
Abstract
Understanding the lattice dynamics and heat transport physics in the lead-free halide double perovskites remains an outstanding challenge due to their lattice dynamical instability and strong anharmonicity. In this work, we investigate the microscopic mechanisms of anharmonic lattice dynamics and thermal transport in lead-free halide double perovskite Cs2AgBiBr6 from first principles. We combine self-consistent phonon calculations with bubble diagram correction and a unified theory of lattice thermal transport that considers both the particle-like phonon propagation and wave-like tunnelling of phonons. An ultra-low thermal conductivity at room temperature (~0.21 Wm−1K−1) is predicted with weak temperature dependence( ~ T−0.34), in sharp contrast to the conventional ~T−1 dependence. Particularly, the vibrational properties of Cs2AgBiBr6 are featured by strong anharmonicity and wave-like tunnelling of phonons. Anharmonic phonon renormalization from both the cubic and quartic anharmonicities are found essential in precisely predicting the phase transition temperature in Cs2AgBiBr6 while the negative phonon energy shifts induced by cubic anharmonicity has a significant influence on particle-like phonon propagation. Further, the contribution of the wave-like tunnelling to the total thermal conductivity surpasses that of the particle-like propagation above around 310 K, indicating the breakdown of the phonon gas picture conventionally used in the Peierls-Boltzmann Transport Equation. Importantly, further including four-phonon scatterings is required in achieving the dominance of wave-like tunnelling, as compared to the dominant particle-like propagation channel when considering only three-phonon scatterings. Our work highlights the importance of lattice anharmonicity and wave-like tunnelling of phonons in the thermal transport in lead-free halide double perovskites.
Similar content being viewed by others
Introduction
Halide perovskite materials, with a general chemical formula ABX3, where A is a monovalent cation (Cs+, methylammonium (CH3NH3+) or formamidinium (CH(NH2)2+)), B is a bivalent cation (Pb2+ or Sn2+), and X is a halide anion (Cl−, Br− or I−), have emerged as a new class of materials with outstanding optoelectronic properties. Generally, lead-based halide perovskites possess unique merits as optoelectronic materials, exhibiting long carrier lifetime and diffusion length, tuneable direct bandgap, high absorption coefficient and low trap density1,2,3. These properties make halide perovskites an ideal option for photovoltaic and optoelectronic devices in recent years. However, lead-based perovskite materials still encounter two crucial challenges, i.e., poor long-term stability and the toxicity of lead elements3. A promising strategy to tackle these issues is to replace the divalent Pb2+/ Sn2+ cation with one monovalent and one trivalent metal cation in the lead halide perovskites to form the so-called lead-free double perovskites A2B+B3+X6 (A = K, Rb, Cs; B+ = Li, Na, K, Rb, Cs, Ag; B3+ = Al, Ga, In, Sb, Bi, Sc, Y, and X = F, Cl, Br, I)3,4. The structural and functional diversity and the long-term stability of lead-free double perovskites render them attractive candidates for optoelectronic devices.
Apart from the optoelectronic properties, halide perovskites also attract research interest in lattice dynamics and thermal transport characteristics, which affect their thermodynamical stability5, opto-electronic and photovoltaic performances6. Particularly, the effects of lattice vibrations on electronic and optoelectronic properties have been widely investigated to slow down the cooling of charge carriers in halide perovskites7,8,9. Besides, the experimentally reported ultra-low thermal conductivity κL10,11,12,13,14,15, high carrier mobilities12,16,17 in conjunction with large Seebeck coefficient16,17 make halide perovskites attractive for thermoelectric applications. A high ZT of 1~2 in hybrid halide perovskite was predicted by ab initio calculations18. Importantly, the ultra-low κL of halide perovskites was attributed to the low phonon group velocities and short phonon lifetimes19 stemming from the complicated crystal structures and extremely strong lattice anharmonicity. Despite the progress in experimentally and theoretically observed thermal properties of halide perovskites12,13,14,15,20, oxide and fluoride perovskites21, an accurate quantitative prediction of their lattice dynamical properties and a comprehensive microscopic understanding of their thermal transport properties are still in infancy.
The outstanding challenges in theoretical understanding of phonon-related properties in halide perovskites can be traced back to the strong lattice anharmonicity, which makes the conventional approaches within the harmonic approximation framework fail22,23,24,25. To overcome the shortcoming of harmonic approximation, the anharmonic phonon renormalization techniques22,23,24,26 have been applied to predict the phase transition temperature and thermal properties of halide perovskites at finite temperatures27,28,29. However, most of works consider only cubic-anharmonicity and the explicit treatment of effects of both cubic- and quartic-anharmonicities on phonon energies and transport channels remains challenging. Also, the particle-like phonon picture in describing thermal transport physics generally breaks down in highly anharmonic materials when the phonon linewidths are larger than the interbranch spacings30,31,32,33. For example, using the conventional first-principles-based Peierls-Boltzmann transport equation (PBTE) method34, Lee et al.12 failed to reproduce the experimentally measured ultra-low κL in all-inorganic halide perovskites. Going beyond the conventional particle-like description of phonons in PBTE, Simoncelli et al.30 explained the experimentally observed lattice thermal conductivity in the complex perovskite CsPbBr3 by further considering wave-like tunneling of phonons. The unified theory of thermal transport adopted in the above literature30 can accurately reproduce the κL; however, it considers only the three-phonon scattering processes within the harmonic approximation treatment (The error cancellation due to phonon renormalization and four phonon scattering rates may occurs in the κL of CsPbBr3, similar to that observed in PbTe35). Therefore, a comprehensive understanding of the lattice dynamics and thermal properties in lead-free halide double perovskites have been rarely reported.
In this paper, we systematically investigate the anharmonic lattice dynamics and thermal transport properties in a benchmark lead-free halide double perovskite Cs2AgBiBr6 at the atomic level. We have applied a state-of-the-art unified theory of thermal transport to predict the lattice thermal conductivity κL in perovskite Cs2AgBiBr6 by considering the isotope-, three- (3 ph) and four-phonon (4 ph) scatterings, anharmonic phonon renormalization from both cubic and quartic anharmonicities, particle-like propagation and wavelike tunneling transport channels. Using the anharmonic phonon renormalization technique, we find that the soft modes are associated with the tilting of the AgBr6 and BiBr6 octahedra units and become hardened with increasing temperature. The cubic-to-tetragonal phase transition temperature of Cs2AgBiBr6 is predicted as ~119–138 K, agreeing well with the experimental value. Using the unified theory of thermal transport, we show that both the 4 ph scattering processes and wavelike tunneling of phonons play a crucial role in predicting the κL of Cs2AgBiBr6. Particularly, considering only the 3 ph scattering, the populations’ conductivity accounts for more than 50% of the total κL over the entire temperature range. By further considering 4 ph scatterings, the coherences’ conductivity ascribing from the wave-like tunneling channel dominates the total κL above ~310 K. This study highlights the importance of wave-like tunneling and quartic anharmonicity in describing heat conduction in Cs2AgBiBr6.
Results and discussion
Lattice dynamics and anharmonicity
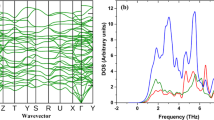
We start by investigating the vibrational properties of double perovskite Cs2AgBiBr6 in its cubic phase (as shown in Fig. 1a, b) using the static harmonic approximation (HA) approach. The calculated harmonic phonon dispersions (represented by dark blue dash lines) and density of states (DOS) of Cs2AgBiBr6 are shown in Fig. 1c. One of the most notable characteristics of the harmonic phonon dispersions in cubic Cs2AgBiBr6 is the occurrence of soft modes with negative phonon energies, which are observed at the Γ and X points in the Brillouin zone. This feature suggests the presence of dynamical instability at low temperatures [see in Fig. 1c]. Upon analyzing the vibrational properties of Cs2AgBiBr6, we have observed that the soft modes at the Γ and X points are associated with the tilting of the AgBr6 and BiBr6 octahedra units, respectively, which was also reported by Klarbring et al.29. By applying a comprehensive anharmonic phonon renormalization technique, these soft modes are found to become significantly hardened with increasing temperature [see anharmonically renormalized phonon dispersions and DOS in Fig. 1c]. The soft modes in Cs2AgBiBr6 are primarily contributed by Br atoms, which is evident from the atom-decomposed partial DOS [see the right panel in Fig. 1c] and atomic participation ratio of Br atoms projected onto the phonon bands [see Supplementary Fig. 1 (See Supplementary Information for additional figures illustrating harmonic and anharmonic phonon properties, along with thermal transport properties, and further discussions)]. Apart from the Br-dominated soft modes, the low-frequency phonon modes (≤2.2 THz), contributed by Cs atoms, also undergo a gradual stiffening [see Fig. 1c and Supplementary Fig. 1]. The temperature-dependent stiffening behavior of low-frequency modes is attributed to the large atomic displacements that arise from the relatively weak bonding interactions [see Supplementary Fig. 2]. In contrast, the high-frequency phonon modes (>2.2 THz) in Cs2AgBiBr6 are predominantly contributed by Ag and Br atoms [see Supplementary Fig. 1] and exhibits a temperature-dependent softening, consistent with experimental observations36.

a Crystal structure of the cubic double perovskite Cs2AgBiBr6, which features two different sub-lattices formed by AgBr6 and BiBr6 octahedra units, respectively, together with Cs atoms. The solid green lines inside the sub-lattice depict a conventional unit cell, and the green, white, blue, and brown spheres represent Cs, Ag, Bi and Br atoms, respectively. b View of the yz plane of the crystal structure. c Calculated anharmonically renormalized phonon dispersions at finite temperatures (from T = 100 to 700 K) compared with the phonon dispersions computed by the harmonic approximation treatment at T = 0 K. The calculated atom-decomposed partial and total phonon densities of states at T = 0, 300, and 700 K are shown on the right panel, respectively. d Calculated temperature-dependent squared phonon frequency of soft modes at the Γ and X points using the SCP and SCPB approximations, respectively. The empty red circle indicates the experimentally measured phase transition temperature (~122 K)36,39,40, and the empty blue pentagon indicates the experimental phonon energies of the soft mode at the Γ point at 135 and 300 K, respectively40. e Calculated temperature-dependent renormalized phonon dispersions along the high-symmetry paths compared with experimental results at 300 K40.
It is worth noting that in some crystals, the phonon energy shifts resulting from 3 ph interaction processes, specifically the bubble diagram (including those in the SCPB approximation), can be ignored due to their minor contributions37 and computational complexity38. In the case of crystalline Cs2AgBiBr6, considering only 4 ph processes, i.e., the loop diagram (SCP approximation), in anharmonic phonon renormalization shows only phonon stiffening [see Supplementary Fig. 3]. Additionally, the anharmonically renormalized phonon energies in Cs2AgBiBr6 are stabilized above 50 K when considering only 4 ph processes [see Fig. 1d]. This value is close to the prediction of Klarbring et al.29 but significantly lower than the experimental value of approximately 122 K36,39,40. The observed discrepancy in the phase transition temperature of Cs2AgBiBr6, between the value computed solely from 4 ph processes and the experimental result, underscores the crucial role of the negative phonon energy shifts from 3 ph processes in accurately determining phonon energies. By incorporating the 3 ph processes in phonon renormalization calculations, it predicts that the cubic-to-tetragonal phase transition of Cs2AgBiBr6 occurs at ~119 K (phonons collapse at X point) and ~138 K (phonons collapse at Γ point) [see in Fig. 1d], in good agreement with experimental measurements36,39,40. Furthermore, in comparison to the SCP approximation, the SCPB approximation provides more accurate predictions for the phonon energies of the soft mode at Γ point, as observed in experiments conducted at 135 and 300 K [see in Fig. 1d]40. To validate out calculations, we compared the phonon dispersions along high-symmetry paths calculated using the SCPB approximation with the experimental results obtained from inelastic neutron scattering measurements40. The comparison shows good agreement between the calculated and experimental values [see Fig. 1e and Supplementary Figs. 4 and 5]. The importance of both 3 ph and 4 ph processes in reproducing the experimental lattice vibrational properties of Cs2AgBiBr6 crystal is highlighted by these results, as has been demonstrated for several other compounds, e.g., BaZrO3, CsPbBr3 and SnSe38,41,42.
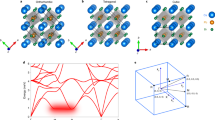
To gain insight into lattice instability, anharmonicity, and phonon energy shift in Cs2AgBiBr6 crystal, we mapped out the 2D potential energy surface (PES)43 of soft modes at the Γ and X points. The PES was plotted along two corresponding normal-mode coordinates (Q1, and Q2) [see Fig. 2a]. The soft modes at the Γ and X points are associated with the out-of-phase and in-phase tilting of the AgBr6 and BiBr6 octahedra units, respectively, as shown in Fig. 2b, c. As anticipated, the minimum energy is located outside the zero-tilt amplitude (\({Q}_{1}={Q}_{2}=0\)) of both soft modes at the Γ and X points, indicating lattice instability or dynamical instability [see Fig. 2a]43. The 2D PES mapping in Fig. 2a reveals that the minimum energy of the soft mode at Γ point is lower than that of soft mode at the X point. This observation suggests the dominant role of the out-of-phase tilting mode, associated with the AgBr6 octahedra, in the cubic-to-tetragonal phase transition of Cs2AgBiBr6. Although the minimum energies exist outside the zero-tilt amplitude for both soft modes, the corresponding minima are relatively shallow, with values of about −0.79 and −0.45 meV∙atom−1, respectively. This observation suggests that the high-temperature cubic phase of Cs2AgBiBr6 crystal is effectively an average structure at relatively low temperatures, similar to the specific phase of some previously reported compounds such as CsSnI3, CsSnBr3 and CsSnCl344,45,46.

a Calculated two-dimensional (2D) potential energy surface of Cs2AgBiBr6 as a function of normal mode coordinates Q1 and Q2. Here the soft modes at the Γ and X points associated with the out-of-phase and in-phase tilts are selected to generate the corresponding configurations. b Crystal structure of Cs2AgBiBr6 with displacement pattern of the soft mode at the Γ point, wherein the red and light blue circles with arrows are depicted for the displacements of AgBr6 and BiBr6 octahedron units (out-of-phase tilting), respectively. c The same as b, but with displacement pattern of the soft mode at the X point and (in-phase tilting). d DFT-calculated double well potential energy surface (solid green disks) of the soft mode at the Γ point as a function of the vibrational amplitude (Q1) in the normal mode coordinate. The blue, yellow and green dash lines show the potential energy surface decomposed to the second, fourth and sixth orders, respectively. The blue, brown and pink solid lines represent the renormalized harmonic potentials at T = 300, 500, and 700 K, respectively. e The same as d, but for soft mode at the X point and the normal mode coordinate (Q2).
To gain a more intuitive understanding of the lattice anharmonicity of Cs2AgBiBr6, we projected the 2D PES onto 1D PES for both the soft modes at Γ and X points [see Fig. 2d, e]. As expected, each soft mode forms a double-well potential, suggesting strong lattice anharmonicity47. Indeed, the anharmonic double-well potentials of soft modes can be decomposed up to the sixth order, although the contribution of the sextic terms is minor [see Fig. 2d, e]. Additionally, the harmonic potential (blue dash line) of soft modes has a negative coefficient, which is responsible for the imaginary frequency [see Fig. 1b], and therefore leads to the failure of the conventional harmonic approximation (HA) treatment22,34,47. It is worth mentioning that although the PESs of the soft modes exhibit anharmonic behavior up to the sixth-order terms, the contribution of these higher-order terms is minor. Therefore, in our anharmonic phonon renormalization calculations, we only consider the anharmonic contributions up to fourth-order terms. The lattice anharmonicity was utilized in the anharmonic phonon renormalization to gradually transform the negative harmonic potentials of the soft modes into positive ones at finite temperatures, such as 300 K, 500 K and 700 K [see Fig. 2d, e]. These positive potentials represent the effective harmonic potential with temperature effects taken into account.
Lattice thermal conductivity
Next, we investigate the effects of phonon energy renormalization and multi-phonon interaction from both cubic and quartic anharmonicities on lattice thermal conductivity κL. To achieve this, we employ a hierarchy of theoretical approaches with increasing accuracy, as depicted in Fig. 3a. Due to the failure of conventional harmonic approximation (HA) treatment in capturing the lattice dynamics of Cs2AgBiBr6, we begin our analysis by examining the κL obtained using the SCP+3 ph model. The populations’ thermal conductivity \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) was computed using the SCP+3 ph model, which takes into account renormalized phonon energies from only quartic anharmonicity and employs only 3 ph scattering processes. The resulting value at 300 K is ~0.38 \({\rm{W}}{{\rm{m}}}^{-1}{{\rm{K}}}^{-1}\), as shown in Fig. 3b. This predicted \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) is consistent with the result reported by Klarbring et al.29, although their method utilized the temperature-dependent effective potential approach based on AIMD simulations. As discussed earlier, the anharmonic phonon renormalization using only quartic anharmonicity can lead to an overestimation of the phonon energies and phase transition temperature of Cs2AgBiBr6. To account for extra phonon energy shifts from cubic anharmonicity, we move up the theory level progressively to the SCPB+3 ph model. As a result, we observe a remarkable decrease in \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) from ~0.38 to ~0.17 Wm−1 K−1 (~55.95% reduction) at 300 K, as demonstrated in Fig. 3b. This observation highlights the significant influence of cubic anharmonicity not only on lattice dynamics but also on phonon transport in crystalline Cs2AgBiBr6. A similar phenomenon has been reported for simple cubic perovskites, e.g., CsSnBr3, BaZrO3 and CsPbBr3, in previous studies28,38,41. To properly account for the anharmonicity of highly anharmonic compounds, it is necessary to include multi-phonon scatterings resulting from quartic anharmonicity25,47. By further incorporating 4 ph scattering processes into the SCPB+3 ph model, the improved SCPB+3,4 ph model predicts a further reduction in \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\). Specifically, the predicted \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) decreases to ~0.11 and ~0.06 Wm−1 K−1 (~34.6 and 51.8% reduction) at 300 and 700 K, respectively [see Fig. 3b]. This highlights the crucial role of 4 ph scattering processes in determining the \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) of highly anharmonic Cs2AgBiBr6. Recent studies have emphasized the importance of coherences’ contributions from non-diagonal terms of heat flux operators, i.e., wave-like tunneling channel, in compounds with ultra-low κL or/and strong anharmonicity30,31,47,48. By incorporating the wave-like tunneling transport channel30,48 through non-diagonal terms of heat flux operators, beyond the phonon-gas model, the SCPB+3,4 ph+OD model results in a substantial increase in κL to 0.21 and 0.16 Wm−1 K−1 (~93.9 and 184.2% enhancement) at 300 and 700 K, respectively. Interestingly, using the SCPB+3,4 ph+OD model reveals that the total κL contributed from the wavelike tunneling channel comprises more than 50% of the total κL above 310 K [see Fig. 3b, c]. In contrast, when considering only 3 ph scatterings, the phonon propagation channel dominates the total κL, accounting for more than 50% of the total heat conduction over the entire temperature range [see Fig. 3c]. The presence of strong 4 ph scatterings triggers the inter-conversion of the dominant role of heat conduction between the particle-like phonon propagation and the wave-like tunneling channels in Cs2AgBiBr6. This observation emphasizes the limitations of the conventional phonon-gas model and highlights the importance of considering higher-order anharmonicity and coherences’ effects in advanced thermal transport models to accurately describe the thermal transport of Cs2AgBiBr6.

a A schematic diagram depicting the prediction accuracy of lattice thermal conductivity using various hierarchical thermal transport models, namely, the HA+3 ph, SCP+3 ph, SCPB+3 ph, SCPB+3,4 ph, and SCPB+3,4 ph+OD models, respectively. The ellipsis indicates the theory of thermal transport beyond the SCPB+3,4 ph+OD model and it may further include anharmonic heat flux operators61. b Calculated temperature-dependent lattice thermal conductivity \({\kappa }_{{\rm{L}}}\) using different levels of thermal transport theory including the SCP/SCPB+3 ph, SCPB+3,4 ph+OD models. The yellow and pink shaded areas show the contributions from phonon and coherence thermal transport channels calculated using the SCPB+3,4 ph+OD model, respectively. c Temperature-dependent populations’, coherences’ and total thermal conductivity calculated using SCPB+3 ph+OD model and the coherences’ contributions, percentage wise, to total thermal conductivity calculated using SCPB+3/3,4 ph models. d Comparison of our predicted lattice thermal conductivity of crystalline Cs2AgBiBr6 with other experimentally measured ultra-low thermal conductivity of all-inorganic halide perovskites at room temperature12,13,14,15,51,52.
The dominant role of coherences’ contributions \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) to the total κL over a wide range of temperatures implies that the heat transfer physics in Cs2AgBiBr6 exhibits similar features to those observed in glasses. We proceed to analyze the temperature dependence of the lattice thermal conductivity of Cs2AgBiBr6 calculated using different levels of theory. The predicted \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) predicted by the SCP+3 ph model follows an extremely weak temperature dependence of \(\sim {T}^{-0.28}\) in the temperature range of 300–700 K [see Fig. 3b]. This observation challenges the conventional understanding that the \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) computed by the lowest-order perturbation theory49 follows a temperature dependence of \(\sim {T}^{-1}\). Using the SCPB+3 ph model, the temperature dependence of the \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) becomes stronger and follows \(\sim {T}^{-0.40}\), which is a result of the softening of phonon frequency. Further including 4 ph scattering processes leads to a significant change in the temperature dependence of the \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\), which is remarkably enhanced to \(\sim {T}^{-0.75}\). This observation is consistent with the findings of Feng et al.50 and Zheng et al.41 By utilizing the SCPB+3,4 ph+OD model, the κL exhibits a weak temperature dependence of \(\sim {T}^{-0.34}\), resembling the experimentally observed out-of-plane temperature-dependent behavior of κL in Cs2PbI2Cl214 [see Fig. 3b].
To validate our computational results, we compared the calculated κL of Cs2AgBiBr6 with the available experimental data for all-inorganic halide perovskites at 300 K12,13,14,15,51,52, as illustrated in Fig. 3d. Our predicted total κL for Cs2AgBiBr6 is within the same order of magnitude (≤0.64 Wm−1 K−1) as the κL values reported for other halide perovskites [see Fig. 3d]. After a thorough examination, we discovered that the experimental κL of halide perovskites tends to decrease as the structural complexity increase. Structural complexity is typically characterized by the size of the unit-cell and the types of elements present in the halide perovskite crystal. An experiment recently reported an ultralow room-temperature κL of 0.31 Wm−1 K−1 in crystalline Cs2SnI651. Given the similarity in structure and phonon dispersions between Cs2SnI6 and Cs2AgBiBr653, we can expect a similar result for Cs2AgBiBr6. Furthermore, the frequency of soft mode at Γ point in Cs2SnI6 is around 0.6 THz, which is higher than that in Cs2AgBiBr6 (~0.4 THz) at 300 K. Therefore, we can expect a lower experimental κL in Cs2AgBiBr6 as compared to Cs2SnI6. An even lower κL value of 0.19 Wm−1 K−1 was observed for the halide perovskite Cs3Bi2I6Cl3, which has a higher structural complexity than Cs2AgBiBr6, in a recent experiment at room temperature15. Therefore, the κL of Cs2AgBiBr6 is expected to be higher than 0.19 Wm−1 K−1. Overall, the predicted total κL of Cs2AgBiBr6 in this study is considered to be reasonably reliable and can be verified by future experimental investigations.
Microscopic mechanisms of populations’ conductivity
To better illustrate the effect of anharmonic phonon renormalization and higher-order phonon scattering processes on the \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) of Cs2AgBiBr6, we present the spectral and cumulative κL in Fig. 4a, b calculated using SCP+3 ph and SCPB+3/3,4 ph models at 300 and 700 K, respectively. For all the three levels of theory, our results consistently show that most of populations’ thermal conductivity \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) in Cs2AgBiBr6 is carried by phonons with a frequency less than 3 THz across the temperature range of 300–700 K [see Fig. 4a, b]. Additionally, we observed that optical phonons with a frequency higher than 1.2 THz play a dominant role in the \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) from the particle-like propagation channel. This behavior is commonly observed in cubic SrTiO3 with strong anharmonicity and low-\({\kappa }_{{\rm{L}}}^{{\rm{P}}}\)22, and is attributed to the suppression of acoustic phonons by highly anharmonic optical modes.

a Calculated spectral and cumulative phonon thermal conductivities at T = 300 K using the SCP+3 ph and SCPB+3/3,4 ph models, respectively. b The same as a, but T = 700 K. c Calculated 3 ph and 4 ph scattering rates at T = 300 K using the SCP+3 ph and SCPB+3/3,4 ph models, respectively. d The same as c, but T = 700 K.
The spectral and cumulative \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) obtained from the SCP+3 ph model show a reduction compared to the results from the SCPB+3 ph model. This decrease in \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) is mainly contributed by the phonons with frequency less than 3 THz [see Fig. 4a, b]. This is because the cubic anharmonicity contributes negative phonon energy shifts, which results in the softening of phonons and enhancement of phonon scattering processes [see Fig. 4c, d]. With the additional 4 ph scattering processes, the reduced \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) obtained by SCPB+3,4 ph relative to SCPB+3 ph is primarily contributed by phonons with a frequency less than 3 THz at both 300 and 700 K. This can be attributed to the inherently strong 4 ph scattering rates of specific modes, such as rattling-like flat modes, which will be demonstrated later.
By examining the group velocity, specific heat, and scattering rates, in computing \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\)34, the reduction in \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) in two improved models (SCPB+3/3,4 ph) relative to the SCP+3 ph model is mainly attributed to the scattering rates [see Fig. 4c, d]. This is due to the fact that the difference in group velocity and specific heat between the SCP and SCPB approximation is negligible in Cs2AgBiBr6, as demonstrated by see Supplementary Figs. 6 and 7. Indeed, previous studies have shown that the group velocity and specific heat of certain crystals are relatively insensitive to phonon energy shifts resulting from lattice anharmonicity25,41,47,54. As shown in Figs. 3b and 4a, b, the inclusion of additional 4 ph scatterings in the calculation of \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) (using the SCPB+3,4 ph model) leads to a significant reduction compared to considering only 3 ph processes (using the SCPB+3 ph model). Specifically, the reduction in \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) is 34.6 and 51.8% at 300 and 700 K, respectively. This can be ascribed to the large 4 ph scattering rates, which are roughly as high as those of 3 ph processes at 300 K, and even become dominant at 700 K, particularly for phonons with frequency less than 2 THz, as shown in Fig. 4c, d.
It is worth noting that both the 3 ph and 4 ph scattering rates decrease with increasing frequency for phonons below 1 THz (shown in the upper left part of Fig. 4c, d). Upon analysis of the distribution of scattering rates and atomic participation ratios [see Fig. 4a, b and Supplementary Fig. 1], we have found that the observed phenomenon can be attributed to the presence of Br-dominated soft modes, and a similar phenomenon was also reported for other compounds, e.g., SrTiO3, PbSe and PbTe22,55. Generally, the ultra-low κL observed in halide perovskites is attributed to the combination of strong scattering rates and small group velocity19,56. The small group velocity with a maximum value of ~2.35 Km∙s-1 observed in Cs2AgBiBr6 may lead to the ultra-low κL[see Supplementary Fig. 6]. However, neglecting the contribution from Br-dominated soft modes in 3 ph scattering processes calculation leads to a significant enhancement of \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\), with a value of ~87 Wm−1 K−1 at 300 K [see Supplementary Figs. 8 and 9]. Similarly, neglecting Cs-dominated modes leads to an increase in \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) to 18 from ~0.17 Wm−1 K−1 calculated using SCPB+3 ph [see Supplementary Figs. 8 and 9]. This illustrates the important role of the Br-dominated soft modes with strong anharmonicity [see Supplementary Figs. 8, 10 and 11] in contributing to the ultra-low \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) of Cs2AgBiBr6. Overall, the strong anharmonicity and resulting high scattering rates are the dominant factors in the ultra-low κL of Cs2AgBiBr6.
To reveal the correlation between the negative phonon energy shift due to cubic anharmonicity and the resulting change in phonon scattering rates, we compare scattering phase space57,58 calculated using the SCP and SCPB approximation at 300 and 700 K, as shown in Fig. 5a, b. The scattering phase space of emission processes computed with SCPB+3 ph is substantially larger relative to the SCP+3 ph results for phonons with a frequency less than 3 THz at 300 and 700 K. This observation suggests that the negative phonon shifts resulting from cubic anharmonicity leads to enhance the scattering phase space and thus stronger phonon scattering rates. Similar observations have been also reported in other crystals such as BaZrO3 and CsPbBr338,54. Similarly, the enhanced scattering rates for phonons with frequencies greater than 3 THz between the SCP+3 ph and SCPB+3 ph models can be attributed to the enhanced scattering phase space of absorption processes. Note that another key quantity in calculating scattering rates, namely, the scattering matrix element, is insensitive to the phonon energy shift due to lattice anharmonicity41.

a Comparison of mode-resolved 3 ph scattering phase space calculated using SCP and SCPB approximations at T = 300 K, respectively. The 3 ph scattering phase spaces are decomposed to the absorption (\(q+{q\text{'}}\to {q\text{'}\text{'}}\)) and emission (\(q\to {q}^{{\prime} }+{q\text{'}\text{'}}\)) processes, respectively. b The same as a, but at T = 700 K. c Mode-resolved 4 ph scattering phase space calculated using SCPB approximation at T = 300 K, wherein the 4 ph scattering phase spaces are decomposed to the recombination (\(q+{q}^{{\prime} }+{q\text{'}\text{'}}\to {q\text{'}\text{'}\text{'}}\)), redistribution (\(q+{q}^{{\prime} }\to {q\text{'}\text{'}}+{q\text{'}\text{'}\text{'}}\)) and splitting (\(q\to {q}^{{\prime} }+{q}^{{\text{'}\text{'}}}+{q\text{'}\text{'}\text{'}}\)) processes, respectively. d The same as c, but at T = 700 K.
To gain a deeper insight into the microscopic origin of strong 4 ph scattering rates in Cs2AgBiBr6, we calculated the scattering phase space for 4 ph processes at 300 and 700 K, respectively, as shown in Fig. 5c, d. We found that the scattering phase space for 4 ph processes, specifically for the redistribution processes (\(q+{q}^{{\prime} }\to {q}^{{\text{'}\text{'}}}+{q\text{'}\text{'}\text{'}}\)), is remarkably strong in Cs2AgBiBr6, with magnitudes comparable to those of 3 ph processes at 300 K and even surpassing them at 700 K [see Fig. 5a–d]. This observation is in line with the magnitude distribution between the 3 ph and 4 ph scattering rates [see Fig. 4c, d] and provides further evidence that the large 4 ph scattering phase space contributes to strong 4 ph scattering rates in Cs2AgBiBr6 [see Fig. 5c, d]. The peak region of the 4 ph redistribution processes in Cs2AgBiBr6 was found to coincide with the partial DOS of Cs atoms [see Figs. 1c and 5c–d], which suggests that Cs atoms play a significant role in the strong 4 ph scattering rates. Additionally, the atom participation ratio calculated in Supplementary Fig. 12 reveals that Cs atoms contribute to flat phonon bands and exhibit rattling-like behaviors (This observation can be attributed to the loose bonding of Cs atoms [see Supplementary Fig. 13]), which are known to result in strong scattering phase space and scattering rates47,59. Flat rattling-like modes contributing to strong 4 ph scatterings have also been observed in other materials such as crystalline AgCrSe260 and Cu12Sb4S13 Tetrahedrites47.
Microscopic mechanisms of coherences’ conductivity
As shown in Fig. 3b, c, the dominant contribution to the total κL above ~310 K is from the coherences’ contributions. Specifically, using the SCPB+3,4 ph+OD model, the coherences’ conductivity \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) accounts for 48.4 and 60.3% of the total κL at 300 and 700 K, respectively. To uncover the microscopic origin of wavelike conduction in crystalline Cs2AgBiBr6, we calculated the phonon lifetime as a function of frequency from 3 ph and 4 ph processes at 300 and 700 K, respectively, as shown in Fig. 6a, b. Recently, a new concept called the Wigner limit in time proposed by Simoncelli et al.48 was used to separate phonons into different thermal transport regimes, mainly including particle-like propagation and wave-like tunneling regimes. The Wigner limit in time is defined as \(\tau ={3N}_{{at}}/{\omega }_{\max }\), where Nat is the number of atoms in the primitive cell and ωmax is the maximum phonon frequency. Using the criterion of Wigner limit in time, phonons with a lifetime above this limit behave in a particle-like propagation and mainly contribute to the populations’ thermal conductivity; on the contrary, phonons behave in a wave-like tunneling and mainly contribute to the coherences’ thermal conductivity.

a Calculated phonon lifetimes as a function of frequency at T = 300 K within SCPB+3/3,4 ph models, respectively. The green solid line depicts the Wigner limit in time48 and can be written as \(\tau ={\left[\Delta {\omega }_{{avg}}\right]}^{-1}\), where \(\Delta {\omega }_{{avg}}\) is the average phonon inter-band spacing and can be defined as \({\omega }_{\max }/3{N}_{{at}}\), where \({\omega }_{\max }\) denotes the highest frequency and \({N}_{{at}}\) denotes the number of atoms in primitive cell. Phonons with lifetime larger than this limit mainly contribute to populations’ thermal conductivity; on the contrary, phonons mainly contribute to coherences’ thermal conductivity. The Cyan dots indicates the Ioffe-Regel limit in time77, i.e., \(\tau =2\pi /\omega\), \(\omega\) is the phonon frequency with unit of cm−1. Phonons with lifetime larger than the Ioffe-Regel limit in time are recognized as well-defined and can be handled by the unified theory of thermal transport30. b The same as a, but T = 700 K.
For the transport regimes in crystalline Cs2AgBiBr6, after considering both 3 ph and 4 ph scattering processes, most phonons have a lifetime below the Wigner limit in time at both 300 and 700 K, highlighting the necessity of evoking wavelike tunneling transport channel [see Fig. 6a, b]. These findings suggest that the coherences’ conductivity from wavelike tunneling channel may dominate over the total thermal conductivity in Cs2AgBiBr6, consistent with the results shown in Fig. 3b, c. When only 3 ph scattering processes are considered, it is shown in Fig. 6a, b that there are many phonons with a lifetime above the Wigner limit in time. This suggests that the wave-like tunneling channel may be as important as the particle-like propagation channel in Cs2AgBiBr6 [see Fig. 3c and Supplementary Fig. 14]. Interestingly, compared with the \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) calculated by the SCPB+3 ph+OD model, further including 4 ph scattering processes, namely, SCPB+3,4 ph+OD, significantly suppresses the particle-like propagation channel. As a result, the wave-like tunneling channel plays a dominant role in thermal transport in Cs2AgBiBr6 [see Fig. 3c]. This indicates the conversion of dominant thermal transport channels between the particle-like phonon propagation and wave-like tunneling channels. These findings emphasize the inadequacy of the conventional phonon-gas model in describing thermal transport in Cs2AgBiBr6. They also highlight the crucial role of comprehensive modeling, such as the SCPB+3,4 ph+OD model, in gaining a deeper understanding of the actual heat transfer mechanism in Cs2AgBiBr6.
To gain a better understanding of microscopic mechanisms behind the coherences’ thermal conductivity \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) in Cs2AgBiBr6, we calculated the 1D-spectral and cumulative mode-specific contributions to coherences’ conductivity at 300 and 700 K, respectively, as shown in Fig. 7a, b. Similar to the spectral populations’ conductivity \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) of Cs2AgBiBr6 in Fig. 4a, b, the majority of the coherences’ conductivity from the wavelike tunneling channel is carried by the phonons with a frequency less than 3 THz at both 300 and 700 K [see Fig. 7a, b]. This can be attributed to the small inter-band spacing (due to dense phonon dispersions [see Fig. 1b]) in conjunction with large linewidths (large scattering rates [see Fig. 4a, b] or strong anharmonicity30) within frequency region below 3 THz48. This phenomenon is consistent with the observation presented in Fig. 6a, b.

a Calculated spectral and cumulative coherences’ thermal conductivity using SCPB+3,4 ph+OD model at T = 300 K. The contribution of phonon mode j in coherence couplings between two phonon modes (q, j) and (q, jʹ) is evaluated by \({c}_{{\boldsymbol{q}}j}/\left({c}_{{\boldsymbol{q}}j}+{c}_{{\boldsymbol{q}}{j}^{{\prime} }}\right)\), where \({c}_{{\boldsymbol{q}}j}\) is the phonon mode-specific heat. b The same as a but at 700 K. c Two-dimensional (2D) modal \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\left({\omega }_{{\boldsymbol{q}}j},{\omega }_{{\boldsymbol{q}}{j}^{{\prime} }}\right)\) of the contributions to the coherences’ thermal conductivity calculated using the SCPB+3,4 ph+OD model at T = 300 K. The diagonal data points \(({\omega }_{{\boldsymbol{q}}j}={\omega }_{{\boldsymbol{q}}{j}^{{\prime} }})\) indicate phonon degenerate eigenstates. d The same as c but T = 700 K. e The same as c but using SCPB+3 ph+OD model. f The same as e but at T = 700 K.
Figure 7c, d show that the contributions to coherences’ thermal conductivity in Cs2AgBiBr6, calculated using SCPB+3,4 ph+OD at 300 and 700 K, can be resolved in terms of the phonon energies, namely, \({\omega }_{q{\boldsymbol{j}}}\) and \({\omega }_{q{\boldsymbol{j\text{'}}}}\), of two coupled phonons. In contrast to the \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) in CsPbBr3 at 50 K30 and La2Zr2O7 at 200 K48, where quasi-degenerate phonon states dominate, the phonons contributing to the \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) of Cs2AgBiBr6 at both 300 and 700 K have relatively different frequencies [see Fig. 7c, d]. This suggests that the contributions to the \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) are driven by the strong anharmonicity and the corresponding heat transfer physics of wave-like coherences in Cs2AgBiBr6 is intrinsically different from that of harmonic glasses. By comparing the contributions to the \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) in Fig. 7c, d, it is observed that couplings between phonons with large frequency differences begin to contribute to the \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) with increasing temperature [for instance, when considering coupling pairs with a frequency difference larger than 2 THz, their coherences’ contribution \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) increases by ~6.9% with the temperature rising from 300 to 700 K]. This is because the scattering rates of phonons, i.e., their linewidths, also increase as the temperature increases. Additionally, a comparison between the contributions to the \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\) calculated using SCPB+3 ph+OD and SCPB+3,4 ph+OD models [see Fig. 7c–f] reveals that the additional 4 ph scattering rates result in more scattered contributions to \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\). This is because the large 4 ph scattering rates allow phonons with relatively large frequency differences to couple, particularly those involving the Cs rattling-like modes. This phenomenon is similar to what was observed in Cu12Sb4S13 Tetrahedrites47.
To this end, the application of the SCPB+3,4 ph+OD model has provided a reliable prediction of the κL in crystalline Cs2AgBiBr6, and has elucidated its microscopic mechanism of heat transport within the temperature range of 300–700 K. However, further improvement may be possible by accounting for (i) the anharmonic contributions of heat flux operators61, (ii) simultaneously evaluating phonon energy shifts and broadening.
In conclusion, we have performed the DFT calculations to investigate the anharmonic lattice dynamics and unravel the microscopic mechanisms of thermal transport in crystalline Cs2AgBiBr6. Our results show that the unstable soft modes correspond to the in-phase and out-of-phase tilting of the AgBr6 and BiBr6 octahedra units in real space. The anharmonic stabilization of the soft modes is observed to occur at ~119–138 K in excellent agreement with experiments. We found that the negative phonon energy shifts arising from cubic anharmonicity plays a crucial role in accurately reproducing experimental phonon dispersions. The accurate reproduction of experimental phonon energies by the SCPB approximation underscores the significance of considering both cubic and quartic anharmonicities in anharmonic phonon renormalization.
With advanced unified theory of thermal transport, the κL of Cs2AgBiBr6 is predicted to be ultra-low, with a value of 0.21 Wm−1 K−1 at 300 K, which can be mainly attributed to the inherently strong scatterings rather than small group velocities. The negative phonon energy shifts from cubic anharmonicity leads to enhanced phonon scattering rates, thus reducing κL in Cs2AgBiBr6. Additionally, our results indicate that the 4 ph scatterings significantly suppress the κL in Cs2AgBiBr6. Through analysis of the phonon scattering phase space for 3 ph and 4 ph processes, we attribute the strong 4 ph scattering rates to the flat phonon modes dominated by Cs atoms.
It is also revealed that the coherences’ conductivity dominates the total κL above 310 K, highlighting the breakdown of the conventional phonon gas model in accurately predicting the κL of Cs2AgBiBr6. The comparison of the results obtained from SCPB+3 ph+OD and SCPB+3,4 ph+OD models shows that the inclusion of 4 ph scattering processes alters the dominance of wave-like tunneling and particle-like phonon propagation channels in thermal transport of Cs2AgBiBr6. Thus, both cubic and quartic anharmonicities are critical in determining not only the anharmonic lattice dynamics but also lattice thermal conductivity. Our study provides insights into the microscopic mechanisms of thermal transport in halide double perovskites with ultra-low κL and strong anharmonicity.
Methods
First-principles calculations
In this work, the ab-initio calculations within the framework of density functional theory (DFT)62 using the Vienna Ab-initio Simulation Package (VASP)63 were performed for structural optimization of Cs2AgBiBr6 crystal, wherein the projector-augmented wave (PAW)64 method was used to treat the Cs(5s25p66s1), Ag(4d105s1), Bi(5d106s26p3) and Br(4s24p5) shells as valence states. The revised Perdew-Burke-Ernzerhof (PBE) version for solids, i.e., PBEsol65, of the generalized gradient approximation (GGA)66 was used for the exchange-correlation functional. The ionic positions and unit cell geometry were fully optimized using a plane-wave cutoff energy of 800 eV and a 10 × 10 × 10 Monkhorst-Pack electronic k-point mesh, with a tight force convergence criterion of 10−5 eV·Å−1 and a tight energy convergence criterion of 10−8 eV. The resulting relaxed lattice constant is a = b = c = 5.602 Å, in good agreement with the experiment (a = 5.639 Å)39,67 for Cs2AgBiBr6 crystal with a \({\boldsymbol{Fm}}\bar{{\boldsymbol{3}}}{\boldsymbol{m}}\) space group. Considering that the long-term dipole-dipole interaction in the polar Cs2AgBiBr6 crystal will lead to the longitudinal (LO) and transverse optical (TO) phonon modes splitting near the Brillouin zone center, the non-analytic corrections are further included in the dynamical matrix and the dielectric tensor ε and the Born effective charges Z of Cs2AgBiBr6 are calculated using the density functional perturbation theory (DFPT)68. The corresponding parameters are computed to be ε∞ = 5.392, \({Z}^{* }\left({\rm{Cs}}\right)=1.356,{Z}^{* }\left({\rm{Ag}}\right)=1.795,{Z}^{* }\left({\rm{Bi}}\right)=4.805,{{Z}^{* }\left({\rm{Br}}\right)}_{\perp }=-0.769,\) and \({{Z}^{* }\left({\rm{Br}}\right)}_{{\rm{II}}}=-3.114\), which are consistent with previous theorectical calculations29. Given the experimental thermal expansion of Cs2AgBiBr6 in the temperature range of 100–300 K is relatively small (~0.53% – from 11.21 to 11.27 Å)39, we have opted not to consider the thermal expansion in the current calculations [For more details, please refer to Supplementary Fig. 15].
The 0-K harmonic interatomic force constants (IFCs) calculation was performed using the finite-displacement approach69, with a 2 × 2 × 2 supercell and 4 × 4 × 4 Monkhorst-Pack electronic k-point mesh, and a plane-wave cutoff energy of 800 eV in VASP. Additionally, the same supercell dimensions and DFT calculation settings were used to map out the potential energy surfaces (PESs). To extract anharmonic IFCs efficiently, the machine-learning-based method called the compressive sensing lattice dynamics method (CSLD) was used70,71, where the compressive technique72 was implemented to collect the physically important terms of the anharmonic IFCs using the limited displacement-force datasets22. Within the framework of the CSLD method, the ab initio molecular dynamics (AIMD) simulation was performed with a 2 × 2 × 2 supercell to generate the random atomic displacements, with a 2 × 2 × 2 Monkhorst-Pack electronic k-point mesh and an energy convergence criterion of 10−5 eV. The AIMD simulation of the NVT ensemble with the Nosé Hoover thermostat at 500 K was ran for 3000-time steps using a 2-fs time step. By disregarding the first 500 steps from the obtained AIMD trajectories, 100 atomic structures were sampled at equal intervals. After that, a random-direction displacement of 0.1 Å for all atoms was imposed to reduce the cross-correlations between the atomic structures. Furthermore, accurate DFT calculations were performed to obtain the training and cross-validation data, on the basis of the sampled atomic structures, with 4 × 4 × 4 Monkhorst–Pack electronic k-point mesh and energy convergence better than 10−8 eV. Finally, using the displacement-force datasets and 0-K harmonic IFCs obtained, the least absolute shrinkage and selection operator (LASSO) technique73 was used to extract the anharmonic IFCs up to the sixth order. The real-space cutoff radii of 8.47 Å, 6.35 Å, 4.23 Å and 3.18 Å were applied for the cubic, quartic, quintic, and sextic IFCs extraction, respectively. We observe a relative fitting error of 6.2% for higher-order IFCs in the context of atomic force. It is noteworthy that this error is exceptionally small, mirroring values consistently reported in the existing literatures22,25,71. In this work, the IFCs estimation was performed by using the ALAMODE package22,37,74.
Anharmonic phonon renormalization
The anharmonic phonon energy renormalization was performed using the self-consistent phonon approximation (SCP) formulated in the reciprocal space26,37, as implemented in the ALAMODE package22,37. Considering only the first-order correction from quartic anharmonicity, i.e., the loop diagram, the resulting SCP equation in the diagonal form neglecting polarization mixing (PM) can be written as
where ωq is the bare harmonic phonon frequency associated with the phonon mode q, and Ωq is the anharmonically renormalized phonon frequency at finite temperatures. The quantity Iq can be defined as
where N, ℏ, n and \({V}^{(4)}\left({q;}-{q;q\text{'};}-{q\text{'}}\right)\) are the total number of sampled phonon wavevectors in the first Brillouin zone, the reduced Planck constant, the Bose-Einstein distribution and the reciprocal representation of 4th-order IFCs, respectively. Note that the off-diagonal terms of the phonon loop self-energy for PM22 were found to be crucial for some crystal systems41,54, thus, we also further included it in the anharmonic phonon energy renormalization calculations. In this work, the q-mesh of the SCP calculations was set to 2 × 2 × 2, which is equivalent to a 2 × 2 × 2 supercell adopted by the real-space-based phonon energy renormalization technique. Meanwhile, the SCP q-mesh of 2 × 2 × 2 was used to correspond to the supercell dimensions of the second-order IFCs extracted in the real space.
Using the above first-order SCP technique that only obtains the renormalized phonon energies resulting from the quartic anharmonicity, i.e., the loop diagram, the anharmonic phonon energies of highly anharmonic compounds may be overestimated38,47. On top of the renormalized phonon energies calculated by the first-order SCP, the renormalized phonon energies arising from the cubic anharmonicity, i.e., the dominant bubble diagram, can be estimated using the following self-consistent equation within quasi-particle approximation38
where \({\sum }_{q}^{{\rm{B}}}[G,{\varPhi }_{3}]\left({\Omega }_{q}\right)\) is the phonon frequency-dependent bubble self-energy, B denotes the bubble diagram, \({\varPhi }_{3}\) is the third-order force constant explicitly included in the anharmonic self-energy calculation. Note that several treatments within quasiparticle (QP) approximation including QP[0], QP[S] and QP-NL can be employed to solve Eq. (3) and obtain the fully renormalized phonon energies due to cubic and quartic anharmonicities38. The QP-NL treatment was used in this work due to its reliable predictions. Additionally, the extra phonon energy shifts arising from the tadpole diagram due to the cubic anharmonicity were found to be small and negligible, which was also reported for many crystals41,47,54. Note that the SCP and SCPB were denoted as the shorthand notation of the first-order SCP calculation and SCP calculation with bubble diagram correction in this work.
Phonon scattering rates and unified thermal transport theory
With the renormalized harmonic (2nd-order) and original anharmonic IFCs (3rd and 4th-order), the phonon scattering rates resulting from phonon-phonon and phonon-isotope interaction processes, as the key ingredients entering the thermal transport equation34, can be calculated using Fermi’s golden rule of perturbation theory50. In this study, the intrinsic multi-phonon interaction mainly includes three-phonon (3 ph) and four-phonon (4 ph) scattering processes and the corresponding expressions within single-mode relaxation time approximation (SMRTA) can be written as25,47,50
where \({\zeta }_{\pm }\) and \({\zeta }_{\pm \pm }\) are defined as
where \({\Gamma }_{q}^{3{ph}}\) and \({\Gamma }_{q}^{4{ph}}\) denote the 3 ph and 4 ph scattering rates, respectively, the phonon mode q is a shorthand notation for a composite index of the wavevector q and phonon branch j, \({V}^{\left(3\right)}\left(q,\pm {q}^{{\prime} },-{q}^{{\text{'}\text{'}}}\right)\) and \({V}^{\left(4\right)}\left(q,\pm {q}^{{\prime} },\pm {q}^{{\text{'}\text{'}}},-{q}^{{\text{'}\text{'}\text{'}}}\right)\) are the reciprocal representation of 3rd- and 4th-order IFCs, respectively57, the delta function \(\delta \left(\varOmega \right)\) describes the selection rules for energy in both 3 ph and 4 ph scattering processes, the Kronecker deltas \({\triangle }_{\pm }\) and \({\triangle }_{\pm \pm }\) are shorthand notation for \({\triangle }_{{\boldsymbol{q}}{\boldsymbol{\pm }}{\boldsymbol{q\text{'}}}{\boldsymbol{-}}{\boldsymbol{q\text{'}\text{'}}}{\boldsymbol{,}}{\boldsymbol{Q}}}\) and \({\triangle }_{{\boldsymbol{q}}{\boldsymbol{\pm }}{{\boldsymbol{q}}}^{{\boldsymbol{{\prime} }}}{\boldsymbol{\pm }}{{\boldsymbol{q}}}^{{\boldsymbol{\text{'}\text{'}}}}{\boldsymbol{-}}{\boldsymbol{q\text{'}\text{'}\text{'}}}{\boldsymbol{,}}{\boldsymbol{Q}}}\), respectively, and account for the selection rules for momentum, where the reciprocal lattice vector Q = 0 indicates the normal (N) process and Q ≠ 0 the Umklapp (U) process.
The phonon scattering term resulting from isotope can be written as75
where the mass variance, \(g\left(i\right)\), is defined as\(g\left(i\right)=\sum _{s}{f}_{s}\left(i\right){\left[1-{M}_{s}\left(i\right)/\bar{M}\left(i\right)\right]}^{2}=\sum _{s}{f}_{s}\left(i\right){\left[\Delta {M}_{s}\left(i\right)/\bar{M}\left(i\right)\right]}^{2}\), where \({f}_{s}\left(i\right)\) and \({M}_{s}\left(i\right)\) are the concentration and mass of the \({sth}\) isotope of atom \(i\), respectively. \(\bar{M}\left(i\right)\) denotes the average mass of the ith atom in the primitive cell. \({e}_{q}\left(i\right)\) denotes the eigenfunction of phonon mode q at the atom i.
Using Matthiessen’s rule, the total phonon scattering rate Γq can be expressed as
The lattice thermal conductivity κL can be calculated by a unified theory of thermal transport incorporating populations’ and coherences’ conductivities/contributions30, and the detailed formula for κL under the SMRTA is given as follows30,47,48:
where the superscripts P and C denote the populations’ and coherences’ contributions, respectively, kB is the Boltzmann constant, T is the temperature, V is the unit-cell volume and υ is the group velocity matix including both the intra- and inter-branch terms76. In Eq. (10), when \(j={j\text{'}}\), it calculates the populations’ contribution (PBTE calculation result) \(\left({\kappa }_{{\rm{L}}}^{{\rm{P}}}\right)\), i.e., particle-like phonon propagation described by the diagonal terms of heat flux operators, otherwise, it gives the coherences’ contribution \(\left({\kappa }_{{\rm{L}}}^{{\rm{C}}}\right)\), i.e., wave-like tunelling of phonons described by the off-diagonal terms of heat flux operators (The terminology, i.e., diffuson, is also used to describe this operator In galsses). The total lattice thermal conductivity κL can be obtained by summing over \({\kappa }_{{\rm{L}}}^{{\rm{P}}}\) and \({\kappa }_{{\rm{L}}}^{{\rm{C}}}\), namely \({\kappa }_{{\rm{L}}}={\kappa }_{{\rm{L}}}^{{\rm{P}}}+{\kappa }_{{\rm{L}}}^{{\rm{C}}}\). In this work, the q mesh for 3 ph scattering processes was set to 12 × 12 × 12, which gives well-converged results for the crystalline Cs2AgBiBr6. Considering the huge computational cost, a q mesh of 8 × 8 × 8 with a scalebroad parameter of 0.06 were used for 4 ph scattering processes. Note that the iterative scheme to PBTE is only applied for 3 ph scattering processes in this work, while the 4 ph scattering processes are treated at the SMRTA level considering the exceptionally large memory demands47,57. Thermal transport calculations including particle-like propagation and wave-like tunneling transport channels were performed by using the ShengBTE58 and FourPhonon57 packages, as well as our in-house code41.
Data availability
Data that support the findings of this study will be available from the corresponding authors upon reasonable request.
Code availability
The open-source codes can be found as following: Alamode is available at https://github.com/ttadano/alamode, ShengBTE is available at https://www.shengbte.org and FOURPHONON is available at https://github.com/FourPhonon/FourPhonon. The in-house codes will be available from the corresponding authors upon reasonable request.
References
Green, M. A., Ho-Baillie, A. & Snaith, H. J. The emergence of perovskite solar cells. Nat. Photonics 8, 506–514 (2014).
Bai, S. et al. Planar perovskite solar cells with long-term stability using ionic liquid additives. Nature 571, 245–250 (2019).
Lei, H., Hardy, D. & Gao, F. Lead‐Free Double Perovskite Cs2AgBiBr6: Fundamentals, Applications, and Perspectives. Adv. Funct. Mater. 31, 2105898 (2021).
Zhang, T., Cai, Z. & Chen, S. Chemical trends in the thermodynamic stability and band gaps of 980 halide double perovskites: A high-throughput first-principles study. ACS Appl. Mater. Interfaces 12, 20680–20690 (2020).
Boyd, C. C., Cheacharoen, R., Leijtens, T. & McGehee, M. D. Understanding degradation mechanisms and improving stability of perovskite photovoltaics. Chem. Rev. 119, 3418–3451 (2018).
Divitini, G. et al. In situ observation of heat-induced degradation of perovskite solar cells. Nat. Energy 1, 1–6 (2016).
Lanigan-Atkins, T. et al. Two-dimensional overdamped fluctuations of the soft perovskite lattice in CsPbBr3. Nat. Mater. 20, 977–983 (2021).
Yang, J. et al. Acoustic-optical phonon up-conversion and hot-phonon bottleneck in lead-halide perovskites. Nat. Commun. 8, 1–9 (2017).
Yang, Y. et al. Observation of a hot-phonon bottleneck in lead-iodide perovskites. Nat. Photonics 10, 53–59 (2016).
Pisoni, A. et al. Ultra-low thermal conductivity in organic–inorganic hybrid perovskite CH3NH3PbI3. J. Phys. Chem. Lett. 5, 2488–2492 (2014).
Wang, Y. et al. Cation dynamics governed thermal properties of lead halide perovskite nanowires. Nano Lett. 18, 2772–2779 (2018).
Lee, W. et al. Ultralow thermal conductivity in all-inorganic halide perovskites. Proc. Natl Acad. Sci. USA 114, 8693–8697 (2017).
Xie, H. et al. All-inorganic halide perovskites as potential thermoelectric materials: dynamic cation off-centering induces ultralow thermal conductivity. J. Am. Chem. Soc. 142, 9553–9563 (2020).
Acharyya, P. et al. Intrinsically ultralow thermal conductivity in Ruddlesden–Popper 2D perovskite Cs2PbI2Cl2: localized anharmonic vibrations and dynamic octahedral distortions. J. Am. Chem. Soc. 142, 15595–15603 (2020).
Acharyya, P. et al. Glassy thermal conductivity in Cs3Bi2I6Cl3 single crystal. Nat. Commun. 13, 1–9 (2022).
Stoumpos, C. C., Malliakas, C. D. & Kanatzidis, M. G. Semiconducting tin and lead iodide perovskites with organic cations: phase transitions, high mobilities, and near-infrared photoluminescent properties. Inorg. Chem. 52, 9019–9038 (2013).
Chung, I. et al. CsSnI3: semiconductor or metal? High electrical conductivity and strong near-infrared photoluminescence from a single material. High hole mobility and phase-transitions. J. Am. Chem. Soc. 134, 8579–8587 (2012).
He, Y. & Galli, G. Perovskites for solar thermoelectric applications: A first principle study of CH3NH3AI3 (A= Pb and Sn). Chem. Mater. 26, 5394–5400 (2014).
Wang, M. & Lin, S. Anisotropic and ultralow phonon thermal transport in organic–inorganic hybrid perovskites: atomistic insights into solar cell thermal management and thermoelectric energy conversion efficiency. Adv. Funct. Mater. 26, 5297–5306 (2016).
Fransson, E. et al. Limits of the phonon quasi-particle picture at the cubic-to-tetragonal phase transition in halide perovskites. Commun. Phys. 6, 173 (2023).
Zhao, Y. et al. Lattice thermal conductivity including phonon frequency shifts and scattering rates induced by quartic anharmonicity in cubic oxide and fluoride perovskites. Phys. Rev. B 104, 224304 (2021).
Tadano, T. & Tsuneyuki, S. Self-consistent phonon calculations of lattice dynamical properties in cubic SrTiO 3 with first-principles anharmonic force constants. Phys. Rev. B 92, 054301 (2015).
Hellman, O., Abrikosov, I. A. & Simak, S. I. Lattice dynamics of anharmonic solids from first principles. Phys. Rev. B 84, 180301 (2011).
Hellman, O., Steneteg, P., Abrikosov, I. A. & Simak, S. I. Temperature dependent effective potential method for accurate free energy calculations of solids. Phys. Rev. B 87, 104111 (2013).
Xia, Y., Pal, K., He, J., Ozoliņš, V. & Wolverton, C. Particlelike Phonon Propagation Dominates Ultralow Lattice Thermal Conductivity in Crystalline Tl 3 VSe 4. Phys. Rev. Lett. 124, 065901 (2020).
Errea, I., Rousseau, B. & Bergara, A. Anharmonic stabilization of the high-pressure simple cubic phase of calcium. Phys. Rev. Lett. 106, 165501 (2011).
Kawano, S., Tadano, T. & Iikubo, S. Effect of Halogen Ions on the Low Thermal Conductivity of Cesium Halide Perovskite. J. Phys. Chem. C. 125, 91–97 (2021).
Wang, X. et al. Cubic halide perovskites as potential low thermal conductivity materials: A combined approach of machine learning and first-principles calculations. Phys. Rev. B 105, 014310 (2022).
Klarbring, J., Hellman, O., Abrikosov, I. A. & Simak, S. I. Anharmonicity and ultralow thermal conductivity in lead-free halide double perovskites. Phys. Rev. Lett. 125, 045701 (2020).
Simoncelli, M., Marzari, N. & Mauri, F. Unified theory of thermal transport in crystals and glasses. Nat. Phys. 15, 809–813 (2019).
Luo, Y., Yang, X., Feng, T., Wang, J. & Ruan, X. Vibrational hierarchy leads to dual-phonon transport in low thermal conductivity crystals. Nat. Commun. 11, 1–10 (2020).
Hanus, R. et al. Uncovering design principles for amorphous-like heat conduction using two-channel lattice dynamics. Mater. Today Phys. 18, 100344 (2021).
Mukhopadhyay, S. et al. Two-channel model for ultralow thermal conductivity of crystalline Tl3VSe4. Science 360, 1455–1458 (2018).
Srivastava, G. P. The physics of phonons (Routledge, 2019).
Xia, Y. Revisiting lattice thermal transport in PbTe: The crucial role of quartic anharmonicity. Appl. Phys. Lett. 113, 073901 (2018).
Cohen, A. et al. Diverging Expressions of Anharmonicity in Halide Perovskites. Adv. Mater. 34, 2107932 (2022).
Tadano, T. & Tsuneyuki, S. Quartic anharmonicity of rattlers and its effect on lattice thermal conductivity of clathrates from first principles. Phys. Rev. Lett. 120, 105901 (2018).
Tadano, T. & Saidi, W. A. First-Principles Phonon Quasiparticle Theory Applied to a Strongly Anharmonic Halide Perovskite. Phys. Rev. Lett. 129, 185901 (2022).
Schade, L. et al. Structural and optical properties of Cs2AgBiBr6 double perovskite. ACS Energy Lett. 4, 299–305 (2018).
He, X. et al. Multiple lattice instabilities and complex ground state in Cs2AgBiBr6. Preprint at https://arxiv.org/pdf/2112.04717 (2021).
Zheng, J. et al. Anharmonicity-induced phonon hardening and phonon transport enhancement in crystalline perovskite BaZrO 3. Phys. Rev. B 105, 224303 (2022).
Aseginolaza, U. et al. Phonon collapse and second-order phase transition in thermoelectric SnSe. Phys. Rev. Lett. 122, 075901 (2019).
Skelton, J. M. et al. Anharmonicity in the High-Temperature C m c m Phase of SnSe: Soft Modes and Three-Phonon Interactions. Phys. Rev. Lett. 117, 075502 (2016).
Buckeridge, J., Scanlon, D. O., Walsh, A., Catlow, C. & Sokol, A. A. Dynamical response and instability in ceria under lattice expansion. Phys. Rev. B 87, 214304 (2013).
Huang, L. & Lambrecht, W. R. Lattice dynamics in perovskite halides CsSn X 3 with X = I, Br, Cl. Phys. Rev. B 90, 195201 (2014).
Da Silva, E. L., Skelton, J. M., Parker, S. C. & Walsh, A. Phase stability and transformations in the halide perovskite CsSnI 3. Phys. Rev. B 91, 144107 (2015).
Xia, Y., Ozoliņš, V. & Wolverton, C. Microscopic Mechanisms of Glasslike Lattice Thermal Transport in Cubic Cu 12 Sb 4 S 13 Tetrahedrites. Phys. Rev. Lett. 125, 085901 (2020).
Simoncelli, M., Marzari, N. & Mauri, F. Wigner formulation of thermal transport in solids. Phys. Rev. X. 12, 041011 (2022).
Lindsay, L., Hua, C., Ruan, X. L. & Lee, S. Survey of ab initio phonon thermal transport. Mater. Today Phys. 7, 106–120 (2018).
Feng, T. & Ruan, X. Quantum mechanical prediction of four-phonon scattering rates and reduced thermal conductivity of solids. Phys. Rev. B 93, 045202 (2016).
Bhui, A. et al. Intrinsically Low Thermal Conductivity in the n-Type Vacancy-Ordered Double Perovskite Cs2SnI6: Octahedral Rotation and Anharmonic Rattling. Chem. Mater. 34, 3301–3310 (2022).
Liu, T. et al. Enhanced control of self-doping in halide perovskites for improved thermoelectric performance. Nat. comm. 10, 1–9 (2019).
Pandey, T., Du, M., Parker, D. S. & Lindsay, L. Origin of ultralow phonon transport and strong anharmonicity in lead-free halide perovskites. Mater. Today Phys. 28, 100881 (2022).
Zheng, J. et al. Effects of high-order anharmonicity on anomalous lattice dynamics and thermal transport in fully filled skutterudite YbFe 4 Sb 12. Phys. Rev. Mater. 6, 093801 (2022).
Tian, Z. et al. Phonon conduction in PbSe, PbTe, and PbTe 1− x Se x from first-principles calculations. Phys. Rev. B 85, 184303 (2012).
Elbaz, G. A. et al. Phonon speed, not scattering, differentiates thermal transport in lead halide perovskites. Nano Lett. 17, 5734–5739 (2017).
Han, Z., Yang, X., Li, W., Feng, T. & Ruan, X. FourPhonon: An extension module to ShengBTE for computing four-phonon scattering rates and thermal conductivity. Comput. Phys. Commun. 270, 108179 (2022).
Li, W., Carrete, J., Katcho, N. A. & Mingo, N. ShengBTE: A solver of the Boltzmann transport equation for phonons. Comput. Phys. Commun. 185, 1747–1758 (2014).
Li, W. & Mingo, N. Ultralow lattice thermal conductivity of the fully filled skutterudite YbFe 4 Sb 12 due to the flat avoided-crossing filler modes. Phys. Rev. B 91, 144304 (2015).
Xie, L., Feng, J. H., Li, R. & He, J. Q. First-Principles Study of Anharmonic Lattice Dynamics in Low Thermal Conductivity AgCrSe 2: Evidence for a Large Resonant Four-Phonon Scattering. Phys. Rev. Lett. 125, 245901 (2020).
Sun, T. & Allen, P. B. Lattice thermal conductivity: Computations and theory of the high-temperature breakdown of the phonon-gas model. Phys. Rev. B 82, 224305 (2010).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864 (1964).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Perdew, J. P., Burke, K. & Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 54, 16533 (1996).
Ning, W. et al. Thermochromic Lead‐Free Halide Double Perovskites. Adv. Funct. Mater. 29, 1807375 (2019).
Baroni, S., De Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515 (2001).
Esfarjani, K. & Stokes, H. T. Method to extract anharmonic force constants from first principles calculations. Phys. Rev. B 77, 144112 (2008).
Zhou, F., Nielson, W., Xia, Y. & Ozoliņš, V. Compressive sensing lattice dynamics. I. General formalism. Phys. Rev. B 100, 184308 (2019).
Zhou, F., Nielson, W., Xia, Y. & Ozoliņš, V. Lattice anharmonicity and thermal conductivity from compressive sensing of first-principles calculations. Phys. Rev. Lett. 113, 185501 (2014).
Candès, E. J. & Wakin, M. B. An introduction to compressive sampling. IEEE Signal Process. Mag. 25, 21–30 (2008).
Nelson, L. J., Hart, G. L., Zhou, F. & Ozoliņš, V. Compressive sensing as a paradigm for building physics models. Phys. Rev. B 87, 035125 (2013).
Tadano, T., Gohda, Y. & Tsuneyuki, S. Anharmonic force constants extracted from first-principles molecular dynamics: applications to heat transfer simulations. J. Condens. Matter Phys. 26, 225402 (2014).
Tamura, S. Isotope scattering of dispersive phonons in Ge. Phys. Rev. B 27, 858 (1983).
Allen, P. B. & Feldman, J. L. Thermal conductivity of disordered harmonic solids. Phys. Rev. B 48, 12581 (1993).
Ioffe, A. F., & A. R. Regel. Non-crystalline, amorphous and liquid electronic semiconductors. In Progress in Semiconductors, Vol. 4, (ed. Gibson A. F.) 237–291 (John Wiley & Sons, New York) (1960).
Acknowledgements
We are thankful for the financial support from the Science and Technology Planning Project of Guangdong Province, China (Grant No. 2017A050506053), the Science and Technology Program of Guangzhou (No. 201704030107), and the Hong Kong General Research Fund (Grants No. 16214217 and No. 16206020). This paper was supported in part by the Project of Hetao Shenzhen-Hong Kong Science and Technology Innovation Cooperation Zone (HZQB-KCZYB2020083). R.G. acknowledges support from the Excellent Young Scientists Fund (Overseas) of Shandong Province (2022HWYQ091), the Taishan Scholars Program of Shandong Province, the Natural Science Foundation of Shandong Province (ZR2022MA011), and the Initiative Research Fund of Shandong Institute of Advanced Technology (2020107R03). G.H. and J.Z. acknowledge funding by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division, under Contract No. DE-AC02-05-CH11231: Materials Project program KC23MP. ChangpengL. acknowledges the support from the Sinergia project of the Swiss National Science Foundation (grant number CRSII5_189924).
Author information
Authors and Affiliations
Contributions
J.Z. contributed to the methodology, software, all calculations, analysis and writing of the original draft. ChangpengL. and ChongjiaL. contributed to analysis and writing of the manuscript. G.H., R.G. and B.H. were responsible for supervision, writing and analysis throughout. All authors reviewed the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41524_2024_1211_MOESM1_ESM.pdf
Supplementary Information for Unravelling Ultralow Thermal Conductivity in Double Perovskite Cs2AgBiBr6 Dominant Wave-like Phonon Tunneling, Strong Quartic Anharmonicity and Lattice Instability
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, J., Lin, C., Lin, C. et al. Unravelling ultralow thermal conductivity in perovskite Cs2AgBiBr6: dominant wave-like phonon tunnelling and strong anharmonicity. npj Comput Mater 10, 30 (2024). https://doi.org/10.1038/s41524-024-01211-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-024-01211-y
