymized samples, the approval from the ethics committee was obtained without the requirement of a specific consent inform from each patient for this study.
DNA libraries were prepared using a standard-size WES kit “SureSelect Human All Exon v6 kit” 60 Mb (Agilent, Santa Clara, CA, USA) for 4002 individuals, or with a CES kit “SureSelect Custom Constitutional Panel 17 Mb kit” (Agilent, Santa Clara, CA, USA) for 999 individuals. Paired-end sequencing was performed in a NovaSeq 6000 S2 Flow Cell (Illumina, San Diego, CA, USA). Bioinformatics analysis for germline SNVs and indel variant calling was performed following GATK Best Practices (Haplotypecaller54) using the GRCh37 human reference genome. Minimum mapping quality threshold was set to 20 (–minimum-mapping-quality 20). Variants were annotated with an in-house pipeline to include information regarding variant-specific predicted effect (impact, conservation), population-specific variant frequencies (e.g. gnomAD), and clinical annotations from medical databases (ClinVar, HGMD, disease-specific).
Selection of pharmacogenes and alleles
First, we selected all pharmacogenes included in CPIC guidelines (https://cpicpgx.org/guidelines/; July 2021; n = 788 alleles) and compiled only those alleles annotated with no function, decreased function, and increased function in the “Allele Clinical Functional Status” column within the “Allele Functionality table”. Alleles annotated as normal function, uncertain function, unclear function, not reported or unknown function, were not considered further. Of note, CACNA1S alleles (c.520 C > T and c.3257 G > A) were considered as actionable despite having “uncertain” allele functional status due to their link to MHS; G6PD functional alleles were defined by I-III /Deficient status; despite the absence of a CYP4F2 CPIC Allele Functionality Table in CPIC guideline for warfarin, based on this guideline, CYP4F2*3 allele was considered for the analysis, while CYP4F2*2 was filtered out43. Second, we filtered out alleles supported by a limited/inadequate level of evidence (this information was only available for CYP2C9, CYP2C19, and DPYD at the “Strength of Evidence” column located in their “Allele Functionality tables”). Third, we identified the alleles that could not be resolved using Exome data (e.g. those defined by intronic or promoter variants, CNVs or HLA alleles). If a high impact allele, critical for pharmacogenetic phenotype group definition, was not available in Exome data (e.g. CYP3A5*3, CYP2C19*17, VKORC1-rs9923231, CYP2D6*5), the gene was deemed as not interpretative by exome sequencing and all alleles of this gene discarded for the analysis. Fourth, CFTR was removed from the analysis because the alleles included in its guideline are disease-causing and thus not within the objectives of preemptive pharmacogenetics. Supplementary Data 1 contains the full list of pharmacogenetic alleles and variants in the 11 genes included in the analysis, as well as a link and accession dates to CPIC “allele/ functionality” tables.
Quality control analysis
All actionable pharmacogenetics alleles defined by SNVs were subjected to QC analysis ensuring a correct homozygous-reference genotype calling using GATK v4 Haplotypecaller54 in “-ERC BP_RESOLUTION” mode. This strategy forces genotyping all relevant genomic loci, regardless of the nucleotide in the reference sequence (reference or alternative variant). Coverage values were extracted from the “depth” (DP) field to assess the fraction of samples that failed to inform at these sites. Alleles containing indel variation were manually curated to prevent genomic coordinate mismatches between called indels and their description. We reviewed all insertions and deletions flanking the described genomic coordinates of clinically actionable indels by searching within reported coordinates plus padded (upstream and downstream) genomic intervals with the length of the actionable indel. Also, QC analysis at the positions for the indels was performed. We extracted mappability scores (24, 36, 40, 50, 75, and 100-mer window sizes) from UCSC Alignability Tracks (http://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeMapability) for all genomic loci included in this study.
Haplotype and phenotype assignment
We obtained the pharmacogenetic information of the 280 CPIC actionable alleles (Supplementary Data 1) by leveraging genotype information obtained after the QC method described in the previous section. We calculated all possible actionable alleles according to the genetic variation and following the “Allele Definition Tables” provided in the CPIC guidelines. Similarly to the procedure described by McInnes et al30, if the defining genetic variation for one star allele was a proper subset of those for another star allele, the matching star allele with the greatest number of variants was reported. This situation was necessary for some alleles in SLCO1B1 (*5 and *15), CYP2B6 (*4, *6, *7, *8, *9, *13, *18, *20, *22, *26, *34 and *36), TPMT (*3A, *3B, *3C), and G6PD (Asahi and A-202A-376G). Python scripts for haplotype and diplotype assignment were used (see “Code availability” section, below). When available, we used parental variation data for phasing (i.e. TPMT *1/*3A or *3B/*3C haplotypes in 22 individuals heterozygous for rs1800460 and rs1142345; all found to be TPMT *1/*3A). For G6PD, located in the X-chromosome, the gender of the individual enabled to differentiate hemizygous males, homozygous females, and compound heterozygous females.
Resulting diplotypes were translated into their corresponding pharmacogenetic phenotypes following the functionality tables provided by CPIC clinical guidelines.
Comparison of population-specific allele frequencies
Statistical analysis to compare the allele frequencies in our Spanish and Latin American subcohorts was performed using χ2 test. GnomAD variation data (number of alleles and minor allele frequencies) was extracted from MyVariant.info database55. Chi-square statistical analysis was performed to compare gnomAD Non-Finish European (NFE) and Latino/Admixed American (AMR) population data to our Spanish and Latin American individuals, respectively. The threshold for statistical significance was 0.001 due to multiple comparisons. Allele frequencies reported in CPIC guidelines were also used in the analysis.
Discovery of novel variants
The variant discovery of novel and known variants (SNV and indel variants) in the 11 selected pharmacogenes was done using GATK v3.6. We selected all non-synonymous coding variants (e.g. missense, frameshift, stop gain, start lost) and those affecting canonical splice sites. A variant was considered as “novel” when it was not present neither in gnomAD (v2.1.1) nor in the SNP database (dbSNP v138-b3756). Novel frameshift, stop gain and start lost variants together with those altering canonical splicing sites were considered LOF. Variants with <20x depth of coverage or <30% Variant Allele Fraction were filtered out. However, novel variants with 10-20x coverage were manually curated.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Pharmacogenetics variation data to reproduce results presented in this work are deposited at the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) and publicly available under accession number PRJEB48632. Novel variants are provided in Supplementary Data 3. Individual-related information (country and exome platform) are available in Supplementary Data 2.
Code availability
Python scripts were created to perform the analyses described in the previous sections: coverage QC, genotyping data to CPIC alleles and phenotype conversion, collecting gnomAD population data for specific variants, all statistical analysis and filtering of the novel variants. Plots displayed in figures were also created using Python (v3.6). All scripts have been uploaded to a GitHub repository: https://github.com/jlanillos/clinAcc_PGx_WES.
References
Coleman, J. J. & Pontefract, S. K. Adverse drug reactions. Clin. Med. 16, 5 (2016).
Schork, N. J. Time for one-person trials. Nature 520, 609–611 (2015).
Zhou, Z.-W. et al. Clinical association between pharmacogenomics and adverse drug reactions. Drugs 75, 589–631 (2015).
Van Driest, S. et al. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin. Pharmacol. Ther. 95, 423–431 (2014).
Bush, W. et al. Genetic variation among 82 pharmacogenes: the PGRNseq data from the eMERGE network. Clin. Pharmacol. Ther. 100, 160–169 (2016).
Kalman, L. et al. Pharmacogenetic allele nomenclature: International workgroup recommendations for test result reporting. Clin. Pharmacol. Ther. 99, 172–185 (2016).
Gaedigk, A., Whirl-Carrillo, M., Pratt, V. M., Miller, N. A. & Klein, T. E. PharmVar and the landscape of pharmacogenetic resources. Clin. Pharmacol. Ther. 107, 43–46 (2020).
Ehmann, F. et al. Pharmacogenomic information in drug labels: European Medicines Agency perspective. Pharmacogenomics J. 15, 201–210 (2015).
Bank, P. et al. Comparison of the guidelines of the clinical pharmacogenetics implementation consortium and the dutch pharmacogenetics working group. Clin. Pharmacol. Ther. 103, 599–618 (2018).
Reisberg, S. et al. Translating genotype data of 44,000 biobank participants into clinical pharmacogenetic recommendations: challenges and solutions. Genet. Med. 21, 1345–1354 (2019).
Stark, Z. et al. Integrating genomics into healthcare: a global responsibility. Am. J. Hum. Genet. 104, 13–20 (2019).
Ji, Y., Si, Y., McMillin, G. A. & Lyon, E. Clinical pharmacogenomics testing in the era of next generation sequencing: challenges and opportunities for precision medicine. Expert Rev. Mol. Diagn. 18, 411–421 (2018).
van der Wouden, C. et al. Implementing pharmacogenomics in europe: design and implementation strategy of the ubiquitous pharmacogenomics consortium. Clin. Pharmacol. Ther. 101, 341–358 (2017).
Ng, D. et al. Assessing the capability of massively parallel sequencing for opportunistic pharmacogenetic screening. Genet. Med. 19, 357–361 (2017).
Londin, E. R. et al. Performance of exome sequencing for pharmacogenomics. Personalized Med. 12, 109–115 (2015).
Lee, M. et al. Repurposing of diagnostic whole exome sequencing data of 1,583 individuals for clinical pharmacogenetics. Clin. Pharmacol. Ther. 107, 617–627 (2020).
Yang, W. et al. Comparison of genome sequencing and clinical genotyping for pharmacogenes. Clin. Pharmacol. Ther. 100, 380–388 (2016).
Carr, D. F. & Pirmohamed, M. Precision medicine in drug safety. Curr. Opin. Toxicol. 23–24, 87–97 (2020).
Zhou, Y., Fujikura, K., Mkrtchian, S. & Lauschke, V. M. Computational methods for the pharmacogenetic interpretation of next generation sequencing data. Front. Pharmacol. 9, 1437 (2018).
Smith, D. M. et al. Pharmacogenetics in practice: estimating the clinical actionability of pharmacogenetic testing in perioperative and ambulatory settings. Clin. Transl. Sci. 13, 618–627 (2020).
Gulilat, M. et al. Targeted next generation sequencing as a tool for precision medicine. BMC Med. Genomics 12, 81 (2019).
Bank, P. C. D., Swen, J. J. & Guchelaar, H.-J. Advances in Pharmacology. Vol. 83, p. 219–246 (Elsevier, 2018).
Lauschke, V. M. & Ingelman-Sundberg, M. Prediction of drug response and adverse drug reactions: from twin studies to Next Generation Sequencing. Eur. J. Pharm. Sci. 130, 65–77 (2019).
Ingelman-Sundberg, M., Mkrtchian, S., Zhou, Y. & Lauschke, V. M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genomics 12, 26 (2018).
Santos, M. et al. Novel copy-number variations in pharmacogenes contribute to interindividual differences in drug pharmacokinetics. Genet. Med. 20, 622–629 (2018).
Twesigomwe, D. et al. A systematic comparison of pharmacogene star allele calling bioinformatics algorithms: a focus on CYP2D6 genotyping. npj Genom. Med. 5, 30 (2020).
Srivastava, S. et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med. 21, 2413–2421 (2019).
Aref-Eshghi, E. et al. Clinical and technical assessment of MedExome vs. NGS panels in patients with suspected genetic disorders in Southwestern Ontario. J. Hum. Genet. 66, 451–464 (2021).
Melbourne Genomics Health Alliance. et al. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur. J. Hum. Genet. 26, 644–651 (2018).
McInnes, G. et al. Pharmacogenetics at scale: an analysis of the UK Biobank. Clin. Pharmacol. Ther. 109, 1528–1537 (2021).
Mulder, D. J. et al. Utilization of whole exome sequencing data to identify clinically relevant pharmacogenomic variants in pediatric inflammatory bowel disease. Clin. Transl. Gastroenterol. 11, e00263 (2020).
Yu, M. H. C. et al. Actionable pharmacogenetic variants in Hong Kong Chinese exome sequencing data and projected prescription impact in the Hong Kong population. PLoS Genet. 17, e1009323 (2021).
Rotger, M. et al. Partial deletion of CYP2B6 owing to unequal crossover with CYP2B7. Pharmacogenet. Genomics 17, 885–890 (2007).
Martis, S. et al. Multi-ethnic cytochrome-P450 copy number profiling: novel pharmacogenetic alleles and mechanism of copy number variation formation. Pharmacogenomics J. 13, 558–566 (2013).
Genome Aggregation Database Consortium. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Relling, M. V. & Klein, T. E. CPIC: clinical pharmacogenetics implementation consortium of the pharmacogenomics research network. Clin. Pharmacol. Ther. 89, 464–467 (2011).
Birney, E., Vamathevan, J. & Goodhand, P. Genomics in healthcare: GA4GH looks to 2022. https://doi.org/10.1101/203554 (2017).
Ross, J. P., Dion, P. A. & Rouleau, G. A. Exome sequencing in genetic disease: recent advances and considerations. F1000Res 9, 336 (2020).
Szustakowski, J. D. et al. Advancing Human Genetics Research and Drug Discovery through Exome Sequencing of the UK Biobank https://doi.org/10.1101/2020.11.02.20222232 (2020).
Park, J. et al. Exome-wide evaluation of rare coding variants using electronic health records identifies new gene–phenotype associations. Nat. Med. 27, 66–72 (2021).
Schwarze, K., Buchanan, J., Taylor, J. C. & Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 20, 1122–1130 (2018).
Alfares, A. et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet. Med. 20, 1328–1333 (2018).
Johnson, J. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for pharmacogenetics-guided warfarin dosing: 2017 update. Clin. Pharmacol. Ther. 102, 397–404 (2017).
Karnes, J. H. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2C9 and HLA‐B genotypes and phenytoin dosing: 2020 update. Clin. Pharmacol. Ther. 109, 302–309 (2021).
Rehm, H. L. et al. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 15, 733–747 (2013).
Lee, S., Wheeler, M. M., Thummel, K. E. & Nickerson, D. A. Calling star alleles with stargazer in 28 pharmacogenes with whole genome sequences. Clin. Pharmacol. Ther. 106, 1328–1337 (2019).
van der Lee, M., Kriek, M., Guchelaar, H.-J. & Swen, J. J. Technologies for pharmacogenomics: a review. Genes 11, 1456 (2020).
Volpi, S. et al. Research directions in the clinical implementation of pharmacogenomics: an overview of US programs and projects. Clin. Pharmacol. Ther. 103, 778–786 (2018).
Weinshilboum, R. M. & Wang, L. Pharmacogenomics: precision medicine and drug response. Mayo Clin. Proc. 92, 1711–1722 (2017).
Tremmel, R. et al. Copy number variation profiling in pharmacogenes using panel-based exome resequencing and correlation to human liver expression. Hum. Genet. 139, 137–149 (2020).
Ingelman-Sundberg, M., Sim, S. C., Gomez, A. & Rodriguez-Antona, C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol. Ther. 116, 496–526 (2007).
Desta, Z. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2B6 and efavirenz‐containing antiretroviral therapy. Clin. Pharmacol. Ther. 106, 726–733 (2019).
Amstutz, U. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin. Pharmacol. Ther. 103, 210–216 (2018).
Poplin, R. et al. Scaling accurate genetic variant discovery to Tens of thousands of samples. https://doi.org/10.1101/201178 (2017).
Xin, J. et al. High-performance web services for querying gene and variant annotation. Genome Biol. 17, 91 (2016).
Sherry, S. T., Ward, M. & Sirotkin, K. dbSNP—database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 9, 677–679 (1999).
Acknowledgements
This work was supported by the project RTI2018-095039-B-I00 from the Agencia Estatal de Investigación (AEI-MCIN 10.13039/501100011033) and co-funded by the European Regional Development Fund (ERDF-EU) and “la Caixa Foundation” INPhiNIT Retaining Doctorate Fellowship Programme (LCF/BQ/DR19/11740015).
Author information
Authors and Affiliations
Contributions
J.L., M.C., P.M., S.A., and C.R.A. designed research. J.L., M.C., and P.M. performed the research and analyzed the data. J.L. contributed with new analytical tools. J.L. and C.R.A. wrote the manuscript draft. All authors reviewed and approved the manuscript draft, contributed to the review process, and approved the completed version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lanillos, J., Carcajona, M., Maietta, P. et al. Clinical pharmacogenetic analysis in 5,001 individuals with diagnostic Exome Sequencing data. npj Genom. Med. 7, 12 (2022). https://doi.org/10.1038/s41525-022-00283-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-022-00283-3
This article is cited by
-
Development of a coding SNP panel for tracking the origin of whole-exome sequencing samples
BMC Genomics (2024)
-
Current status of the analytical validation of next generation sequencing applications for pharmacogenetic profiling
Molecular Biology Reports (2023)
