
Abstract
Polymers play an integral role in various applications, from everyday use to advanced technologies. In the era of machine learning (ML), polymer informatics has become a vital field for efficiently designing and developing polymeric materials. However, the focus of polymer informatics has predominantly centered on single-component polymers, leaving the vast chemical space of polymer blends relatively unexplored. This study employs a high-throughput molecular dynamics (MD) simulation combined with active learning (AL) to uncover polymer blends with enhanced thermal conductivity (TC) compared to the constituent single-component polymers. Initially, the TC of about 600 amorphous single-component polymers and 200 amorphous polymer blends with varying blending ratios are determined through MD simulations. The optimal representation method for polymer blends is identified, which involves a weighted sum approach that extends existing polymer representation from single-component polymers to polymer blends. An AL framework, combining MD simulation and ML, is employed to explore the TC of approximately 550,000 unlabeled polymer blends. The AL framework proves highly effective in accelerating the discovery of high-performance polymer blends for thermal transport. Additionally, we delve into the relationship between TC, radius of gyration (Rg), and hydrogen bonding, highlighting the roles of inter- and intra-chain interactions in thermal transport in amorphous polymer blends. A significant positive association between TC and Rg improvement and an indirect contribution from H-bond interaction to TC enhancement are revealed through a log-linear model and an odds ratio calculation, emphasizing the impact of increasing Rg and H-bond interactions on enhancing polymer blend TC.
Similar content being viewed by others
Introduction
The global energy crisis has continued to escalate due to the conflict between the limited availability of fossil energy and the ever-increasing demand for it. Approximately 50% of the world’s final energy consumption in 2021 was related to thermal energy, including industrial processes, buildings, and agriculture1. Therefore, the efficient use of thermal energy is essential to overcome the global energy crisis. Moreover, inefficient heat dissipation in applications like high-power electronics, solar photovoltaics, and semiconductor lasers can significantly degrade the efficiency and lifespan of these devices2,3,4,5. Thus, research on thermal transport in relevant materials is crucial in tackling the global energy crisis and improving existing technologies.
As a widely used constituent in thermal applications, polymers have advantages like low costs, high electrical resistivity, lightweight, excellent mechanical flexibility, and corrosion resistance6,7. However, the thermal conductivity (TC) of most amorphous polymers is low, on the order of 0.1–0.5 W m−1 K−1 due to the disordered atomic arrangement8. Higher TC fillers, such as aluminum oxide, boron nitride, graphite, and carbon nanotubes are often introduced into the polymer matrix for industrial applications6,9,10,11,12,13,14. However, increasing the intrinsic TC of the polymer matrix remains a crucial factor as it significantly affects the TC of the composite and enables the composite to retain the benefits of polymer materials, such as low cost and lightweight. Effective medium theory calculations have shown that polymer composites with a TC exceeding 20 W m−1 K−1 will require the TC of the polymer matrix to increase to 1 W m−1 K−1,15.
In amorphous polymer, the total TC can be divided into contributions from primarily two parts, bonded intramolecular (e.g., covalent bonding force along the polymer chain) and non-bonded inter-molecular (e.g., van der Waals (vdW), hydrogen bond, and electrostatic) interactions. The bonded interactions are reported to dominate the effective heat transfer over other contributions from non-bonded interactions8,16,17,18,19,20,21. Thus, one effective route to change the intrinsic TC of amorphous polymers is through modifying the heat transfer path through bonded interactions, e.g., polymer chain alignment through stretching22,23,24,25, template-assisted growth26, etc., which enhances the heat flow along the bonded backbone. For instance, Shen et al. fabricated ultra-drawn polyethylene nanofibers with a high TC of ~104 W m−1 K−1,24. Later, Xu et al. produced drawn polyethylene films with TC of 62 W m−1 K−1,25, scaling up from individual nanofibers to more macroscopic thin films. However, the high values of TC resulting from the alignment of polymer chains are restricted to the chain orientation direction, and it necessitates specialized manufacturing techniques (e.g., mechanical stretching) that are usually not suitable for conventional processing methods (e.g., solution casting). Another strategy to modify the TC of existing polymers is to engineer the non-bonded inter-chain interactions, e.g., \({\rm{\pi }}-{\rm{\pi }}\) stacking27, electrostatic interaction enhanced by ionization28, and blending polymers with strong inter-molecular interactions19,29,30. Among them, polymer blend provides higher tunability and larger design space, potentially allowing a higher possibility of finding high TC polymers.
Polymer blend refers to a mixture of two or more different polymers and allows for combining the properties of different materials into a new material with targeted performance31. Normally, the TC of a binary polymer blend will change monotonically as the proportion of one component varies, and this simple rule of mixtures has been demonstrated in various studies29,32,33,34,35. However, there are several notable exceptions where the TC of polymer blends exceeds the bounds predicted by the simple rule of mixtures. For instance, in 2014, Guo et al.33 found that the TC of a binary polymer blend of bulk-heterojunction films is lower than the pure phase when the volumetric fraction of the added polymer is below 35%, which is potentially related to phase segregation when the added polymers are minor phases. In 2015, Kim et al.19 reported a sharp increase in TC of a blend of poly(N-acryloyl piperidine) (PAP) and poly(acrylic acid) (PAA), reaching over 1.5 W m−1 K−1, much higher than those of the individual components, when the molar ratio of PAP:PAA was 30:70, and they also observed a nonmonotonic relation in TC when mixing two other polymer blends. The authors attributed this improvement of PAP-PAA blend to the homogenous thermal transport network created by the inter-chain hydrogen bonds between the two polymers. However, later in 2016, this large enhancement cannot be reproduced by Xie et al.29, where the TC of the polymer blends simply followed the rule of mixtures, and near the 30:70 mixing ratio of PAP and PAA, the blends were found to be phase-separated. Nevertheless, among single-component polymers, those with capacities to form intra- and inter-chain hydrogen bonds were still discovered to have higher TC compared to those without29. The TC enhancement from hydrogen bond network formation was further verified by Mehra et al.36 in 2017 by inserting water molecules. In addition to experiments, molecular dynamics (MD) simulations are another powerful tool for uncovering the physics of thermal transport in polymer blends. For example, in Wei et al.17 tuned the inter- and intra-chain interaction strengths in a polymer blend model in MD simulation and found that the increase of inter-chain interactions could lead to an increase of TC because of the stretched polymer chain conformation of the major phase. In 2019, the simulation results from Bruns et al.37 showed that PAP-PAA blends were always phase-separated and TC-invariance over the whole range of mixing ratios. However, they also reported the improved TC of poly(acrylamide)(PAM)-PAA blends due to the stronger H-bonded contact (compared with PAP-PAA blends), which further resulted in the formation of short PAM bridges cross-linking PAA monomers.
For polymer blends, despite various and occasionally conflicting outcomes that have been reported, the strengthening of intra- and inter-molecular bonding is still considered an effective means of improving thermal transport, either through directly increasing the inter-chain thermal transport or indirectly impacting the chain configuration (e.g., increased radius of gyration). However, there are still considerable unknowns surrounding the physics of thermal transport in polymer blends. For example, how can blending alter the interactions both within and between polymer chains? How do the interaction variations lead to changes in the conformation of the chains and, subsequently the TC? What are the mechanisms behind the improvement of thermal transport in polymer blends when the simple rule of mixtures does not apply?
To answer these questions and identify high TC polymer blends, traditional experimental trial-and-error methods, and case-by-case studies are both time-consuming and expensive. MD simulations offer an accelerated alternative for investigating thermal transport in polymer blends, but due to the enormous chemical space of such blends—for instance, even considering only 500 single-component polymers, there are 124,750 distinct binary blends possible without taking into account the full range of blending ratios between 0 and 1—these simulations still require considerable computational resources. The application of machine learning (ML) has proven to be a promising means of efficiently scanning large target spaces through data-driven approaches, with successful recent examples in the realm of polymer research and property predictions38,39,40,41,42, which gives rise to the emerging field of polymer informatics (PI). However, due to the limitation of data and polymer representation methods, most of the research in PI has focused on single-component homopolymers43,44,45,46,47 and copolymers48,49,50, leaving the chemical space of polymer blends largely unexplored51. Recently, Liang et al.52 built up a polymer blend compatibility database using extracted experimental literature data and presented an ML method to predict polymer blend compatibility as a classification task, which is the only known research on polymer blend informatics, as of yet, relying solely on repeating units and composition information of polymer blends. While this work is undoubtedly a great start, there is still much research to be done in the field of polymer blend informatics, especially concerning physical properties. One significant challenge is the lack of labeled data on polymer blends. Fortunately, a combination of MD simulation and active learning (AL) presents a promising solution. AL is an ML framework that enables the algorithm to identify the most informative samples for the task at hand and utilize them to update the ML model53. This approach reduces the number of data samples required to train the model, making it much more efficient and cost-effective, especially in material optimization and design processes where the data acquisition process is very expensive54,55,56,57. By combining MD simulations with AL, researchers can obtain high-quality data from standardized simulation protocols to train their models while also reducing computational costs to find target materials.
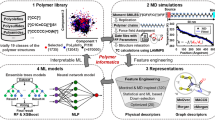
In this work, we employ a high-throughput MD simulation technique in conjunction with AL, as shown in Fig. 1, to unearth polymer blends exhibiting higher TC when compared to individual single-component polymers. In the initial phase, TC values are computed for selected amorphous single-component polymers and polymer blends through MD simulations. Different methods for representing polymer blends are also compared using this initial labeled dataset. Additionally, an AL framework, combining MD simulation with an ML classifier, is applied to explore an unlabeled virtual dataset containing roughly 550,000 potential polymer blends for which TC values are unknown. This AL approach significantly expedites the discovery of high-performance polymer blends concerning TC. Furthermore, a statistical investigation into the relationship between TC, the radius of gyration, and hydrogen bonding underscores the critical roles played by inter- and intra-chain interactions in the thermal transport mechanisms of amorphous polymer blends.

First, thermal conductivities (TC) are calculated for a selection of amorphous single-component polymers and polymer blends from a large dataset of unlabeled polymers. These calculations are performed using MD simulations, which are considered the oracle in the context of AL. Starting with these initial labeled training data, the objective is to tackle a binary classification problem of identifying polymer blends in the unlabeled pool that exhibit higher TC values compared to their individual single-component polymers, denoted as \({\rm{T}}{{\rm{C}}}_{{\rm{blend}}} > \max \{{\rm{T}}{{\rm{C}}}_{1}\,,{\rm{T}}{{\rm{C}}}_{2}\}\). TC of the polymer blend is denoted as \({\rm{T}}{{\rm{C}}}_{{\rm{blend}}}\) and TC of the single-component polymer is denoted as \({\rm{T}}{{\rm{C}}}_{1}\) or \({\rm{T}}{{\rm{C}}}_{2}\). Then a pool-based AL framework that utilizes a random forest machine learning classifier in conjunction with a certainty-based acquisition function is employed to iteratively select high-performance polymer blends from the unlabeled pool. This AL approach, combined with MD simulation greatly accelerates the process of identifying high-performance polymer blends in terms of TC. The specific methods for carrying out MD simulations and TC calculations are described in the Methods section, while details regarding the labeled data and the unlabeled polymer dataset are provided in the Dataset section.
Results and discussions
Dataset
In this work, five datasets are leveraged to explore the TC of amorphous single-component homopolymers and binary polymer blends. Relationships between the five datasets are shown in Fig. 2a. Dataset 1 encompasses TC values calculated through MD simulations performed on 608 randomly chosen amorphous single-component homopolymers. This dataset serves as the foundation for understanding the TC in a broad range of polymer chemical space. Dataset 2 is a subset of Dataset 1 using stratified sampling. Building upon our prior research on high TC single-component homopolymers58, Dataset 2 is intentionally designed into two balanced parts. Half of the entries (17) are randomly selected from the single-component polymers in Dataset 1 that display comparatively low TC values (<0.4 W m−1 K−1), while the remaining half of entries (18) are randomly chosen from those with higher TC values (≥0.4 W m−1 K−1). This selection strategy ensures a balanced dataset comprising both high and low TC values. Dataset 3 comprises 216 TC values of binary polymer blends, with the constituent polymers from Dataset 2. These blends have three different blending ratios (1:5, 1:1, and 5:1) and constitute the initial dataset to facilitate an investigation into the polymer TC variation induced by blending. Dataset 4 is a combination of Dataset 1 and Dataset 3, specifically used to train a regression model aiming to identify the most effective representation method for polymer blends. The data distributions of Datasets 1 to 4 regarding TC are shown in Fig. 2b, c. Lastly, Dataset 5 comprises an extensive database of ~550,000 polymer blends, generated by considering all possible combinations of two different polymers from the single-component polymer dataset (Dataset 1) with the three different blending ratios. This large dataset serves the purpose of virtual screening in our study. Notably, to address the specific problem defined in our work, which focuses on determining whether a blend exhibits a higher TC compared to its constituents, blends in Dataset 5 exclusively consider single-component polymers with known TC values (Dataset 1). Details of TC calculation using MD simulation for both single-component polymers and polymer blends can be found in the Methods section. Besides, the data distribution in the chemical space of Dataset 1, 2, 3, and 5 can be found in the next section on polymer blend representation.

a Datasets relationship and data distribution of b Dataset 1 to 3 and c Dataset 4 in the target TC space.
Polymer blend representation
The numerical representation of polymers holds paramount importance in PI, as ML models rely on numerical inputs to quantify the relationships between structures and properties. Thus, prior to training the ML model, it is essential to establish a numerical representation for polymer blends. However, it is worth noting that the majority of PI research has primarily focused on single-component homopolymers, leaving the chemical space of polymer blends largely unexplored. For the representation of single-component polymers, two well-established methods exist: Morgan fingerprints (MF)59,60 and polymer embeddings (PE)47. Both approaches have demonstrated excellent performance in representing single-component homopolymer structures47,59,60. Details of the two representation methods are introduced in the Methods section. In this study, we propose two blending methods as variations of these conventional single-component polymer representations for describing polymer blends (Fig. 3): weighted summation (Fig. 3a) and weighted concatenation (Fig. 3b). The weight used in these variations corresponds to the mixing ratio. Weighted summation involves element-wise summation of the weighted vectors of the two constituent polymers’ descriptors, while weighted concatenation entails concatenating the two weighted vectors. In this way, four methods are available to represent polymer blends, each involving different combinations of single-component polymer representations (MF and PE) and blending methods (weighted summation and concatenation). For simplicity, we will refer to them as MF-WS (MF with Weighted Summation), MF-WC (MF with Weighted Concatenation), PE-WS (PE with Weighted Summation), and PE-WC (PE with Weighted Concatenation).

Polymer blend representation methods a weighted summation and b weighted concatenation.
To determine the optimal method for representing polymer blends, a random forest (RF) regression model is trained on the combined dataset (Dataset 4). Based on the coefficient of determination (R2) and mean square error (MSE) on the test set, as reported in Table 1, PE-WS demonstrates the highest performance among the four methods investigated. The prediction parity plot in Fig. 4 also revealed a strong agreement between the model’s predictions and the ground truth values using PE-WS as the representation method. In general, PE outperforms MF in both blending methods, suggesting the superiority of PE in polymer representation, as also demonstrated in ref. 59. No significant difference is observed between different blending methods. Additionally, these high-dimensional polymer blend fingerprints were visualized in a 2D space using the t-SNE method in Fig. 5. These points were color-coded based on their TC values. Notably, the PE-WS method, shown in Fig. 5a, exhibited a clearer structure-property relationship than the other three methods, where polymers with similar structures tended to exhibit similar properties. However, the t-SNE plots can only provide qualitative analysis and comparison of different representations. To qualitatively verify the superiority of PE-WS in capturing the structure-property relationship, we calculate Spearman’s rank correlation coefficient between the pairwise Euclidean distances of polymer representations in the chemical space and the corresponding pairwise absolute differences in TC values. As shown in Supplementary Fig. 1, PE-WS exhibits the highest Spearman’s rank correlation coefficient of 0.546 among the four representations, and the coefficients of PE-WC, MF-WS, and MF-WC are only 0.463, 0.331, and 0.348, respectively. This indicates that PE-WS can capture a stronger monotonic relationship between chemical structure similarity and TC variation.

a PE-WS, b PE-WC, c MF-WS, and d MF-WC. All the x-axes represent the predicted TC in units of W m−1 K−1 and all the y-axes represent the ground-truth (MD-calculated) TC in units of W m−1 K−1.

a PE-WS, b PE-WC, c MF-WS, and d MF-WC. All data points are colored by their corresponding TC values, as shown in the color bar.
Detailed analysis, as illustrated in Fig. 4c, d, reveals the difficulty of MF to accurately distinguish polymers with TC values ranging from 0.3 to 0.5 W m−1 K−1, consistently leading to over- or under-estimations. This limitation is not isolated but further corroborated by observations in Supplementary Fig. 1 and Fig. 5, where MF’s challenges in distinguishing certain chemical structures are highlighted. Specifically, Supplementary Fig. 1c, d showcase distinct peaks at lower normalized pairwise distances, with a notable sharp peak near 0 and multiple peaks between 0 and 0.2. These peaks suggest a significant limitation of MF in differentiating between chemical structures that result in a wide range of TCs. This issue is further visualized in Fig. 5c, d, which depicts circular clusters where polymers, despite having varied TC values (represented by different colors), are aggregated closely in the chemical space. Such clustering indicates a deficiency in MF’s ability to discriminate among diverse chemical structures effectively. The root of this deficiency in MF is identified as its multiplicity—a condition where a single fingerprint feature may represent multiple different substructures, thereby limiting the capacity of MF to accurately represent and differentiate complex chemical structures of polymers60. This contrasts sharply with the performance of PE, which is inspired by the word2vec concept47,61. PE has shown a superior ability to capture and distinguish the subtle chemical structures that influence variations in TC, providing a more nuanced and reliable prediction of polymer properties. This comparison not only highlights the limitations inherent in the use of MF for predicting polymer TC but also emphasizes the promise of alternative embedding techniques like PE for enhancing the precision and reliability of such predictions, which is consistent with our previous finding for polymer representations47,59. Therefore, after careful comparison, PE-WS was selected as the preferred approach for representing polymer blends for the remainder of this study. Under PE-WS, the data distribution in the chemical space of Dataset 1, 2, 3, and 5 is shown in Supplementary Fig. 2, revealing that Dataset 5 complements Dataset 1 well within the chemical space and Dataset 2 encompasses a diverse range as a stratified sampled subset of Dataset 1.
AL for high-performance polymer blends
In our study, we address a binary classification problem where the objective is to identify polymer blends with TC higher than their constituent single-component homopolymers from a large unlabeled pool of polymer blends (see Fig. 2, Dataset 5). In this binary classification task, we assign a label of ‘1’ to the high-performance polymer blends that show higher TC than their constituent single-component polymers, while assigning a label of ‘0’ to those with TC lower than or equal to their constituent single-component polymers. To efficiently identify higher-performance polymer blends, we adopt a pool-based AL framework, as shown in Fig. 1, which naturally fits our iterative approach. AL is an ML approach that strategically selects informative data points from an unlabeled pool to query for labels53,55,62. Pool-based AL is a specific variant of AL, where the unlabeled data is organized into a pool, and the model selectively samples data points from this pool for labeling based on certain acquisition functions, enabling more efficient and targeted data selection for training53. In the pool-based AL framework of our binary classification task, the initial step involves training a classifier using the available labeled training data (Dataset 4). This classifier is then utilized to screen the vast pool of unlabeled candidate polymer blends (Dataset 5) using different acquisition functions. In this study, the goal is to efficiently identify targeted candidates from the pool with fewer runs of MD simulations for labeling, which is the most time-consuming step. Thus, we emphasized prediction precision over general accuracy—aiming to maximize the identification of true positive candidates and thereby reduce false positive instances that would lead to unnecessary MD simulations. To achieve this objective, we utilize an acquisition function that prioritizes those with the highest predicted scores, which is a predicted probability assigned to each candidate by the binary classifier, ranging from 0 to 1, indicating the likelihood of being a polymer blend of label ‘1’. This certainty-based (exploitation) acquisition strategy is complemented by random acquisition for comparative purposes. By employing a batch querying process, we also effectively mix certainty-based sampling with random sampling in each iteration. This hybrid approach is designed not merely to contrast certainty-based with random acquisition but to establish a refined balance, enabling us to explore the broader chemical space while focusing on candidates with the greatest potential. The selected batch candidates are then presented to an oracle, in this case, the MD simulation, which provides the labels for these candidates, i.e., the corresponding TC values. The labeled data is then added to the existing dataset, and this process iteratively continues until we achieve a satisfactory proportion of high-performance polymer blends in the combined dataset (Dataset 4), which is set to be 5%.
Table 2 shows the iterative results of the AL process. We start with Dataset 4 version 0 (V0), comprising 824 polymer blends, among which 23 are identified as high-performance blends (labeled as ‘1’). Utilizing the classifier trained on V0, we select 16 candidate blends from the pool (Dataset 5) based on the highest prediction scores from the binary classifier. The score indicates the model’s certainty that the given polymer blend belongs to the class of label ‘1’. Five of these selected candidates are confirmed to be high-performance polymer blends through verification using the MD simulation oracle, suggesting a success rate of 31.25%. In contrast, we conducted a random selection of 82 candidates to demonstrate the efficacy of the classifier, and only six of them are indeed high-performance blends based on the MD simulation result, corresponding to a success rate of 7.31%. These results suggest the effectiveness of our initial dataset-trained model in identifying desired candidates. With three iterations of data expansion, we observe a gradual increase in the proportion of high-performance polymer blends, ultimately reaching almost twice the proportion found in the initial dataset (from 2.79% in V0 to 5.04% in V3). The statistics of the iterative data expansion process are presented in Table 2. Notably, certainty-based sampling consistently achieves a success rate of around 30%, in contrast to random sampling, which attains only around 7%. Details of the model training are described in the Methods section.
To further validate the efficiency of the AL approach and to compare different acquisition strategies within our pool-based AL framework, we conduct a virtual experiment using Dataset 4-V3, which contains a total of 992 data points. This dataset represents the largest collection of polymer blends we have after iterative data expansion through the previous AL. The primary objective of this virtual experiment is to find an acquisition strategy that can find a specific number of high-performance polymer blends within the shortest time (smallest number of MD simulations). To initiate the experiment, we randomly select 10 polymers from Dataset 4-V3. We pretend to have knowledge of the labels (‘0’ or ‘1’) for only these 10 polymer blends. Subsequently, we train an RF classifier on these 10 data points. Next, we employ one of the acquisition methods to iteratively select the next top 4 candidates as recommended by the RF classifier among the remaining 982 candidates. The labels of the recommended candidates are then revealed and added to the training set, and a new classifier is trained on the expanded training dataset. Predictions are subsequently made on the remaining 978 data points. This iterative process continues until we successfully identify a total of 20 candidates labeled as ‘1’. The model training and prediction procedure in this virtual experiment mirrors that of the actual AL experiments conducted on Datasets 4 and 5. To account for the effects of random variability, for each acquisition method, the experiment is repeated 10 independent times. In each run, different randomly selected initial datasets are used to reduce the impact of randomness in initial dataset selection on model performance. Further details about the model can be found in the Methods section.
The three different acquisition functions tested are certainty-based acquisition, uncertainty-based acquisition, and balanced acquisition. The certainty-based acquisition involves choosing the next four experiments based on the highest predicted probability. This approach tends to favor candidates that exhibit chemical similarity to the top performers within the training dataset and emphasizes exploitation in the chemical space covered by the training data. In comparison, the uncertainty-based acquisition selects the next four experiments that demonstrate the greatest uncertainty. In our binary classification problem, we measure uncertainty by looking at how close the predicted probability (ranging from 0 to 1) is to the threshold value of 0.5. If the predicted probability is ~0.5, the model is more uncertain, and the uncertainty-based acquisition prioritizes that data point. This strategy emphasizes the exploration of chemical space beyond that covered by the training dataset. As a compromise between exploitation and exploration, a balanced acquisition strategy that mixes certainty-based and uncertainty-based acquisition is employed. At every iteration, this strategy involves the selection of two candidates using the certainty-based acquisition method to exploit the model’s current knowledge, and simultaneously, two candidates are chosen based on uncertainty-based acquisition to explore less certain regions of the dataset63. Apart from these three strategies, random acquisition is also conducted as the baseline, randomly selecting four new candidates at each iteration.
The result in Fig. 6 shows the average number of experiments needed to identify 1 to 20 polymer blends with higher TC than the constituent single-component polymers (i.e., label ‘1’). Certainty-based and balanced sampling strategies display similarly high efficacy, outperforming both uncertainty-based sampling and random sampling. Notably, the initial phase of the experiment reveals a sharper increase for both certainty-based and balanced methods, indicative of the model’s quick identification of regions rich in high-performance polymer blends from the training dataset. However, as the model exhausts the chemical space covered by the initial training set in later iterations, the pace of discovery for certainty-based sampling slows. In contrast, while the discovery rate for balanced sampling also decelerates, it does so to a lesser extent, highlighting the benefits of incorporating exploratory elements to mitigate the limitations inherent in purely exploitative strategies in AL. Nonetheless, within the context of this virtual experiment, integrating exploration does increase the likelihood of encountering iterations without success, culminating in a performance for balanced sampling that is either on par with or slightly inferior to certainty-based sampling over time. The trajectory for uncertainty-based sampling initially mirrors that of random sampling, implying that early explorations beyond the limitations of the training data are somewhat akin to a random search, yielding minimal success. This trend shifts with an uptick in success rates following several rounds of data acquisition, eventually plateauing. This suggests that while uncertainty-based sampling initially thrives on navigating a diverse chemical space, the necessity for such broad exploration diminishes as the model develops a more comprehensive understanding of the chemical space. Interestingly, balanced sampling exhibits the greatest variability in performance, particularly as the search extends towards identifying larger numbers of high-performance polymer blends (e.g., from 15–20), as indicated by the error bars in Fig. 6. This variability underscores the potential of balanced sampling not only to match but occasionally surpass the success rates of certainty-based sampling, albeit with a higher degree of stochasticity. This reflects the nuanced trade-offs between exploration and exploitation strategies in AL, underscoring the complex interplay between achieving average success rates and the possibility of exceptional outcomes in specific instances.

This average is computed across 10 different runs, and the standard deviation is represented by the error bars, which is the mean ± standard deviation. Four different acquisition functions are compared: uncertainty (dark blue), certainty (green), balanced (light blue), and random (red) sampling.
However, within the context of this AL framework, a consideration arises concerning the inherent uncertainty associated with our oracle, the MD simulation. Notably, different initial structures can lead to slight variations in the calculated TC of the simulated amorphous polymers58. To mitigate this uncertainty, we conduct an ensemble of MD simulations on the top-10 predicted polymer blend candidates (based on the predictions from the classifier trained on the Dataset 4-V3 data) and their respective constituent single-component polymers. Three different initial structures are generated for simulation for all the single-component polymers and polymer blends. The validation outcome, as shown in Fig. 7, underscores the effectiveness of the ML model, as eight out of these ten candidates indeed exhibit the desired high-performance TC characteristics, with only candidates #1 and #9 being the exceptions. Detailed information on the 10 polymer blend candidates and the TC values can be found in Supplementary Tables 1 and 2.

The simulations were performed using three distinct initial structures for each candidate. The mean TC value is displayed, accompanied by error bars, which is the mean ± standard deviation.
Molecular-level origin of blend TC enhancement
This validation result motivates a deeper inquiry into the underlying factors influencing polymer blend TC enhancement. As stated earlier in the Introduction section, polymer TC can be decomposed into two primary contributors: intra-chain covalent bond interactions, which dictate heat transfer within polymer chains, and inter-chain non-bonding interactions, such as hydrogen bonds8,16,17,18,19,20,21. The former can be related to the chain conformation change, which can be characterized by the radius of gyration (Rg). In light of this, we pinpoint two specific features for detailed examination of the trajectory from the MD simulations: (1) the radius of gyration (Rg) and (2) the hydrogen bond.
The Rg of a molecule offers a measure of its spatial extension. Specifically, the squared Rg characterizes the average squared distance between any point within the polymer coil and its center of mass. The Rg holds significance in quantifying the spatial extension of the polymer coil and is found to be closely intertwined with TC16,17. The hydrogen bond is previously shown to exert an important influence on TC19,29,36. The distance between atoms participating in a hydrogen bond can be employed to assign the hydrogen bond strength64, which can be characterized by the radial pair distribution function (RDF) analysis. Therefore, for the leading 10 candidates alongside their corresponding constituent single-component polymers, we compute the values of Rg and the RDF of atoms participating in the hydrogen bond, aiming to explore the TC characteristics within various polymer blend configurations. The average Rg is computed for both polymer blends and corresponding single-component polymers for all three different initial structures, as outlined in Supplementary Table 2. For the quantification of hydrogen bond strength, we analyze the position and magnitude of the first primary peak in the RDF near radius = 2Å, which is a common checkpoint of the hydrogen bond presence65. Both the occurrence of a peak at the smaller radius and larger RDF intensity, indicate a stronger hydrogen bond. A detailed comparison of RDF is shown in Supplementary Fig. 3. Notably, the forcefield utilized in this study (GAFF2) does not explicitly model hydrogen bonds as separate entities. The electrostatic component effectively captures the essence of hydrogen bonding through the attraction between the donor and acceptor atoms. This, combined with torsional angle parameters that account for polarization and charge transfer effects, provides a robust framework for understanding hydrogen bond dynamics66,67. The integration of force fields like GAFF2 with RDF analysis has been validated in numerous studies for its efficacy in conformational and hydrogen bond analysis, making it an optimal choice for this work64,68,69.
Table 3 presents a comparative analysis of the ten validation cases, outlining the changes in TC, Rg, and H-bond strength of the blends relative to their constituent single-component polymers. The arrows in Table 3 indicate that the direction (either increase or decrease) of Rg or H-bond change aligns with the direction of TC change. As a result, eight out of ten Rg values exhibit changes in the same direction as TC, while seven out of ten H-bond interaction changes show the same direction as TC. Only the TC change of one candidate (#8) out of 10 is not captured by either Rg change or H-bond interaction change. Therefore, both strategies, enhancing Rg and improving H-bond interactions, have the potential to result in increased TC in polymer blends.
However, it’s important to note that drawing statistically significant conclusions about the relationships between Rg, H-bond strength, and TC only based on the ten selected candidates could be limiting. To address this potential limitation, our study extends the analysis to encompass a broader dataset. This expanded dataset comprises all 387 polymer blends for which TC was calculated after the AL iterations, along with the 109 associated single-component polymers. By calculating Rg values and RDF on this larger dataset, we aim to establish a more data-driven and statistically robust observation of these complex relationships between Rg, H-bond strength, and TC.
Expanding our analysis to encompass the entire set of 387 polymer blends, we derive three binary parameters, building on the observation from Table 3:
-
(1)
TC Improvement (denoted as ‘A’): This parameter assesses whether the TC of a given polymer blend surpasses that of both its constituent single-component polymers. This binary parameter is assigned ‘1’ to denote a positive improvement and “0” otherwise.
-
(2)
Rg Improvement (denoted as ‘B’): This parameter evaluates whether the Rg of the polymer blend exceeds that of any of its constituent single-component polymers. A value of ‘1’ means an Rg improvement, while ‘0’ denotes the absence of such enhancement.
-
(3)
H-bond Improvement (denoted as ‘C’): The H-bond improvement parameter offers two different scenarios:
-
H-bond Strength Improvement: This case is indicated by the presence of an H-bond-related peak in the RDF at a smaller radius within the blend, compared with the same H-bond in the single-component polymers. Here, ‘the same H-bond’ means an H-bond formed between the same H-bond acceptor and donor. A complete list of such acceptors and donors and the corresponding chemical structures considered in this work are shown in Supplementary Table 3. An ‘1’ is assigned if this peak appears at a smaller radius within the blend, implying an improvement in H-bond strength; otherwise, a ‘0’ is assigned.
-
H-bond Formation: We consider an H-bond between a proton acceptor and proton donator as newly formed within the blending system if a peak emerges in the RDF of the blend within the radius of 2.72 Å, whereas there is no such peak in the corresponding single-component polymers systems. 2.72 Å is the criterion employed to determine the formation of H-bonds65. A ‘1’ is allocated to indicate the formation of a new H-bond and a ‘0’ if no such bond emerges.
Notably, either an H-bond strength improvement or the formation of a new H-bond leads to an overall H-bond improvement (C=1).
A three-way contingency table (see Table 4) is used to summarize the 387 polymer blend data and show the cross-classification of these data by the levels of three categorical variables. To explore the complex relationships among the three binary parameters, we employ the log-linear model, which is a statistical technique used to analyze the relationships between categorical variables within a multi-dimensional contingency table70. The log-linear model offers an advantage over other statistical techniques, such as the logistic model, standing out for its capacity to estimate relationships among any of the variables without designating a single variable as the response70. This flexibility allows us to investigate interactions involving any of the derived binary parameters—TC improvement, Rg improvement, and H-bond improvement. The log-linear model works by examining the expected cell frequencies in a contingency table and comparing them with the observed frequencies. This comparison helps us identify whether the observed frequencies deviate significantly from what would be expected if the variables were independent. In our case, we use the log-linear model and hierarchical model selection method to assess whether the improvements in TC, Rg, and H-bond strength are independent or if there are statistically significant interactions between them.
We use a likelihood ratio test to measure the reduction in fit of smaller models (null hypothesis, H0) relative to the larger models (alternative hypothesis, Ha). The degrees of freedom (df) for residual, deviance, log-likelihood, and p value columns in Supplementary Table 4 correspond to the goodness-of-fit test for each model compared with the saturated model (Eq. 1). As is shown in Supplementary Table 4, all the reduced models are not significantly worse fit than the saturated model, since all the p values >0.05. Thus, all the reduced models are preferred relative to the saturated one because they have fewer parameters.
We continue to test further reductions with the likelihood ratio test following the hierarchical model selection method. As demonstrated in Supplementary Table 5, the initial assumption considers homogeneous associations (AB, AC, BC) as the alternative hypothesis (Ha), while all three scenarios of conditional independence (one-step reduced models of the homogeneous association model) are regarded as the null hypothesis (H0). If H0 is not rejected (p value > 0.05), it is subsequently treated as Ha and tested against its one-step reduced models. As a result, the reduced model (C, AB) is ultimately preferred, which indicates that C is jointly independent of A and B. The model structure is shown in Eq. 2.
This result indicates that, based on the current observed data, variable C is independent of both A and B, but variables A and B are not independent of each other. In other words, there is no significant association between H-bond improvement and either TC or Rg improvement according to the 387 polymer blend data. However, a significant association between TC and Rg improvement is observed from the data. This emphasizes the substantial impact of increasing Rg on enhancing polymer blend TC, while the role of H-bond interactions requires further discussion.
To further quantify the impact of Rg and H-bond on TC, we calculate the marginal and conditional odds ratio based on Table 4. Details of the calculation can be found in Supplementary Equations (1) to (6). The only statistically significant odds ratio is the conditional odds ratio \({\hat{\theta }}_{{AB}(C=1)}=2.87\), as shown in Eq. S4. This indicates that, when variable C is equal to 1, the odds of variable A being 1 are 2.87 times higher when variable B is also 1 compared to when B is 0. Moreover, since the odds ratio is greater than 1, it suggests a positive association between variables A and B when C is 1. That is to say, given the existence of H-bond improvement (C = 1), the odds that a polymer blend with the existence of Rg improvement (B;= 1) exhibits TC improvement (A = 1) are estimated to be 2.87 times higher than the odds that a polymer blend with no Rg improvement (B = 0) exhibits TC improvement (A = 1). This further confirms the significant role of Rg improvement in enhancing polymer blend TC and suggests that H-bond improvement can also contribute to TC enhancement indirectly.
The observed correlation between increased Rg and enhanced TC in polymer blends highlights the role of extended polymer chain structures in improving thermal transport through stronger bonding interactions. The presence of H-bonding could also enhance Rg or the spatial extension of the polymer chains, thereby potentially enhancing thermal transport along the chains. However, it is critical to underscore that, despite this strong correlation, the thermal transport efficiency in polymer blend systems cannot be exclusively linked to the presence of an increased Rg. The overall thermal transport efficiency in amorphous polymer blends is influenced by a multitude of factors, including but not limited to, the interfacial thermal resistance between different polymer components, the distribution and connectivity of polymer chains, and the inherent TC of the constituent polymers. In this study, related topics like the miscibility of polymer blends and cross-linking are not considered, and we focus only on the relative change of TC compared to the constituent polymers. Further research is needed to more thoroughly decipher the mechanisms that govern thermal transport in polymer blend systems.
To summarize, in the realm of polymer materials, where versatility meets practicality, our work studies the TC of polymer blends in the field of polymer informatics. Through high-throughput MD simulations and AL, we have shown that polymer blends for higher TC can be accelerated compared to random search. By screening a large chemical space, we identified high-performance polymer blends, with TC surpassing that of their constituent single-component polymers. Furthermore, our investigation into the intricate relationship between TC, Rg, and hydrogen bonding sheds light on the mechanisms governing thermal transport in amorphous polymer blends. It is found that improved Rg has a strong correlation with the enhanced TC via blending and H-bond improvement can also contribute to the enhanced TC. In conclusion, the strategies and results from this work can be useful for extending the chemical space of current polymer informatics research to polymer blends and can contribute to the automated design of high-performance polymer blends in thermal transport and other applications.
Methods
MD simulation—calculation of TC, R g, and RDF
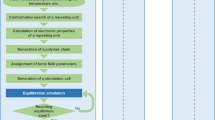
The overall process of generating TC data for amorphous polymer blends through high-throughput MD simulation is illustrated in Fig. 1. This process comprises two primary steps: the generation and optimization of amorphous structures and the calculation of TC using non-equilibrium molecular dynamics (NEMD). The procedure for calculating TC in amorphous single-component polymers is identical to amorphous polymer blends, with the exception that the blending ratio during chain mixing is set to 0:6 (or 6:0).
To initiate the process, we begin with the SMILES notation obtained from PoLyInfo71 for the two constituent polymers. Using a Python pipeline built on PYSIMM72, we generate the initial structure of the amorphous polymer blend. This involves creating a polymer chain for each of the constituent polymers through a polymerization process, with a fixed number of ~600 atoms per chain. This number has been determined to be adequate for TC calculations73. Subsequently, we replicate these chains based on the predetermined blending ratio (1:5, 3:3, and 5:1), resulting in a system comprising six chains in total, contained within a simulation box. It’s worth noting that for single-component polymers, the blending ratio is effectively 0:6 or 6:0. Additionally, we assign GAFF2 (General AMBER Force Field 2)69 forcefield parameters to the polymer system and generate an input script for MD simulations using the large-scale atomic-molecular massively parallel simulator (LAMMPS)74. Periodic boundary conditions are applied in all spatial directions. After the initialization, optimization of the system proceeds in stages. Initially, we perform simulations with electrostatic interactions disabled and Lennard-Jones interactions truncated at 0.300 nm. Under these conditions, the system undergoes a simulation in the NPT ensemble at 100 K for 2 ps, employing a time step of 0.1 fs. Subsequently, the system is heated from 100 K to 1000 K over 1 ns using the NVT ensemble and then simulated in the NPT ensemble for an additional 50 ps at 0.1 atm and 1000 K. Following this, the system experiences a 1 ns NPT simulation at 1000 K, allowing the pressure to rise from 0.1 atm to 500 atm. During this 1 ns NPT simulation, a time step of 1 fs is utilized, and SHAKE constraints75 are applied. The polymer system, now optimized, undergoes an annealing process with electrostatic interactions enabled. Electrostatic interactions are computed using the Particle–Particle–Particle–Mesh-based Ewald sum method. For Lennard-Jones interactions, a cutoff of 0.800 nm is applied. In the annealing process, the system is initially simulated in an NPT ensemble at 1 atm and 1000 K for 2 ps, with a time step of 0.1 fs. Following this, the system is gradually cooled from 1000 K to 300 K at a rate of 140 K(ns)−1 within an NPT ensemble at 1 atm. Subsequently, an additional NPT run at 300 K and 1 atm is conducted for 8 ns to further relax the annealed system with a time step of 1 fs and the application of SHAKE constraints. During this step, Rg is monitored for each polymer chain, and the average Rg value over the 8 ns duration is taken as the definitive representation of the polymer chain’s Rg. The Rg values of the same single-component polymers in the blend are also averaged to determine the final Rg value of the constituent single-component polymers in the blend system. These steps collectively lead to the attainment of the final amorphous state of the polymer blend system, as shown in Fig. 1.
Each obtained amorphous polymer blend system, initially confined within a cubic box, is then replicated three times to create a cuboid shape, as illustrated in Fig. 1. The dimensions vary slightly, ~9.900 × 3.300 × 3.300 nm3, depending on the specific polymer blends due to density differences. This extended system is used for TC calculations through NEMD simulations. During the NEMD simulation, the system operates under an NVE ensemble for 5 ns with a 0.25 fs time step. SHAKE constraints are omitted to preserve atoms’ natural vibrations, which is vital for thermal transport analysis. Langevin thermostats are applied near the cuboid ends (Fig. 1), with the heat source at 320 K and the heat sink at 280 K, each applied in a 0.500 nm thick region. Fixed regions at both ends prevent system drift and heat flow across boundaries. The measured heat flux is determined by tracking the heat added to and removed from the Langevin heat baths and the temperature gradient is derived by a linear fit on the temperature profile along the direction of heat transport within the cuboid. TC is computed from the heat flux and temperature gradient via Fourier’s law. Data from the last 4 ns is divided into 8 intervals, each yielding a TC value. The final TC output is the average of these 8 values. The accuracy and reliability of our NEMD calculations were further affirmed through a comparative analysis with equilibrium molecular dynamics (EMD) simulations and experimentally measured values for three widely studied amorphous polymers, as depicted in Supplementary Fig. 4. While minor differences are observed from Supplementary Fig. 4, they are deemed acceptable given the inherent uncertainties of MD simulations (both EMD and NEMD) and the potential variability in experiments (median is used to represent experimental TCs)58. Importantly, our research primarily focuses on the relative changes in TC due to polymer blending rather than the absolute TC values. Therefore, as long as the relative trend in TC changes can be accurately captured, these small variances do not impair the validity of our findings concerning the effects of polymer blending.
To evaluate the hydrogen bond formation within the blend system, the RDF of atoms related to hydrogen bonds is determined prior to the NEMD step. Following the final NPT run before NEMD step, an additional NPT run is performed, lasting 0.08 ns at 300 K and 1 atm, with a time step of 1 fs, and the RDF of hydrogen bond-related atoms is calculated. Involved atoms are listed in Supplementary Table 3. This pipeline facilitates the high-throughput generation of amorphous polymer blend structures and TC calculation with minimal human intervention, resulting in expedited AL iterations and high-performance polymer blend design.
Polymer representations
As a widely employed molecular representation in chemoinformatics and computational chemistry, MF encodes structural information about chemical compounds. These fingerprints, generated using the Morgan algorithm76, are binary bit vectors that denote the presence or absence of specific substructural features within a molecule. A chemoinformatics package RDKit (https://www.rdkit.org/) is utilized to generate the MF of polymers, where the radius is set as 2, and the number of bits of the generated MF is set as 1024. Compared with MF, PE has proved to be more informative for quantifying structure-property relationships. It is a 300-dimension continuous vector derived by following the mol2vec model61 framework. Specifically, a polymer is broken down into a sequential arrangement of substructures. Then, a specific substructure from this sequence is selected as the target, and a single-layer neural network is employed to predict the surrounding context substructures associated with it. Notably, each substructure within the sequence serves as the target substructure once per training epoch. Upon completion of training, the neural network’s weights are utilized as the PE. The PE generated in this work follows the process described in ref. 47.
A RF regression model is trained on Dataset 4 to determine the optimal representation method for polymer blends. Dataset 4 is first randomly split into training and test sets with a ratio of 80%:20%. The RF regressor from the scikit-learn library is utilized, and five-fold cross-validation is performed on the training dataset to select the major hyperparameters of the model ‘n_estimators’ and ‘max_depth’. Variable ‘n_estimators’ is searched over a range of 10 evenly spaced numbers within the interval [200, 2000], and variable ‘max_depth’ is searched over a range of 11 evenly spaced numbers within the interval [10, 110]. After the model is trained using the selected best hyperparameters on the training dataset, the performance of the model on the hold-out test dataset is used as the criteria to evaluate the representation effectiveness.
Active learning
In the AL workflow, an RF classification model is iteratively trained on Dataset 4, which contains all the labeled data. This iterative process involves updating both Dataset 4 and Dataset 5 by transferring selected candidates from Dataset 5 to Dataset 4. These candidates are labeled using MD simulation before being added to Dataset 4 and subsequently removed from Dataset 5. In each iteration, the RF classifier from the scikit-learn library is utilized, and two-fold cross-validation is performed on Dataset 4 to select the major hyperparameters of the model ‘n_estimators’ and ‘max_depth’. Variable ‘n_estimators’ is searched over a range of 20 evenly spaced numbers within the interval [100, 1000], and variable ‘max_depth’ is searched over a range of 11 evenly spaced numbers within the interval [10, 110]. After training the classifier with the selected optimal hyperparameters on the updated Dataset 4, it is employed to assess the candidates in the updated Dataset 5. Each candidate is assigned a predicted probability, ranging from 0 to 1, indicating the likelihood of being a high-performance polymer blend. Subsequently, the top k candidates with the highest predicted probabilities are chosen for subsequent MD testing and labeling. The specific value of k for each iteration is presented in Table 2. This is the so-called certainty-based sampling. To assess and showcase the classifier’s prediction performance, we also conduct a random sampling from Dataset 5 before the certainty-based sampling during every iteration. Since a batch of data points (from both certainty-based sampling and random sampling) is queried and added to the training data pool in every iteration, the AL process involves a hybrid acquisition method that is more balanced between exploration and exploitation.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its supplementary information files or will be available for download from https://github.com/Jiaxin-Xu/PolymerBlendTC-ActiveLearning upon publication.
Code availability
The code for the active-learning process will be available for download from https://github.com/Jiaxin-Xu/PolymerBlendTC-ActiveLearning upon publication. Other codes can be available upon reasonable request from the authors.
References
IEA, Heating. https://www.iea.org/reports/heating. (2022).
Pop, E. Energy dissipation and transport in nanoscale devices. Nano Res. 3, 147–169 (2010).
Xing, W., Xu, Y., Song, C. & Deng, T. Recent advances in thermal interface materials for thermal management of high-power electronics. Nanomaterials 12, 3365 (2022).
Elavarasan, R. M. et al. “Pathways toward high-efficiency solar photovoltaic thermal management for electrical, thermal and combined generation applications: a critical review”. Energy Convers. Manag. 255, 115278 (2022).
Ning, C.-Z. Semiconductor nanolasers and the size-energy-efficiency challenge: a review. Adv. Photonics 1, 014002 (2019).
Chen, H. et al. “Thermal conductivity of polymer-based composites: fundamentals and applications”. Prog. Polym. Sci. 59, 41–85 (2016).
Feng, C.-P. et al. Recent advances in polymer-based thermal interface materials for thermal management: A mini-review. Compos. Commun. 22, 100528 (2020).
Wei, X., Wang, Z., Tian, Z. & Luo, T. Thermal transport in polymers: a review. J. Heat Transfer 143, 072101 (2021).
Yang, Y. Thermal conductivity. In: Physical properties of polymers handbook (ed. Mark, J. E.) 155–163 (Springer, New York, NY, 2007).
Chen, Y.-M. & Ting, J.-M. Ultra high thermal conductivity polymer composites. Carbon 40, 359–362 (2002).
Yang, X. et al. A review on thermally conductive polymeric composites: classification, measurement, model and equations, mechanism and fabrication methods. Adv. Compos. Hybrid. Mater. 1, 207–230 (2018).
Li, C. et al. Polymer composites with high thermal conductivity optimized by polyline-folded graphite paper. Compos. Sci. Technol. 188, 107970 (2020).
Li, C. et al. Enhancement of thermal conductivity for epoxy laminated composites by constructing hetero-structured GF/BN networks. J. Appl. Polym. Sci. 140, e53252 (2023).
Sheng, Y. et al. Multiscale modeling of thermal conductivity of hierarchical CNT-polymer nanocomposite system with progressive agglomeration. Carbon 201, 785–795 (2023).
Henry, A. Thermal transport in polymers. Annu. Rev. Heat. Transf. 17, 485–520 (2014).
Zhang, T. & Luo, T. Role of chain morphology and stiffness in thermal conductivity of amorphous polymers. J. Phys. Chem. B 120, 803–812 (2016).
Wei, X., Zhang, T. & Luo, T. Chain conformation-dependent thermal conductivity of amorphous polymer blends: the impact of inter- and intra-chain interactions. Phys. Chem. Chem. Phys. 18, 32146–32154 (2016).
Luo, T. & Chen, G. Nanoscale heat transfer – from computation to experiment. Phys. Chem. Chem. Phys. 15, 3389–3412 (2013).
Kim, G.-H. et al. High thermal conductivity in amorphous polymer blends by engineered interchain interactions. Nat. Mater. 14, 295–300 (2015).
Guo, Y., Zhou, Y. & Xu, Y. Engineering polymers with metal-like thermal conductivity—present status and future perspectives. Polymer 233, 124168 (2021).
Qian, X., Zhou, J. & Chen, G. Phonon-engineered extreme thermal conductivity materials. Nat. Mater. 20, 1188–1202 (2021).
Choy, C. L., Luk, W. H. & Chen, F. C. Thermal conductivity of highly oriented polyethylene. Polymer 19, 155–162 (1978).
Choy, C. L., Wong, Y. W., Yang, G. W. & Kanamoto, T. Elastic modulus and thermal conductivity of ultradrawn polyethylene. J. Polym. Sci. Part B Polym. Phys. 37, 3359–3367 (1999).
Shen, S., Henry, A., Tong, J., Zheng, R. & Chen, G. Polyethylene nanofibres with very high thermal conductivities. Nat. Nanotechnol. 5, 251–255 (2010).
Xu, Y. et al. Nanostructured polymer films with metal-like thermal conductivity. Nat. Commun. 10, 1771 (2019).
Singh, V. et al. High thermal conductivity of chain-oriented amorphous polythiophene. Nat. Nanotechnol. 9, 384–390 (2014).
Xu, Y. et al. “Molecular engineered conjugated polymer with high thermal conductivity”. Sci. Adv. 4, eaar3031 (2018).
Shanker, A. et al. High thermal conductivity in electrostatically engineered amorphous polymers. Sci. Adv. 3, e1700342 (2017).
Xie, X. et al. Thermal conductivity, heat capacity, and elastic constants of water-soluble polymers and polymer blends. Macromolecules 49, 972–978 (2016).
Mu, L. et al. Molecular origin of efficient phonon transfer in modulated polymer blends: effect of hydrogen bonding on polymer coil size and assembled microstructure. J. Phys. Chem. C. 121, 14204–14212 (2017).
Yong, W. F. & Zhang, H. Recent advances in polymer blend membranes for gas separation and pervaporation. Prog. Mater. Sci. 116, 100713 (2021).
Agari, Y., Ueda, A., Omura, Y. & Nagai, S. Thermal diffusivity and conductivity of PMMA/PC blends. Polymer 38, 801–807 (1997).
Guo, Z. et al. Thermal conductivity of organic bulk heterojunction solar cells: an unusual binary mixing effect. Phys. Chem. Chem. Phys. 16, 26359–26364 (2014).
Taraghi, I. et al. Thermally and electrically conducting polycarbonate/elastomer blends combined with multiwalled carbon nanotubes. J. Thermoplast. Compos. Mater. 34, 1488–1503 (2021).
Duda, J. C., Hopkins, P. E., Shen, Y. & Gupta, M. C. Thermal transport in organic semiconducting polymers. Appl. Phys. Lett. 102, 251912 (2013).
Mehra, N., Mu, L., Ji, T., Li, Y. & Zhu, J. Moisture driven thermal conduction in polymer and polymer blends. Compos. Sci. Technol. 151, 115–123 (2017).
Bruns, D., de Oliveira, T. E., Rottler, J. & Mukherji, D. Tuning morphology and thermal transport of asymmetric smart polymer blends by macromolecular engineering. Macromolecules 52, 5510–5517 (2019).
Xie, S. “Perspectives on development of biomedical polymer materials in artificial intelligence age”. J. Biomater. Appl. 37, 1355–1375 (2023).
Martin, T. B. & Audus, D. J. Emerging trends in machine learning: a polymer perspective. ACS Polym. Au. https://doi.org/10.1021/acspolymersau.2c00053 (2023).
Xu, P., Chen, H., Li, M. & Lu, W. New opportunity: machine learning for polymer materials design and discovery. Adv. Theory Simul. 5, 2100565 (2022).
Cencer, M. M., Moore, J. S. & Assary, R. S. Machine learning for polymeric materials: an introduction. Polym. Int. 71, 537–542 (2022).
Patra, T. K. Data-driven methods for accelerating polymer design. ACS Polym. Au 2, 8–26 (2022).
Ma, R., Zhang, H. & Luo, T. Exploring high thermal conductivity amorphous polymers using reinforcement learning. ACS Appl. Mater. Interfaces 14, 15587–15598 (2022).
Kuenneth, C. et al. Bioplastic design using multitask deep neural networks. Commun. Mater. 3, 96 (2022).
Wu, S. et al. Machine-learning-assisted discovery of polymers with high thermal conductivity using a molecular design algorithm. Npj Comput. Mater. 5, 1–11 (2019).
Kuenneth, C. et al. Polymer informatics with multi-task learning. Patterns 2, 100238 (2021).
Ma, R. & Luo, T. PI1M: a benchmark database for polymer informatics. J. Chem. Inf. Model. 60, 4684–4690 (2020).
Kuenneth, C., Schertzer, W. & Ramprasad, R. Copolymer informatics with multitask deep neural networks. Macromolecules 54, 5957–5961 (2021).
Boublia A. et al. Multitask neural network for mapping the glass transition and melting temperature space of homo- and co-polyhydroxyalkanoates using σProfiles molecular inputs. ACS Sustain. Chem. Eng. https://doi.org/10.1021/acssuschemeng.2c05225 (2022).
Tao, L., Arbaugh, T., Byrnes, J., Varshney, V. & Li, Y. Unified machine learning protocol for copolymer structure-property predictions. STAR Protoc. 3, 101875 (2022).
Chen, L. et al. Polymer informatics: current status and critical next steps. Mater. Sci. Eng. R. Rep. 144, 100595 (2021).
Liang, Z. et al. Machine-learning exploration of polymer compatibility. Cell Rep. Phys. Sci. 3, 100931 (2022).
Settles, B. “Active learning literature survey”. Computer Sciences Technical Report 1648, University of Wisconsin–Madison (2009).
Oftelie, L. B. et al. Active learning for accelerated design of layered materials. Npj Comput. Mater. 4, 1–9 (2018).
Smith, J. S., Nebgen, B., Lubbers, N., Isayev, O. & Roitberg, A. E. Less is more: sampling chemical space with active learning. J. Chem. Phys. 148, 241733 (2018).
Zhang, Y. & Lee, A. A. Bayesian semi-supervised learning for uncertainty-calibrated prediction of molecular properties and active learning. Chem. Sci. 10, 8154–8163 (2019).
Rakhimbekova, A. et al. Efficient design of peptide-binding polymers using active learning approaches. J. Control. Release 353, 903–914 (2023).
Ma, R. et al. Machine learning-assisted exploration of thermally conductive polymers based on high-throughput molecular dynamics simulations. Mater. Today Phys. 28, 100850 (2022).
Ma, R., Liu, Z., Zhang, Q., Liu, Z. & Luo, T. Evaluating polymer representations via quantifying structure–property relationships. J. Chem. Inf. Model. https://doi.org/10.1021/acs.jcim.9b00358 (2019).
Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010).
Jaeger, S., Fulle, S. & Turk, S. Mol2vec: unsupervised machine learning approach with chemical intuition. J. Chem. Inf. Model. 58, 27–35 (2018).
Mosqueira-Rey, E., Hernández-Pereira, E., Alonso-Ríos, D., Bobes-Bascarán, J. & Fernández-Leal, Á. Human-in-the-loop machine learning: a state of the art. Artif. Intell. Rev. 56, 3005–3054 (2023).
Bondu, A., Lemaire, V. & Boulle, M. Exploration vs. exploitation in active learning: a bayesian approach. The 2010 International Joint Conference on Neural Networks (IJCNN) 1–7. IEEE, Barcelona, Spain, 2010).
Grabowski, S. J. Ab initio calculations on conventional and unconventional hydrogen bondsstudy of the hydrogen bond strength. J. Phys. Chem. A 105, 10739–10746 (2001).
Zahn, S., Wendler, K., Delle Site, L. & Kirchner, B. Depolarization of water in protic ionic liquids. Phys. Chem. Chem. Phys. 13, 15083–15093 (2011).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
He, X., Man, V. H., Yang, W., Lee, T.-S. & Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 153, 114502 (2020).
Zhang, T. et al. Role of hydrogen bonds in thermal transport across hard/soft material interfaces. ACS Appl. Mater. Interfaces 8, 33326–33334 (2016).
Vassetti, D., Pagliai, M. & Procacci, P. Assessment of GAFF2 and OPLS-AA general force fields in combination with the water models TIP3P, SPCE, and OPC3 for the solvation free energy of druglike organic molecules. J. Chem. Theory Comput. 15, 1983–1995 (2019).
Agresti, A. Categorical data analysis. (John Wiley & Sons, 2012).
Polymer Database(PoLyInfo) - DICE: national institute for materials science. https://polymer.nims.go.jp/en/.
pysimm: a python package for simulation of molecular systems | Elsevier Enhanced Reader. https://doi.org/10.1016/j.softx.2016.12.002 (2017)
Wei, X. & Luo, T. Chain length effect on thermal transport in amorphous polymers and a structure–thermal conductivity relation. Phys. Chem. Chem. Phys. 21, 15523–15530 (2019).
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 117, 1–19 (1995).
Ryckaert, J.-P., Ciccotti, G. & Berendsen, H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Morgan, H. L. The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. J. Chem. Doc. 5, 107–113 (1965).
Acknowledgements
We would like to acknowledge funding support from the U.S. National Science Foundation (CDSE-2102592).
Author information
Authors and Affiliations
Contributions
J.X. and T.L. conceived the idea and initiated this project. J.X. designed and trained the model. All authors have contributed to the writing of the paper and the analysis of the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, J., Luo, T. Unlocking enhanced thermal conductivity in polymer blends through active learning. npj Comput Mater 10, 74 (2024). https://doi.org/10.1038/s41524-024-01261-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-024-01261-2
