
Abstract
Deep learning (DL) models currently employed in materials research exhibit certain limitations in delivering meaningful information for interpreting predictions and comprehending the relationships between structure and material properties. To address these limitations, we propose an interpretable DL architecture that incorporates the attention mechanism to predict material properties and gain insights into their structure–property relationships. The proposed architecture is evaluated using two well-known datasets (the QM9 and the Materials Project datasets), and three in-house-developed computational materials datasets. Train–test–split validations confirm that the models derived using the proposed DL architecture exhibit strong predictive capabilities, which are comparable to those of current state-of-the-art models. Furthermore, comparative validations, based on first-principles calculations, indicate that the degree of attention of the atoms’ local structures to the representation of the material structure is critical when interpreting structure–property relationships with respect to physical properties. These properties encompass molecular orbital energies and the formation energies of crystals. The proposed architecture shows great potential in accelerating material design by predicting material properties and explicitly identifying crucial features within the corresponding structures.
Similar content being viewed by others
Introduction
A central challenge in the field of materials science involves the use of both experience and theory to explore the compositions and structures of materials with specific properties and subsequently validating them through experimentation. Unfortunately, the research and development of materials is a time-consuming endeavor that often relies on serendipity. Materials informatics (MI) has emerged as a rapidly growing interdisciplinary field toward addressing these challenges. This concept employs data-driven methods to extract practical knowledge regarding materials and their related physicochemical phenomena from experimental and computational data, thus ultimately accelerating the discovery of superior materials1,2,3,4.
The majority of MI approaches comprise three key components5. The first component comprises datasets containing information regarding the structure of the materials, measurement results directly related to these structures, and physical properties relevant to the material development goals. The second component, that is, representation, quantitatively describes the data instances in the first component, collecting a primitive description of materials for identification and analogic inference. The final component is a system that utilizes machine learning or data mining algorithms (either a single approach or a combination of approaches) to extract knowledge from the materials datasets for specific purposes, such as predicting properties or identifying new material compositions and structures.
Traditionally, materials have been characterized based on their elemental compositions and structures. Researchers have primarily relied on their knowledge and experience, often referred to as tacit knowledge, to predict certain properties of hypothetical materials with specific compositions and structures. Computational chemistry approaches based on quantum mechanics, particularly density functional theory (DFT) simulations, can be used to theoretically verify the compositions and structures of these materials through in-silico computational experimentation. However, despite providing accurate information on the physical properties of hypothetical materials, computational experiments have certain limitations. For example, the vast number of potential hypothetical materials renders the design of materials with desired physical properties time-consuming and expensive due to the exhaustive calculations required. Moreover, researchers require specialized and detailed knowledge to narrow down the potential compositions and material structures.
Unlike traditional approaches, MI approaches initially involve the conversion of primitive data descriptions into appropriate representations that can be used for mathematical reasoning and inference. In particular, MI systems are given the task of estimating qualitative and quantitative between materials based on this transformed representation, allowing them to uncover potential patterns in the material data6,7,8. The development of material representation (i.e., the design of material descriptors or methods for learning material representation from data) play a crucial role in MI approaches. This is because the effectiveness of an MI algorithm highly depends on the material representation, as it directly affects the algorithm’s performance and facilitates the explanation and interpretation of the inference process and prediction results9. Recent advancements in automated experiments and high-performance computers have helped acquire substantial experimental and computational data. Consequently, there is a growing need for the development of explainable and interpretable MI methods to enhance our understanding of physical and chemical phenomena.
Recently, various deep learning (DL)-based MI approaches have been developed to address the challenges associated with material representation and to predict physical properties10,11,12,13. A typical example is the DL architecture that uses a continuous-filter convolution layer with filter-generation networks to handle atomistic systems and accurately predict the properties of molecular and crystalline materials10. Another example is the convolutional neural network based on crystal graphs, which can predict material properties with an accuracy comparable to that of DFT calculations while also providing atomic-level chemical insight12. In addition to the aforementioned approaches, researchers have developed various other DL architectures to encode the local chemical environments of atoms and improve the prediction accuracy by integrating different types of material descriptors, applying graph neural networks (GNNs), and utilizing many-body tensor representations11,13. Furthermore, several studies have incorporated prior knowledge to construct neural network models that ensure the relationship between structures and properties of materials is learned accurately14,15,16.
However, a significant challenge confronted by both traditional and DL-based machine learning approaches is the issue of interpretability. Machine learning models often prioritize including all available information rather than selecting an interpretable representation to improve prediction accuracy. The relationship between material representation and its properties is complex and nonlinear, and as a result, machine learning models acting as “black boxes” that do not explicitly reveal correlations. Although statistical evaluations based on existing data often exhibit high prediction accuracies, estimating their predictive capability for new materials is difficult. Gaining a comprehensive understanding machine learning to clarify underlying physicochemical phenomena also remains challenging.
Numerous studies have aimed to enhance model interpretability by incorporating additional information or features. For instance, graph convolutional networks use SMILES strings to represent molecules as inputs, which helps identify crucial fingerprint fragments and facilitating interpretation17,18. Even with these advancements, these networks still require assistance in accurately predicting the properties of molecular and crystalline materials due to the absence of 3D structural information. Message-passing neural network-based models (MPNNs)19,20,21 employ heuristic bonding information to capture atomic interactions but encounter several challenges with long-range interactions, feature interpretability, global information representation, and scalability when handling large molecule/crystal datasets. With the aim of addressing these limitations, recent works have turned to transformer-based networks15,22,23,24,25,26,27,28, which utilize attention mechanisms16. These networks offer a promising avenue by modeling interatomic reactions between constituent atoms through attention scores, which indicate the significance of each atom in learning the representation of other atoms. Various pooling methods like max or average pooling14,15,28,29,30,31,32,33are then employed to derive a comprehensive representation of the entire structure. However, extracting meaningful structure–property relationships from these transformer-based networks remains challenging and non-trivial.
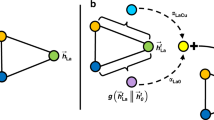
In this study, we propose an interpretable DL architecture that incorporates the attention mechanism to predict material structure properties and provide meaningful information about the structure–property relationships. The proposed architecture initiates by learning the representation of local structures of atoms within a material structure through the recursive application of attention mechanisms to the local structures of the neighboring atoms (Fig. 1a). The local structure of an atom includes the atom itself (central atom), its neighboring atoms, and the arrangement of the neighboring atoms around the central atom. Finally, the material structure representation is derived from the representations of these local structures of the atoms. This architecture utilizes the attention mechanism to incorporate information about the geometrical arrangement of neighboring atoms into the representations of local structures. Moreover, it quantitatively measures the degree of attention given to each local structure from a global perspective when determining the representation of the material structure (Fig. 1b). Consequently, through the training of the model with a specific target property, our approach aids in the interpretation of the structure-property relationships in materials.

Schematics of (a) the learning recursive representation of a local structure (central atom and its neighboring atoms) within the molecular structure of phenol (C6H5OH), and (b) measurement of the global attention given to a local structure when determining representation of the molecular structure. The direction and size of each arrow indicate the degree of attention given to other atoms when establishing the representation of the local structure of a particular atom.
Results
SCANN framework
We introduce a DL architecture called the self-consistent attention neural network (SCANN). The SCANN focuses on representing material structures from local structures of atoms with learned weights, thus facilitating the prediction and interpretation of material properties. The key objective of SCANN is to recursively learn consistent representations of these local structures within the material (as shown in Fig. 1a), which are then appropriately combined to obtain an overall representation of the material structure.
In this study, each material structure S in a dataset \(\mathcal{D}\) is represented using the atomic numbers and the corresponding coordinates of its M atoms. By employing Voronoi tessellation, a set of neighboring atoms \({\mathcal{N}}_{i}\) is identified for each atom ai in the structure S. Then, a vector \({\bf{g}}_{ij}^{0}\) is defined as the geometrical influence of a neighboring atom aj on atom ai (1 ≤ j ≠ i ≤ M) based on the Euclidean distance and Voronoi solid angle between them (Supplementary Section IVA). The use of the Voronoi method here clearly determines the neighboring atoms in a local structure based on material domain knowledge and in alignment with the logical design of our method.
SCANN employs an embedding layer to express the atomic information of each atom ai in S by an h-dimensional vector \({\bf{c}}_{i}^{0}\). Hereinafter, we denote the matrix \({\bf{C}}^{0}={[{\bf{c}}_{i}^{0}]}_{1\le i\le M}\) as \({[{\bf{c}}_{i}^{0}]}_{1\le i\le M}=[{\bf{c}}_{1}^{0},{\bf{c}}_{2}^{0},...,{\bf{c}}_{M}^{0}]\). The SCANN architecture comprises of a series of L local attention layers and a global attention layer, each utilizing attention mechanisms16 to represent the local structures within a material structure and the material structure itself, respectively. The layer-wise design of the local attention layers enables SCANN to iteratively learn and enhance the consistency of local structure representations, thereby providing insights regarding long-range interactions between these local structures (Supplementary Section IVB). For instance, the representation vector \({\bf{c}}_{i}^{l+1}\) of the local structure \(\{a_{i},{\mathcal{N}}_{i}\}\) at the (l + 1)th local attention layer can be derived as follows:
where \({\bf{c}}_{i}^{l}\) is the central atom at layer lth, \({\bf{C}}_{{\mathcal{N}}_i}^{l}={[{\bf{c}}_{j}^{l}]}_{{a_j}\in {{\mathcal{N}}_i}}\) denotes its neighboring local structures, and the geometrical influence of the neighboring atoms \({\bf{G}}_{{\mathcal{N}}_i}^{l}={[{\bf{g}}_{ij}^{l}]}_{a_j\in {\mathcal{N}}_i}\). Herein, the local attention layer employed key-query attention16 \({\rm{Attention}}\,({\bf{q}}_{i}^{l},{\bf{K}}_{{\mathcal{N}}_i}^{l})\), in which the query vector \({\bf{q}}_{i}^{l}\) and key matrix \({\bf{K}}_{{\mathcal{N}}_i}^{l}\) are defined as \({\bf{c}}_{i}^{l}{\bf{W}}_{\bf{q}}^{l}\) and \(({\bf{C}}_{{\mathcal{N}}_i}^{l}\times {\bf{G}}_{{\mathcal{N}}_i}^{l}){\bf{W}}_{\bf{k}}^{l}\), respectively.
Previous studies have commonly represented a material structure by a combination of its local structures, typically using operators such as summation or pooling. However, these operators either assume equal contribution from all local structures (sum and average-pooling)14,15,28,29,30 or focus on a single specific local structure (max- and min-pooling)31,32,33, which can hinder the elucidation of structure-property relationships. To address this issue, SCANN represents a material structure as a linear combination of the representation vectors of its local structures, with the global attention (GA) scores of each local structures serving as the coefficients (Supplementary Section IVC). We preserve the structural information of S from all representations of its local structures obtained at the final local attention layer to produce CL, where \({\bf{C}}^{L}={[{\bf{c}}_{i}^{L}]}_{1\le i\le M}\). The global attention layer subsequently learns a suitable representation of the material structure by considering representations of its constituent local structures. This enables the accurate prediction of material properties:
where \({\bf{Q}}^{g}={\bf{C}}^{L}{\bf{W}}_{\bf{q}}^{g}\) and \({\bf{K}}^{g}={\bf{C}}^{L}{\bf{W}}_{\bf{k}}^{g}\) are the query and key matrices, \({\boldsymbol{\alpha }}^{g}=[{\alpha }_{1}^{g},{\alpha }_{2}^{g},\cdots,{\alpha }_{M}^{g}]\) are the corresponding GA scores of each local structure ci. Herein, the global attention layer employed key-query attention SAttention; however, instead of the softmax function, a weighting function ρ(. ) is applied to the attention matrix \({\bf{A}}={{\bf{Q}}^{g}}^{\top}{\bf{K}}^{g}\) to evaluate the GA scores. In detail, we obtain \(s_{j}=\sum_{i=1}^{M}[{\bf{A}}(1-{\bf{I}})]_{i,j}\) as the sum of each column j within the attention matrix A (the identity matrix is denoted as I). Then, we define \(\rho ({\bf{A}})={\rm{softmax}}\,([s_1,s_2,...,s_M])\). Subsequently, this approach can measure the amount of attention (GA scores) that should be given to a local structure by summing all corresponding directional pairwise attention scores from other local structures (Fig. 1b).
Consequently, the physical property yS of the material structure S can be predicted from the learned representation xS with fully connected layers FS, as follows:
This design of the SCANN architecture, particularly the inclusion of a fully connected layer, is tailored to capture the complex and nonlinear relationships between their representations and properties. Furthermore, the GA scores αg of the local structures, obtained from the global attention layer, help indentify key factors that contribute to understanding the structure–property relationships of the material. A comprehensive depiction of the proposed SCANN architecture is presented in Fig. 2.

The SCANN is formed by stacking an embedding layer and local attention layers to learn the representations of various local structures in a material. In the readout stage, a global attention layer is used to assess the attention scores of these local structures. The attention score indicates the degree of attention that should be paid to a local structure to accurately represent the material and predict its physical property. The material representation is linearly combined based on the representations of its local structures with their corresponding attention scores. Fully connected (FC) layers are applied to the material representation to estimate the property of the material.
Experimental design
In this study, we implement two versions of deep learning models using the proposed architecture, each trained independently on different datasets with distinct target properties, with the aim of evaluating the architecture’s performance in predicting target properties and its ability to provide information regarding the structure-property relationships (interpretability) across five molecular and crystal structure datasets (Table 1). The properties of these datasets are determined through quantum mechanical calculations using DFT. The predictive capability is evaluated by partitioning the data into train-validation-test sets. The models are then trained on the training set and optimized to minimize the mean absolute error (MAE) on the validation set. The MAEs of the predictions for the target properties on the test sets are reported for comparison with other models in the literature. Hereafter, the implemented models based on the SCANN architecture, trained with the respective datasets, will be referred to as SCANN models. A detailed explanation regarding the used datasets is given in Supplementary Section IVE.
We evaluated the predictiveness of the SCANN models based on a comparison with seven DL models using the QM96 and Materials Projects34,35 (version MP 2018.6.1) datasets. Among the compared models, SchNet30, MEGNet35, and CGCNN12 utilize graph neural networks to represent molecules or crystals as atomistic graphs. Cormorant14 and SE(3)-Trans15 are GNN variants that incorporate physical constraints, such as covariant or equivalence principles, on the 3D coordinates of atoms. Conversely, ALIGNN36, the current leading network, uses an additional line graph where bonds serve as nodes, and edges convey angular information between bonds in addition to the atomistic graph. This enables ALIGNN to represent the geometrical arrangement of triplets of atoms in a molecule or crystal.
Furthermore, the interpretability of the SCANN models is assessed by examining the relationship between the learned GA scores of the local structures and the corresponding results from first-principles calculations. The results demonstrate the ability of the SCANN models to provide valuable information regarding the structure–property relationships of materials in four scenarios: the local structures and HOMO/LUMO molecular orbitals (QM96 and Fullerene-MD37), the deformation energy ΔU and the deformation of the Pt/graphene structures (Pt/graphene-MD37), and the derived crystal formation energy and the substitution atom species and sites of SmFe12-based compounds (SmFe12-CD38).
Evaluation of the predictive performance
Train–validation–test splits are performed in an 80:10:10 ratio to evaluate the predictive capability of SCANN in predicting five physical material properties (EHOMO, ELUMO, Egap, α, and Cv) in the QM9 dataset. Six DL methods with the MAE of the predictions derived from the models are also employed for comparison. The evaluation process is repeated five times to obtain an average MAE for the test set, thereby providing a robust assessment of the predictive capabilities of the models14,15. Similar to previous practices30,35,36, our study employs train–validation–test configurations of 60000–5000–4239 for the MP 2018.6.1 dataset.
Table 2 presents the average MAE scores obtained from five training runs of the SCANN models, alongside the scores of competing models on the QM9 dataset. The ALIGNN outperforms all competing models across the four properties under consideration. In comparison, the MAEs of the SCANN models are 2 to 2.5 times higher than those of ALIGNN. Despite this gap, SCANN exhibits competitive performance relative to other remaining models, particularly in predicting EHOMO, ELUMO, and Egap. For a comprehensive analysis of the predictive capabilities of these models, please see Supplementary Section IIIA. The predictiveness of the SCANN models on the MP 2018.6.1 dataset is detailed in Table 3. Similar to the result on the QM9 dataset, the ALIGNN model yields the highest prediction accuracy among all competitors. Meanwhile, the MAEs of the SCANN models for predicting the formation energy (ΔE) and band gap (Eg) achieve 29 meV atom−1 and 260 meV, which are 24% and 19% higher than those of ALIGNN, respectively. Notably, the SCANN model has a comparable result with the MEGNet model and exhibits enhancements over the CGCNN and SchNet models regarding ΔE predictions. For Eg, the SCANN model outperforms the CGCNN and the MEGNet models by 32% and 21%, respectively.
Incorporating conventional prior knowledge (e.g., several dozen atomic features and bonding information between atoms12,35,36) or adding physical constraints (e.g., equivalencies, covariates, and equations14,15,39) to the learning process for structure representations can enhance the prediction accuracies. For instance, the ALIGNN model outperformed all competitor models by introducing additional angular information between triplets of atoms, while others only considered two-body interactions (distances and/or bond valences). To improve the descriptiveness for the geometrical structure of molecules or crystals, we develop another version of the SCANN, the SCANN+, with minor adjustments to the original one by employing the Voronoi solid angle embedding layer and updating the geometrical information through multiple LocalAttention layers (Supplementary Section IVD). The additional updates significantly improve the predictive power of the method; the SCANN+ models outperform all other competitors, except for the ALIGNN model, in predicting the electronic properties (EHOMO, ELUMO, and Egap on the QM9 dataset; ΔE and Eg on the MP 2018.6.1 dataset) that are sensitive to the geometrical structure of molecules or crystals (Tables 2 and 3).
However, these strategies introduce higher dimensionality and may bring consequent biases into the model by favoring certain materials, overlooking others, or oversimplifying complex phenomena due to constraints or potential inaccuracies in the heuristic information assigned during the training phase. Consequently, such issues could hamper the clear understanding of structure-property relationships, which is the primary objective of this study. Additionally, for the QM9 dataset, the widely accepted “chemical accuracy" thresholds are 43 meV for the three energy-related properties, EHOMO, ELUMO, and Egap; 0.1 Bohr3 for the isotropic polarizability α; and 0.05 cal mol−1 K−1 for the heat capacity Cv at 298 K40. Notably, the SCANN models demonstrated a prediction error of 41 meV for EHOMO, 34 meV for ELUMO and 0.05 cal mol−1 K−1 for Cv, indicating that chemical accuracy thresholds were achieved for these properties.
In practice, exceeding the threshold for chemical accuracy by increasing the model’s complexity is unnecessary, particularly when the data employed are derived via DFT calculations. Such an approach can lead to overfitting or biases, thereby potentially sacrificing model interpretability and the elucidation of underlying chemical principles. Therefore, we investigate the relationship between the structures of the molecules in the QM9 dataset and their properties (EHOMO and ELUMO) by using the GA scores obtained from the SCANN models instead of those from the more complex SCANN+ model, which possesses higher dimensionality and more parameters (Supplementary Section IID).
Supplementary Section IIIA presents the evaluation of SCANN’s predictive capabilities on three in-house-developed material datasets, demonstrating its broad adaptability and high accuracy in diverse prediction scenarios. The remarkable prediction accuracy of SCANN confirms its practical applicability and suggests that the interpretation derived from the attention scores provides valuable insights into key structure–property relationships for the investigated material properties. In the following sections, we examine the correspondence between the obtained GA scores of the local structures and the corresponding results from first-principles calculations to assess the interpretability of the SCANN models (Supplementary Section IIE–G).
Correspondence between the learned attentions of local structures and the molecular orbitals of small molecules
For the small molecules in the QM9 dataset, the SCANN models demonstrate a remarkable correspondence between the obtained GA scores of the local structures and molecular orbitals results obtained via DFT calculations. As a representative example, Fig. 3 shows the comparison between the GA scores of the local structures and the HOMO/LUMO orbitals obtained from DFT calculations for four molecules. Notably, an apparent correspondence between the relative GA scores of the local structures and the HOMO orbitals of the dimethyl butadiene molecule (cis-2,3-dimethyl-1,3-butadiene) is evident (Fig. 3a). Furthermore, the GA scores of the local structures can be easily linked to the interpretation that dimethyl butadiene readily undergoes the Diels–Alder reaction. Similarly, the correspondence between the HOMO orbital and the GA scores of the local structures is apparent for the thymine molecule (5-methyl pyrimidine-2,4 (1H,3H)-dione), which is a nucleobase in DNA (Fig. 3b).

Correspondence between obtained GA scores and molecular orbitals of four molecules: (a) dimethyl butadiene, (b) thymine, (c) methyl acrylate, and (d) dimethyl fumarate. For each molecule, the left side of the figure illustrates the wave function of the HOMO (a, b), or the LUMO (c, d), as calculated using the DFT approach. The isosurfaces with positive and negative values of the wave functions are represented by blue and red lobes, respectively. The right-side figures display the GA scores of the local structures derived from the SCANN models for interpreting the corresponding molecular orbitals. The coloration of atoms indicates their estimated GA scores, while the coloration of links between them does not signify the sign or nodes of the molecular orbital wave functions.
Moreover, similar correspondences are confirmed between the GA scores of the local structures and the LUMO orbitals obtained from the DFT calculations for methyl acrylate (methyl prop-2-enoate) and dimethyl fumarate (dimethyl(2E)-but-2-enedioate). Methyl acrylate is a reagent that is commonly used in the synthesis of various pharmaceutical intermediates41, whereas dimethyl fumarate has been proposed to exhibit immunomodulatory properties without causing significant immunosuppression42; thus, it has been evaluated as a potential treatment for COVID-1943. The apparent correspondence between the LUMO orbitals and the GA scores of the local structures of these two molecules (Fig. 3c, d) further highlight that the attention scores of the SCANN model provide valuable insights to interpret the structure–property relationships of molecules. Further investigations show that the obtained GA scores from the SCANN+ models are almost consistent with those of the SCANN models for these molecules (Supplementary Fig. 1).
All carbon, nitrogen, and oxygen atomic sites in the QM9 dataset were statistically analyzed for a systematic evaluation of the GA scores obtained by the SCANN models. Since the GA scores of atomic sites were normalized to 1, the relative GA scores were calculated based on the average GA score of the sp3-hybridized carbon atoms in each molecule. Molecules without any sp3-hybridized carbon atoms were excluded (Fig. 4). The analysis of the GA scores for the HOMO energy reveals that the influence on HOMO follows the order of oxygen, nitrogen, and carbon. Specifically, sp3-hybridized carbon sites have a lower influence compared to sp2-hybridized or sp-hybridized carbon sites (Fig. 4a). These findings align with the electronegativity and bonding characteristics of the elements. Oxygen and nitrogen exhibit strong electronegativity and electron-rich regions in π-bonds, leading to a more significant electron density shift and higher HOMO energy localized around oxygen, nitrogen, and carbon sites with double or triple bonds.

Statistics of the relative GA scores for EHOMO (a) and ELUMO (b) for all carbon, nitrogen, and oxygen atomic sites in the molecular structures of the QM9 dataset, calculated based on the average GA score of sp3-hybridized carbon atoms in each molecule. Gray, blue, and red lines and filled regions represent the statistics for carbon, nitrogen, and oxygen sites, respectively.
In contrast, the GA scores for the LUMO energy show no significant difference among the three elements. This observation is consistent with the understanding that unoccupied orbitals primarily influence the LUMO energy, resulting in a less pronounced difference in electronegativity compared to its impact on the HOMO energy (Fig. 4b).
Correspondence between the learned attentions of local structures and molecular orbitals of fullerene molecules
To further evaluate the interpretability of the proposed method, the correspondence between the obtained GA scores of the local structures and the molecular orbitals obtained from DFT calculations for fullerene molecules is examined. Supplementary Fig. 2 shows the GA scores of the local structures for the HOMO and LUMO energies of the C60 molecule (Ih symmetry). In this case, the target molecule has a truncated icosahedral structure composed of 20 hexagons and 12 pentagons, with all carbon atoms exhibiting equivalent local structures. The SCANN model estimates identical GA scores for all local structures of the C60 molecule, thus indicating its ability to handle large and symmetric molecules.
As the number of carbon atoms in the fullerene molecule increases, the symmetry of the C70 (D5h symmetry) and C72 (D6h symmetry) molecules becomes slightly broken, and the local structures of the carbon atoms in these molecules are no longer equivalent. Figure 5 demonstrates the significant correspondence between the GA scores of the local structures and the HOMO and LUMO results obtained from DFT calculations for the C70 and C72 molecules. The GA scores of the local structures in the C70 and C72 molecules exhibit a five-fold (top view) and six-fold (top view) symmetry upon the prediction of the HOMO energy, respectively. These results align with the structural symmetry and degenerate HOMO orbitals of the two fullerene molecules. Notably, the C70 molecule possesses an additional 10-carbon ring, forming a plane symmetry, resulting in a planar symmetry of its HOMO with the node situated on that ring’s plane. The SCANN model reveals a clear correspondence between the HOMO of the C70 molecule and the GA scores of the local structures (Fig. 5a), along with the LUMO and their corresponding GA scores. Furthermore, the shapes of LUMO and HOMO of the C72 molecule exhibit a perfect correspondence with the GA scores of the local structures obtained using the SCANN models (Fig. 5b). Compared to C60, the C72 molecule has an additional ring of 24 carbon atoms with six-fold symmetry, consisting of 12 pairs of carbon–carbon bonds in five-membered carbon rings. The high GA scores of the local structures in the ring indicate the localization of the LUMO of the C72 molecule on the ring. In contrast, the HOMO orbitals are located on two opposite sides of the ring and are also captured by the local structures with high GA scores. This evaluation experiment provides further confirmation that SCANN-derived GA scores offer valuable insights for understanding the structure–property relationship, even for large molecules.

Correspondence between obtained GA scores and the molecular orbitals of (a) C70 and (b) C72. For each molecule, the left side of the figure illustrates the wave functions of the degenerate HOMO (bottom) and LUMO (top) orbitals, as calculated by the DFT approach. The isosurfaces with positive and negative values of the wave functions are represented by the blue and red lobes, respectively. The figure on the right displays the GA scores of local structures obtained using the SCANN model for the corresponding property. The coloration of atoms indicates their estimated GA scores, while the coloration of links between them does not signify the sign or nodes of the molecular orbital wave functions.
Correspondence between the learned attentions of local structures and structural deformation in Pt/graphene
Figure 6a presents the GA scores of the local structures obtained by the SCANN model for predicting the deformation energy of a system comprising a platinum atom adsorbed on a graphene flake. The deformation energy is defined as the difference between the total energy of the deformed and optimized structures. A detailed examination of the obtained GA scores reveals that local structures with high GA scores possess relatively elongated carbon–carbon bonds (Fig. 6b). Additionally, the carbon atoms that form high local curvatures upon the formation of a convex from the planar structure of the sp2 hybridization bonding network received high GA scores (Fig. 6c).

a Visualization of the GA scores obtained from the SCANN model for the Pt/Graphene system with a deformation. The coloration of atoms indicates their estimated GA scores, while the coloration of links between them does not signify the sign or nodes of the molecular orbital wave functions. Structural visualizations of the high attention local structures during the deformation: (b) elongated carbon--carbon bond, and (c) convexed carbon--carbon configuration. The distance from the carbon atom to the adjacent carbon atom (in Å) is highlighted to show the distortion caused by the deformation.
The results obtained from the experiment on the system where a platinum atom was adsorbed on a graphene flake reveal that the GA scores obtained by the SCANN model exhibit a high correspondence with the observed structural deformations. In particular, the high GA scores for the increased carbon–carbon bond lengths and the convexed carbon atoms align well with the contribution to the deformation energy, as determined by DFT calculations. This finding indicates that the GA scores generated by SCANN are reliable indicators of structural deformations in such systems, demonstrating the model’s capability to capture and interpret the underlying material instability. These results validate the usefulness of SCANN in understanding and predicting structural deformations in materials, particularly in cases involving the interaction of different elements or adsorption onto surfaces.
Correspondence between the learned attentions of local structures and stability of SmFe12-based crystal structures
The SCANN model’s ability to predict the formation energy of SmFe12-based crystal structures is evaluated by analyzing the GA scores of atomic sites. The focus is on understanding the effects of substituting Fe sites with other elements on the derived formation energy and the stabilization of the crystal structure, as well as the influence of the elemental substitution on the formation energies of other Sm and Fe sites. It should be noted that the GA scores of the local structures are normalized to ensure that the sum of the attention scores of all local structures in the crystal structure is equal to one.
For instance, Fig. 7a shows the GA scores of the local structures obtained using the SCANN model for predicting the formation energies of the SmFe12, SmFe11Mo, SmFe11Co, and SmFe11Al crystal structures. For the optimized SmFe12 crystal structure, all Fe sites receive identical GA scores, indicating a symmetric cage of Fe atoms surrounding the Sm atoms. Additionally, the negligible difference in GA scores between the Sm and Fe sites suggests that when analyzing the formation energy of the SmFe12 crystal structure, greater attention should be given to the Fe sites rather than the Sm sites. This implies that Sm atoms are comfortably placed within the cage of Fe atoms in the SmFe12 crystal structure.

a Visualization of the GA scores estimated by the model for atomic sites in the SmFe12, SmFe11Mo, SmFe11Co, and SmFe11Al crystal structures. b Correlation between the ratio of GA scores of the substitution sites to the minimum GA scores among the Fe sites and the formation energy, as calculated via the DFT approach, in crystal structures substituted by a single type of element.
For the crystal structures with Mo substitutions, the GA scores of the Mo and Sm sites are estimated to be the same as those of the Fe sites. However, for crystal structures with Co or Al substitutions, the GA scores of the Co and Al sites are significantly higher than those of Fe sites. The GA score results for the three crystal structures indicate that Mo substitution has little effect on the cage of Fe atoms, whereas the Sm sites become nonnegligible in interpreting the formation energy of the SmFe11Mo crystal structure. This suggests that Sm atoms are less comfortably placed within the Fe and Mo atom cages in the substituted crystal structure. By contrast, for crystal structures substituted with Co or Al, the GA scores of the Co and Al sites are significantly higher than those of Fe sites, indicating that the Co and Al sites should be the central focus of attention when interpreting the formation energy of the SmFe11Co and SmFe11Al crystal structures, respectively. Moreover, the GA scores of Fe sites exhibit a slight decrease, indicating that the Fe atoms become more comfortably placed in the substituted crystal structures.
To validate the aforementioned interpretation, the ratio of the GA scores of the substitution sites to the minimum GA scores among the Fe sites was calculated for each crystal structure. Subsequently, the relationship between this ratio and the estimated formation energies of the structures was investigated using DFT calculations. Figure 7b shows that the crystal structures substituted with a single type of element can be divided into two groups: one with Cu, Zn, and Mo substitutions, and the other with Al, Ti, Co, and Ga substitutions. Interestingly, it was observed that crystal structures with higher local structure GA scores for the substitution sites possess lower formation energies, whereas those with lower local structure GA scores for the substitution sites possess higher formation energies. These results highlight the potential of the SCANN model in estimating the local structure GA scores for a rational discussion of SmFe12-substituted crystal structures and their formation energies. While additional first-principles calculations are necessary for each specific crystal structure to fully understand the relationship between the substitution elements, the substitution sites, and the crystal structure stability, these results indicate the potential usefulness of SCANN. The local structure GA score provides valuable information and indicates key focus points for understanding the stability of crystalline material structures. Thus, this study offers valuable insights that can contribute to the development of more efficient and effective methods for designing crystal material structures.
Discussion
This study proposes SCANN, an attention-based DL architecture designed for material dataset analysis. SCANN leverages attention mechanisms to learn from material datasets, predict material properties, and interpret the underlying characteristics of material structures. By applying attention recursively to neighboring local structures, SCANN learns representations of atomic local structures in a self-consistent manner. The architecture then combines these local structure representations to create a comprehensive representation of the entire material structure, enabling precise property predictions. During the learning process, global attention scores are estimated, indicating the importance of each local structure in representing the overall material structure. Experimental results based on five molecular and crystalline material structure datasets demonstrated the excellent predictive capability of SCANN for different material properties. Furthermore, an in-depth qualitative analysis of the global attention scores of local structures revealed that the trained models can extract essential information from material datasets, facilitating a deeper understanding of the structure–property relationships in both molecular and crystalline materials. The ability of the proposed architecture to interpret the attention scores can aid in identifying critical features and accelerating the material design process.
However, it is important to acknowledge that the interpretability of attention scores in DL models is still a subject of debate and lacks clear guidelines44,45,46. Several factors need to be considered, such as the correlation analysis of attention scores, alternative interpretability metrics, and counterfactual analysis, to validate meaningful explanations of the relationships. Additionally, the quantification and assessment of uncertainty in attention score estimation are essential. Despite these challenges, the findings of this study demonstrate the potential of attention mechanisms in uncovering valuable information that can provide a the better understanding of structure–property relationships in materials.
Methods
Characterization of material structure
Given a material structure S with the property of interest \(y_S\in {\mathbb{R}}\) containing M atoms (\({\mathcal{A}}_S=\{a_1,a_2,\cdots,a_M\}\)), we consider the structure S as a geometrical arrangement of M local structures. Each local structure consists of a central atom, its neighboring atoms, and their arrangement around the central atom. To determine the neighboring atoms and segment each material structure into local structures, we employ the definition of O’Keeffe47,48 instead of the assumption about chemical bonds between the atoms in the structure. According to O’Keeffe’s definition, all atoms at these atomic sites share Voronoi polyhedron faces with the atomic site of an atom under consideration (the central atom of the local structure) and are regarded as neighboring atoms. Subsequently, the local structures of the neighboring atoms are referred to as the neighboring local structures. By incorporating the information from the Voronoi polyhedron faces, we assess the geometrical influences of neighboring atoms on the central atoms for conveying the structural information of structure S to SCANN for learning the appropriate representation of S.
For each atom ai in the structure S, by using the Voronoi tessellation, we can determine \({\mathcal{N}}_i\subset{\mathcal{A}}_S\), which contains N atoms whose atomic sites share Voronoi polyhedron faces with an atomic site of ai. Subsequently, the geometrical influence of a neighboring atom \(a_j\in {\mathcal{N}}_i\) on atom ai is represented by a vector \({\bf{g}}_{ij} \in {\mathbb{R}}^{h}\), which is defined by the element-wise multiplication of the Euclidean distance dij (Å) and Voronoi solid angle48 θij ∈ [0, 4π] information between the atoms, as follows:
where DE(dij) is a distance embedding layer representing the distance dij as an h-dimensional vector (Supplementary Section IIB). As a result, for each atom ai, we obtain a matrix \({\bf{G}}_{{\mathcal{N}}_i}^0={[{\bf{g}}_{ij}^0]}_{a_j\in {\mathcal{N}}_i}\) representing the geometrical influences of the neighboring atoms of atom ai. Each row of the matrix consists of a vector \({\bf{g}}_{ij}^0\) that represents the geometrical influence of atom aj on atom ai.
Local structure representation
Similar to other DL architectures, the SCANN employs an embedding layer (Supplementary Section IIA) to express the atomic information of each atom ai in S as an h-dimensional vector \({\bf{c}}_i^0\) (\(\in {\mathbb{R}}^{h}\)). Through training, the vector representation \({\bf{c}}_i^0\) is updated and refined to represent the atom more appropriately for accurately predicting property yS of material structure S.
To learn representations for local structures in material structure S, a local attention layer that utilizes the atomic and geometrical arrangement of atomic sites is proposed. The design of the local attention layer is based on the dot-product key-query attention16, \({\rm{Attention}}\,({\bf{q}},{\bf{K}})={\rm{softmax}}\,({\bf{q}}^{\top }{\bf{K}})\,{\bf{K}}\), where \({\bf{q}}\in {\mathbb{R}}^{h}\) and \({\bf{K}}\in {\mathbb{R}}^{h\times h}\) denote the query vector and key matrix, respectively. In addition, SCANN consists multiple local attention layers to iteratively update the representation of local structures in a layer-wise manner; the (l+1)th local attention layer uses the representations of local structures constructed from the lth layer as inputs. As a result, this design enables SCANN to efficiently capture information on long-range interaction between local structures in the material structure.
For instance, the representation \({\bf{c}}_i^{l+1}\) (\(\in {\mathbb{R}}^h\)) of local structure \(\{a_i,{\mathcal{N}}_i\}\) at the (l + 1)th local attention layer is derived from the representation vectors in the preceding layer of itself (\({\bf{c}}_i^l\)), its neighboring local structures (\({\bf{C}}_{{\mathcal{N}}_i}^l=[{\bf{c}}_j^l]_{a_j\in {\mathcal{N}}_i}\)), and the geometrical influence of the neighboring atoms \({\mathcal{N}}_i\) on atom ai (\({\bf{G}}_{{\mathcal{N}}_i}\)) as follows:
where \({\bf{q}}_i^l={\bf{c}}_i^l{\bf{W}}_{\bf{q}}^l\) and \({\bf{K}}_{{\mathcal{N}}_i}^l=({\bf{C}}_{{\mathcal{N}}_i}^l\times {\bf{G}}_{{\mathcal{N}}_i}^l){\bf{W}}_{\bf{k}}^l\); \({\bf{W}}_{\bf{k}}^l,{\bf{W}}_{\bf{q}}^l\in {\mathbb{R}}^{h\times h}\) are learnable parameters of the local attention layer and are shared between local structures. For the SCANN model, the geometry influences at each layer is kept the same as initial layer \({\bf{g}}_{ij}^l={\bf{g}}_{ij}^0\). The detailed implementation of the local attention layer is described in Supplementary Section IIC.
Owing to the application of multiple local attention layers, the attention information regarding a target property between local structures in a material structure S can be passed through the attention relationships between neighboring local structures. In our evaluation experiments, SCANN models employ L local attention layers, with the value of L tailored to optimize performance for each dataset, as explained in Supplementary Section IIIB. Consequently, we preserve the structural information of S from the representations of all its local structures obtained from the final local attention layer, to produce CL, where \({\bf{C}}^L=[{\bf{c}}_i^L]_{a_i\in {\mathcal{A}}_S}\).
Material structure representation
To represent a material structure S, simple operators, such as the sum or pooling operator, are typically applied to integrate the representations of all local structures in S. However, such operators consider that either the contribution of each local structure to the final structure representation is equal (sum and average-pooling operator)14,15,28,29,30 or that the property depends on only the specific local structures in the material structure whereas the others have zero impact (max- and min-pooling operators)31,32,33. Therefore, designing appropriate combination operators for specific target properties is challenging and requires prior hypotheses regarding the structure–property relationships. To overcome this problem, SCANN again utilizes the dot-product key-query attention16 to coherently learn the representation of local structures and integrate them into the representation of material structure in a target-dependent manner.
An attention mechanism-based layer, called the global attention layer, is proposed to quantitatively model the attention distribution required across each constituent local structure. This layer aims to obtain a more appropriate representation for the entire structure S. The global attention layer is designed to learn an optimal representation of structure S from data, which subsequently facilitates the construction of a highly accurate predictive model for the target property yS. The representation vector xS of the structure S is formulated by aggregating the representations of all the constituent local structures according to the obtained global attention (GA) scores, as shown below:
where \({\bf{A}}={{\bf{Q}}^g}^{\top}{\bf{K}}^g\in {\mathbb{R}}^{M\times M}\), which \({\bf{Q}}^g={\bf{C}}^L{\bf{W}}_{\bf{q}}^g\) and \({\bf{K}}^g={\bf{C}}^L{\bf{W}}_{\bf{k}}^g\) are the query and key matrices, respectively; further, \({\bf{W}}_{\bf{k}}^g,{\bf{W}}_{\bf{q}}^g\in {\mathbb{R}}^{h\times h}\) are the learnable parameters of the global attention layer. A weighting function ρ(.) is applied to the attention matrix A to evaluate the GA scores paid to the local structures. As a result, we obtain \(\rho ({\bf{A}})={\rm{softmax}}\,([s_1,s_2,\cdots,s_M])\), in which \(s_j=\sum_{i=1}^M[{\bf{A}}(1-{\bf{I}})]_{i,j}\) is the sum of each column j within the attention matrix A (the identity matrix is denoted as I).
The function ρ(.) is designed based on the assumption that heightened attention should be allocated to local structures whose representations are crucial for accurately representing the other local structures in S. This attention allocation allows a the precise prediction of target property yS. In essence, a local structure that garners higher cumulative attention scores from all the other local structures should be prioritized when representing material structure S. As a result, the degree of attention to a local structure \(\{a_i,{\mathcal{N}}_i\}\) in S is quantitatively modeled by summing all the attention received from other local structures. For a detailed implementation of the global attention layer, please refer to Supplementary Section IID.
Consequently, the physical property yS of the material structure S can be predicted from the learned representation xS, as shown below:
where \(F_S:{\mathbb{R}}^h\to {\mathbb{R}}\) is represented by two fully connected (FC) layers. The weight matrices and bias vectors of the network are learned by training the prediction model.
Furthermore, the GA scores \({\boldsymbol{\alpha}}^g=[\alpha_1^g,\alpha_2^g,\cdots,\alpha_M^g]\), which describe the degree of attention given to the corresponding local structures for representing S, are used to reveal critical aspects that help interpret the structure–property relationship of interest. Notably, the attention to local structures discussed here signifies the amount of information these local structures contribute to appropriately represent S for the accurate prediction of yS.
SCANN+
The SCANN+ introduced the embedding vector for the Voronoi solid angle θij as follows:
where DE(dij) and AE(θij) are the distance and angle embedding layers corresponding to the distance dij and angle θij of an h-dimensional vector (Supplementary Section IIB). In addition, the geometry influences between the neighbor \({\bf{c}}_j^l\) and the center \({\bf{c}}_i^l\) are updated based on the following formulation:
where ⊕ denotes the concatenating vectors and \(F_g:{\mathbb{R}}^h \to {\mathbb{R}}^h\) is a fully connected (FC) layer.
Model training
The training of the DL model using the proposed architecture begins with the initialization of all learnable parameters. All weighting matrices such as \({\bf{W}}_{\bf{q}}^l\), \({\bf{W}}_{\bf{k}}^l\), \({\bf{W}}_{\bf{q}}^g\), and \({\bf{W}}_{\bf{k}}^g\) are initialized to random matrices using Glorot Uniform49, while the entries of all bias vectors are initialized to zero. The dropout layer and attention dropout16 are applied in the local attention layers with a rate of 0.1 for better regularization.
In the training process, all parameters of the proposed DL model are updated by minimizing a loss function using Adam optimization50 with a scheduled learning rate decay ranging from 5 × 10−4 to 10−4. To predict the physical property yS of a material structure S in training dataset \({\mathcal{D}}\), the loss function is defined as follows:
Each model is trained on an individual Tesla A100-PCIe graphics processing unit (GPU) with a memory capacity of 40 gigabytes. Remarkably, there are fewer than one million parameters in SCANN, primarily affected by the configuration settings of the number of LocalAttention layers (L). Supplementary Table II contains the epoch-wise time cost with a batch size 128 for the QM9 dataset. SCANN excels in term of training times per epoch and is distinguished by its commendable memory efficiency, and it is thus highly suitable for practical deployment.
Dataset information
QM96
This computational dataset comprises of data of 133,885 drug-like organic molecules composed of C, H, O, N, and F. However, 3054 files were removed due to the questionable geometric stability14 that 130,831 molecules remained were used for the experiments. Five physical properties from the QM9 dataset are used as targets for evaluating the predictive capability of the SCANN models. These properties include the energy of the highest occupied molecular orbital (EHOMO), the energy of the lowest unoccupied molecular orbital (ELUMO), the gap between the energies (Egap = ELUMO − EHOMO), the isotropic polarizability (α), and the heat capacity at 298 K (Cv). In the experiment, the predictive capability of the SCANN models is compared with that of recent state-of-the-art DL models14,30,35,39.
Fullerene-MD37
This is an in-house-developed computational material dataset comprising the data of three well-known fullerene molecules, viz. C60 (Ih), C70 (D5h), and C72 (D6h). It includes optimized structures and 3000 deformed structures obtained from molecular dynamics simulations (1000 structures for each molecule). The HOMO (EHOMO) and LUMO (ELUMO) energies of these structures are determined using DFT calculations, similar to the approach used in the QM9 dataset. Experiments are performed on this dataset to evaluate the predictive capability of the SCANN models for HOMO and LUMO energies and to assess the interpretability of the models’ predictions for these properties. A distinctive feature of all structures in this dataset is that they only contain carbon atoms. Furthermore, due to the symmetric nature of fullerene molecules, the local structures within each molecule are highly similar with only minor differences. Therefore, this dataset allows for a precise evaluation of the interpretability of the SCANN model. In the evaluation experiment using this dataset, SCANN models pre-trained on the QM9 dataset are applied to train the prediction models for the HOMO and LUMO energies of the fullerene molecules.
Pt/graphene-MD37
This dataset is also an in-house-developed computational material dataset representing a system composed of a platinum atom adsorbed on a graphene flake terminated by hydrogen atoms51,52. It consists of data from approximately 21,000 optimized and deformed structures generated through molecular dynamics simulations. The adsorption energies of these structures are determined using DFT calculations, similar to the approach used in the QM9 dataset. There are two main purposes of the experiments conducted on this dataset is twofold–to evaluate the predictive performance of the SCANN models for deformation energies of the structures (ΔU) and to assess the interpretability of the models’ predictions for these deformation energies. The structural characteristic of this dataset is the presence of a two-dimensional honeycomb network of carbon atoms forming the graphene flake. Although the local structures of each carbon atom in the system exhibit slight distortions from the ideal sp2 hybridization structure52, this dataset enables a quantitative evaluation of the interpretability of the SCANN models in terms of the distortion of the honeycomb network on the graphene surface.
SmFe12-CD38
This dataset is an in-house-developed computational material dataset containing the data of crystalline magnetic materials. It comprises the data from 3307 optimized structures of SmFe12-based compounds, along with their corresponding formation energies (ΔE) as the target properties. The dataset was generated by introducing partial substitutions of Mo, Zn, Co, Cu, Ti, Al, and Ga into the iron sites of the original SmFe12 structure, which exhibits notable magnetic properties. Subsequently, the structures were optimized, and their formation energies were assessed using DFT calculations. A detailed explanation regarding the DFT calculation method used to create this dataset can be found in a previous work38. By using this dataset, the predictive capability of the SCANN models for the formation energies of the structures (ΔE) and the interpretability of the models are quantitatively evaluated to investigate the structural stability of the SmFe12-based structures.
MP-2018.6.134,35
This dataset is a time-versioned snapshot of the Materials Project dataset. Since solid-state datasets are continuously updated, we specifically chose the MP version used by previous works to facilitate a direct comparison of model performance with the literature. We specifically utilized the Materials Project version that was updated until June 1, 2018, which encompasses 69,239 crystal structures. Two physical properties from the material dataset are used as targets for evaluating the predictive capability of the SCANN models. These properties include the DFT-computed formation energy per atom (ΔE) and the band gaps (Eg). In the experiment, the predictive capability of the SCANN models is compared with that of recent state-of-the-art DL models.
Data availability
All the computational material datasets related to this article have been deposited to a Zenodo repository37.
Code availability
The Python implementations for training the SCANN models and predicting physical properties have been deposited to a GitHub repository53.
References
Agrawal, A. & Choudhary, A. Perspective: materials informatics and big data: realization of the “fourth paradigm" of science in materials science. APL Mater. 4, 053208 (2016).
Ramprasad, R., Batra, R., Pilania, G., Mannodi-Kanakkithodi, A. & Kim, C. Machine learning in materials informatics: recent applications and prospects. Npj Comput. Mater. 3, 54 (2017).
Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O. & Walsh, A. Machine learning for molecular and materials science. Nature 559, 547–555 (2018).
Siriwardane, E. M. D., Zhao, Y., Perera, I. & Hu, J. Generative design of stable semiconductor materials using deep learning and density functional theory. Npj Comput. Mater. 8, 164 (2022).
Ward, L. & Wolverton, C. Atomistic calculations and materials informatics: a review. Curr. Opin. Solid State Mater. Sci. 21, 167–176 (2017).
Ramakrishnan, R., Dral, P. O., Rupp, M. & Von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. data 1, 1–7 (2014).
Himanen, L., Geurts, A., Foster, A. S. & Rinke, P. Data-driven materials science: status, challenges, and perspectives. Adv. Sci. 6, 1900808 (2019).
Zhao, Y. et al. Physics guided deep learning for generative design of crystal materials with symmetry constraints. Npj Comput. Mater. 9, 38 (2023).
Rupp, M., Tkatchenko, A., Müller, K.-R. & Von Lilienfeld, O. A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108, 058301 (2012).
Schütt, K. T. et al. How to represent crystal structures for machine learning: towards fast prediction of electronic properties. Phys. Rev. B 89, 205118 (2014).
Karamad, M. et al. Orbital graph convolutional neural network for material property prediction. Phys. Rev. Mater. 4, 093801 (2020).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Rahaman, O. & Gagliardi, A. Deep learning total energies and orbital energies of large organic molecules using hybridization of molecular fingerprints. J. Chem. Inf. Model. 60, 5971–5983 (2020).
Anderson, B., Hy, T. S. & Kondor, R. Cormorant: covariant molecular neural networks. NeurIPS 32, 14537–14546 (2019).
Fuchs, F. B., Worrall, D. E., Fischer, V. & Welling, M. Se(3)-transformers: 3d roto-translation equivariant attention networks. NeurIPS 33, 1970–1981 (2020).
Vaswani, A. et al. Attention is all you need. NeurIPS 30, 6000–6010 (2017).
Duvenaud, D. K. et al. Convolutional networks on graphs for learning molecular fingerprints. NeurIPS 28, 2224–2232 (2015).
Wu, Z. et al. Moleculenet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018).
Fung, V., Zhang, J., Juarez, E. & Sumpter, B. G. Benchmarking graph neural networks for materials chemistry. Npj Comput. Mater. 7, 84 (2021).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. ICML 70, 1263–1272 (2017).
Yang, K. et al. Analyzing learned molecular representations for property prediction. J. Chem. Inf. Model. 59, 3370–3388 (2019).
Chen, P., Jiao, R., Liu, J., Liu, Y. & Lu, Y. Interpretable graph transformer network for predicting adsorption isotherms of metal-organic frameworks. J. Chem. Inf. Model. 62, 5446–5456 (2022).
Cao, Z., Magar, R., Wang, Y. & Barati Farimani, A. MOFormer: self-supervised transformer model for metal-organic framework property prediction. J. Am. Chem. Soc. 145, 2958–2967 (2023).
Kang, Y., Park, H., Smit, B. & Kim, J. A multi-modal pre-training transformer for universal transfer learning in metal–organic frameworks. Nat. Mach. Intell. 5, 309–318 (2023).
Korolev, V. & Protsenko, P. Accurate, interpretable predictions of materials properties within transformer language models. Patterns 4, 100803 (2023).
Das, K., Goyal, P., Lee, S.-C., Bhattacharjee, S. & Ganguly, N. Crysmmnet: multimodal representation for crystal property prediction. PMLR 216, 507–517 (2023).
Gunning, D. et al. XAI–Explainable artificial intelligence. Sci. Robot. 4, eaay7120 (2019).
Moran, M., Gaultois, M. W., Gusev, V. V. & Rosseinsky, M. J. Site-net: using global self-attention and real-space supercells to capture long-range interactions in crystal structures. Digit. Discov. 2, 1297–1310 (2023).
Pham, T.-L. et al. Learning materials properties from orbital interactions. J. Phys. Conf. Ser. 1290, 012012 (2019).
Schütt, K. T., Sauceda, H. E., Kindermans, P.-J., Tkatchenko, A. & Müller, K.-R. Schnet–a deep learning architecture for molecules and materials. J. Chem. Phys 148, 241722 (2018).
Wu, Z. et al. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 32, 4–24 (2021).
Schweidtmann, A. M. et al. Physical pooling functions in graph neural networks for molecular property prediction. Comput. Chem. Eng. 172, 108202 (2023).
Keyulu, X., Weihua H., Leskovec, J. & Jegelka, S. How Powerful are Graph Neural Networks? In Proceedings of the 7th International Conference on Learning Representations (ICLR 2019), New Orleans, LA, USA, 2019.
Jain, A. et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mater. 31, 3564–3572 (2019).
Choudhary, K. & DeCost, B. Atomistic line graph neural network for improved materials property predictions. Npj Comput. Mater. 7, 185 (2021).
Vu, T.-S. & Chi, D. H. Fullerene structures and Pt absorbed on Graphene structures with HOMO, LUMO and Total energy properties. Zenodo, https://zenodo.org/record/7792716 (2023).
Nguyen, D.-N., Kino, H., Miyake, T. & Dam, H.-C. Explainable active learning in investigating structure-stability of SmFe12−α−βXαYβ structures X, Y {Mo, Zn, Co, Cu, Ti, Al, Ga}. MRS Bull. 48, 31–44 (2022).
Hirn, M., Mallat, S. & Poilvert, N. Wavelet scattering regression of quantum chemical energies. Multiscale Model. Simul. 15, 827–863 (2017).
Faber, F. A. et al. Prediction errors of molecular machine learning models lower than hybrid dft error. J. Chem. Theory Comput. 13, 5255–5264 (2017).
Ohara, T. et al. Acrylic acid and derivatives. Ullmann’s Encyclopedia of Industrial Chemistry 1–21 (Wiley Online Library, 2020).
Schulze-Topphoff, U. et al. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. PNAS 113, 4777–4782 (2016).
Mantero, V. et al. Covid 19 in dimethyl fumarate treated patients with multiple sclerosis. J. Neurol. 268, 2023–2025 (2021).
Jain, S. & Wallace, B. C. Wallace. Attention is not Explanation. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume1 (Long and Short Papers), 3543–3556, Association for Computational Linguistics, Minneapolis, Minnesota.
Wiegreffe, S. & Pinter, Y. Attention is not not Explanation. In Proceedings of the 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP), 11–20, Association for Computational Linguistics, Hong Kong, China.
Grimsley, C., Mayfield, E. & R.S. Bursten, J. Why attention is not explanation: Surgical intervention and causal reasoning about neural models. LREC 12, 1780–1790 (2020).
O’Keeffe, M. A proposed rigorous definition of coordination number. Acta Crystallogr. A: Found. Adv. 35, 772–775 (1979).
Pham, T. L. et al. Machine learning reveals orbital interaction in materials. Sci. Technol. Adv. Mater. 18, 756 (2017).
Glorot, X. & Bengio, Y. Understanding the difficulty of training deep feedforward neural networks. PMLR 9, 249–256 (2010).
Kingma, D. & Ba, J. Adam: A Method for Stochastic Optimization. In Proceedings of the 3rd International Conference on Learning Representations (ICLR 2015), San Diego, CA, USA, 2015.
Chi, D. H. et al. Electronic structures of Pt clusters adsorbed on (5,5) single wall carbon nanotube. Chem. Phys. Lett. 432, 213–217 (2006).
Dam, H. C. et al. Substrate-mediated interactions of Pt atoms adsorbed on single-wall carbon nanotubes: Density functional calculations. Phys. Rev. B 79, 115426 (2009).
Vu, T. S. Python implementation of Self-Consistent Atention-based Neural Network - SCANN. GitHub, https://github.com/sinhvt3421/scann--material (2023).
Acknowledgements
This work was supported by the JSPS KAKENHI Grants 20K05301, JP19H05815, 20K05068, and JP23H05403; and the JST-CREST Program (Innovative Measurement and Analysis) JPMJCR2235, Japan.
Author information
Authors and Affiliations
Contributions
T.-S.V., M.-Q.H., D.-N.N., T.T., and H.-C.D. conceived and designed the experiments. T.-S.V., M.-Q.H., V.-C.N., and H.-C.D. performed the experiments. T.-S.V., D.-N.N., H.K., and H.-C.D. analyzed the data. T.-S.V., D.-N.N., and H.-C.D. contributed the materials and analysis tools. T.-S.V., M.-Q.H., and H.-C.D. wrote the paper. T.-S.V., M.-Q.H., D.-N.N., Y.A., T.T., H.T., H.K., T.M., K.T. and H.-C.D. reviewed and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41524_2023_1163_MOESM1_ESM.pdf
Supplementary Information: Towards understanding structure–property relations in materials with interpretable deep learning
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vu, TS., Ha, MQ., Nguyen, DN. et al. Towards understanding structure–property relations in materials with interpretable deep learning. npj Comput Mater 9, 215 (2023). https://doi.org/10.1038/s41524-023-01163-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-023-01163-9
