
Abstract
While machine learning has emerged in recent years as a useful tool for the rapid prediction of materials properties, generating sufficient data to reliably train models without overfitting is often impractical. Towards overcoming this limitation, we present a general framework for leveraging complementary information across different models and datasets for accurate prediction of data-scarce materials properties. Our approach, based on a machine learning paradigm called mixture of experts, outperforms pairwise transfer learning on 14 of 19 materials property regression tasks, performing comparably on four of the remaining five. The approach is interpretable, model-agnostic, and scalable to combining an arbitrary number of pre-trained models and datasets to any downstream property prediction task. We anticipate the performance of our framework will further improve as better model architectures, new pre-training tasks, and larger materials datasets are developed by the community.
Similar content being viewed by others


Introduction
In recent years, software development of automated density functional theory (DFT) calculation workflows has led to the emergence of large open-source databases of materials and their simulated properties1,2,3. However, due to computational restraints, not all properties are computed for all materials in these databases. For example, at the time of writing, the Materials Project (MP)1 contains 144,595 inorganic materials, but only 76,240 electronic bandstructures, 14,072 elastic tensors, and 3402 piezoelectric tensors. Many studies have thus trained supervised machine learning (ML) models on materials for which property data is available, subsequently screening the remaining materials orders of magnitude faster than DFT. After identifying promising materials with ML-based screenings, these materials are studied more rigorously with DFT and/or experiment. Example applications wherein ML-based screenings led to successful simulated or experimental validation include photovoltaics, superhard materials, batteries, hydrogen storage materials, ferroelectrics, shape memory alloys, dielectrics, and more4.
To handle the data types encountered in materials, ML approaches in materials science generally involve statistical learning models using hand-crafted, application-dependent descriptors as input5,6 or graph neural networks (GNNs) directly using materials’ atomic structures as input7,8. While the latter models have shown superior performance likely by more faithful representation of atomic structures9, their large number of trainable parameters requires on the order of 104 data examples to sufficiently reduce overfitting relative to descriptor-based methods6. Acquiring 104 data examples can be impractically expensive, limiting our ability to build predictive ML models, e.g., for experimental data and complex systems like layered materials, surfaces, and materials with point defects. Similarly, generating large amounts of data is infeasible for rare materials behaviors and phases like high-temperature superconductors or spin liquids. Developing predictive ML models to effectively handle data scarcity in materials science is thus a pervasive challenge with practical significance for a range of technologies.
Several approaches have been applied in materials science to reduce the large data requirement of neural networks. Many of these approaches can be classified as regularizing neural networks to perform well across multiple relevant tasks—similar to how humans use background knowledge to learn from few examples.
One such regularization technique commonly employed in materials science is pairwise transfer learning (TL), wherein parameters of a model pre-trained on a data-abundant source task (e.g., predicting formation energy) are used to initialize training on a data-scarce, downstream task (e.g., predicting experimental bandgaps)7,10,11,12,13,14,15,16. A well-known obstacle for TL is catastrophic forgetting, which is the tendency of a model to forget relevant information from the source task when adapting to the data-scarce task and subsequently overfitting17,18. To avoid catastrophic forgetting, early layers of the pre-trained model are typically frozen while later layers are fine-tuned, i.e., updated with a reduced learning rate15. However, TL suffers from several limitations; its success is contingent on the existence of a source task with many data examples and high similarity to the downstream task. Additionally, TL only allows information to be leveraged from a single task, and the source task from which to transfer from is not generally known a priori19,20. TL from a source task dissimilar to the downstream task can even lead to worse performance than training a model on the downstream task from scratch, a phenomenon known as negative transfer17,21. Previous studies in materials science have thus either transferred from the largest available source task7, transferred from lower to higher fidelity data of the same property12,13,14, conducted brute-force experiments on different source tasks11,22, or engineered new source tasks not directly relevant for a materials application but serving as generalizable pre-training tasks10,23.
Other techniques, such as multitask learning (MTL), can leverage information across many tasks. MTL has already been used to improve model performance by jointly predicting formation energies, bandgaps, and Fermi energies with a single model24. However, MTL models are in general difficult to train; determining task groupings for joint training without detriment to performance (i.e., without negative transfer from task interference) is an open research question25,26. Furthermore, optimal groupings are sensitive to hyperparameters like learning rate and batch size27. This sensitivity arises because MTL models must overcome imbalanced task gradient magnitudes and conflicting task gradient directions during training28,29,30. Also, MTL models frequently suffer from catastrophic forgetting when adapted to new tasks18.
In this work, to overcome the aforementioned limitations of TL and difficulties of MTL, we propose a mixture of experts (MoE) framework for materials property prediction. By construction, our framework can leverage information from an arbitrary number of source tasks and pre-trained models to any downstream task, does not experience catastrophic forgetting or task interference across source tasks as in MTL, and automatically learns which source tasks and pre-trained models are the most useful for a downstream task in a single training run. Our framework consistently outperforms pairwise TL on a suite of data-scarce property prediction tasks; emits interpretable relationships between source and downstream property prediction tasks; and provides a general, modular framework to combine complementary models and datasets for data-scarce property prediction. The generality of our approach also makes it compatible with any new source tasks, model architectures, or datasets which may be developed in the future.
Results and discussion
Pairwise transfer learning
Pairwise transfer learning involves using all or a subset of parameters from a pre-trained model to initialize training on a data-scarce, downstream task. We know fundamental rules of quantum chemistry generalize across materials and properties, i.e., the periodic table and Schrodinger’s equation are universal. However, the final mapping from fundamental physics to a specific property depends heavily on the property. For example, while both formation energy and electronic band gap can be obtained from DFT, computing formation energy requires comparing to a relevant reference state, while computing bandgaps requires comparing band edge positions. Thus, after pre-training a model on a source task for TL (and MoE), we only re-used a subset of the pre-trained model parameters to produce generalizable features of an atomic structure. We let these pre-trained parameters define a feature extractor, E(⋅). Specifically, the extractor takes in an atomic structure x and outputs a feature vector E(x) describing the structure. Predictions of a scalar property of any atomic structure is then produced by passing the feature vector E(x) through a property-specific head neural network, H(⋅). Putting it together, predictions \(\hat{y}\) are produced as \(\hat{y}=H(E(x))\).
Similar to refs. 24 and 31, we let the extractor E(⋅) be the atom embedding and graph convolutional layers of a crystal graph convolutional neural network (CGCNN)8. These layers produce a representation of a crystal from its constituent atom types and pairwise interatomic distances. Our head H(⋅) is a multilayer perceptron. Specific hyperparameters of the architecture can be found in Supplementary Table 1. In our pairwise TL experiments, we found it beneficial to extract from and fine-tune the last graph convolutional layer when transfer learning to a downstream task (see Supplementary Figs. 2 and 3). We applied these design choices to all TL and MoE experiments in the rest of this paper.
The mixture of experts framework
MoEs were first introduced more than three decades ago32 and have since been studied as a general-purpose neural network layer notably for tasks in natural language processing33. MoE layers consist of multiple expert neural networks and a trainable gating network which, often conditionally, routes inputs through the experts. The output of the MoE layer is then computed by aggregating outputs of all the activated experts. A result of the MoE layer’s gating mechanism is that large parts of the model can be inactive on a per-example basis, enabling massive increases in model capacity and performance without concomitant increases in training cost. Interestingly, in natural language processing, it has also been shown that the experts tend to automatically become highly specialized based on syntax and semantics33.
Formally, a MoE layer consists of m experts \({E}_{{\phi }_{1}},...,{E}_{{\phi }_{m}}\) parameterized by ϕ1, . . . , ϕm and a gating function G(x, θ, k) which takes in trainable parameters θ and produces a k-sparse, m-dimensional probability vector. In our work, since each expert is responsible for producing a feature vector describing a material, we refer to each expert as an extractor. For simplicity, we also chose to make our gating function independent of the model input (i.e., which material we are making a property prediction for), so we have G(x, θ, k) = G(θ, k). For a given input x, we denote the output of the ith extractor as \({E}_{{\phi }_{i}}(x)\) and the ith output of G(θ, k) as Gi(θ, k). The output f of our MoE layer is a feature vector produced by a mixture of extractors, i.e.,
where ⨁ is an aggregation function.
We experimented with letting the aggregation function be concatenation or addition, comparing performance with end-to-end learning of a weighted ensemble of different fine-tuned CGCNN predictions. Table 1 reports the mean absolute error (MAE) of each method on three data-scarce tasks: predicting piezoelectric moduli34, 2D exfoliation energies35, and experimental formation energies36,37. These tasks consisted of 941, 636, and 1709 data examples, respectively. A special sampling procedure was used when partitioning test splits for the experimental formation energy dataset (see Model training in “Methods”). None of the aggregation methods consistently outperformed the others on the three tasks, so we opted for addition. An advantage of this choice is that the model’s feature dimensionality becomes independent of the number of feature extractors. As a proof-of-concept, our extractors \(\{{E}_{{\phi }_{i}}(\cdot )\}\) are CGCNNs each pre-trained on a different materials property dataset with at least 104 examples. All datasets were acquired through Matminer38.
Ourgating mechanism is parameterized with \(\theta \in {{\mathbb{R}}}^{m}\) and a hyperparameter \(k\in {{\mathbb{N}}}^{+}\) controlling sparsity, where \({{\mathbb{N}}}^{+}\) denotes natural numbers greater than zero. Our gating function G(θ, k) is as follows:
Before applying the Softmax function, we only keep the top k values of θ. Mathematically, this is equivalent to setting the rest of the values to −∞, assigning the corresponding extractors a gating value of 0 after applying the Softmax. To encourage the model to focus on the most relevant extractors, we followed ref. 39 and added a regularization term P to the training loss:
Here, \({{{\boldsymbol{a}}}}=G(\theta ,k)\in {{\mathbb{R}}}^{(m\times 1)}\) is a vector of probability scores assigned to each extractor, and λ is a hyperparameter weighting the regularization term. We set λ = 0.01. Intuitively, \({({{{{\boldsymbol{a}}}}}^{T}{{{\boldsymbol{a}}}}-1)}^{2}\ge 0\) with equality if and only if a concentrates all probability mass on a single extractor (wherein aTa = 1).
While we chose the extractors \({E}_{{\phi }_{1}},...,{E}_{{\phi }_{m}}\) to be CGCNNs, this choice can be much more flexible. For example, extractor outputs could be embeddings from different layers of a single CGCNN or other graph neural networks7,8,40, hand-crafted featurizers6, language models41, or generative models42 trained by single-task, multitask, supervised, unsupervised, semi-supervised, or self-supervised learning. This ability to combine different extractor architectures is distinct from TL or MTL, where single architectures must be used across all tasks. In addition, extractors can be trained on any dataset which serves as generalizable pre-training data. These might include materials properties from the Materials Project1, the Open Quantum Materials Database43, JARVIS3, or AFLOW2; text from scientific journal abstracts41; or data generated for unsupervised or self-supervised learning10,23,42.
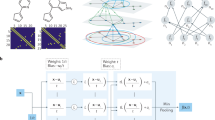
A schematic summarizing our framework is shown in Fig. 1. The first phase of our approach is to pre-train separate models on different source tasks. In our case, these source tasks involve prediction of scalar properties from large datasets, where, following ref. 6, we defined ‘large’ as consisting of more than 104 examples (Fig. 1a). A complete list of our source tasks and the resulting pre-trained model performances are enumerated in Supplementary Table 2. Because each extractor is pre-trained separately, there is no possibility of task interference during pre-training as in MTL. The second phase is to combine and adapt the separate models to a downstream, data-scarce task (Fig. 1b). Our adaptation process consisted of training a randomly initialized head while fine-tuning the last layer of each extractor towards the new task.

a First, separate machine learning models, each consisting of a feature extractor and a classification or regression head, are trained on separate learning tasks. b Next, the pre-trained experts are adapted to a downstream learning task along with a newly initialized head.
MoE outperforms TL from the best pre-trained model
We examined whether transferring information from multiple pre-trained models with the MoE framework could outperform transferring from a single pre-trained model. Specifically, we evaluated five different methods (visually described in Fig. 2) on the same three downstream, data-scarce tasks as before: predicting piezoelectric modulus34, 2D materials’ exfoliation energies35, and experimental formation energies36,37.

The experimental formation energy task is used for demonstration.
The first baseline method, termed STL, was traditional single-task learning on the data-scarce target task from a randomly initialized model (i.e., without pre-training).
The next baseline method, termed Best TL-(3), first involved humans selecting three source tasks for each downstream task. These selections represented our best effort at intuitively picking the most relevant tasks to the downstream tasks without using ML. The rationale behind the chosen source tasks is described below. Next, pairwise TL from each chosen source task was conducted independently, and the best model performance is reported.
The third method, termed MoE-(3), used the same three source tasks as Best TL-(3), but in a single training run under the MoE framework with hyperparameter k = 3.
The fourth method, termed MoE-(18), used all eighteen available source tasks with hyperparameter k = 18, challenging an MoE model to automatically learn which extractors were most useful for the downstream task at hand. This ability to automatically discover task relationships is critical when relationships between source and downstream tasks are counterintuitive or unknown. For example, we may lack or have an incorrect scientific understanding for a particular property. Or, if source tasks are unsupervised or self-supervised, relationships to properties may not be justifiable with domain knowledge.
As a baseline for MoE-(18), the fifth method explored was Best TL-(18). This method followed the same procedure as Best TL-(3) but used all available source tasks instead of three human-chosen ones. We emphasize that Best TL-(18) is not scalable to a large number of source tasks since each source task requires training an additional model.
The human-chosen source tasks for Best TL-(3) and MoE-(3) were picked as follows. We let MP formation energies be a pre-training task across all three downstream tasks since it is the largest dataset available in Matminer. For predicting piezoelectric modulus, we transferred from MP bulk moduli1,44, since piezoelectric tensors are derived in part from elastic tensors34, and MP bandgaps1, since a nonzero band gap is required to maintain an electric polarization. For predicting 2D materials’ exfoliation energies, we transferred from JARVIS formation energies, since these are also thermodynamic quantities and come from the same data source3, and JARVIS shear moduli, since small elastic constants have been suggested as a signature of weak van der Waals bonding and low exfoliation energies45. For predicting experimental formation energies, we transferred from JARVIS3,45 and MP perovskite formation energies46.
While STL unsurprisingly performed the worst of all five approaches, we found that MoE performed as well or better than transferring from the best individual pre-trained model (see Table 2). Specifically, MoE-(3) consistently outperformed Best TL-(3), and MoE-(18) performed as well or better than Best TL-(18). Of note is that MoE-(3) and MoE-(18) did not overfit on the data-scarce tasks despite utilizing three and eighteen times more fine-tunable parameters than TL.
Benchmarking MoE
To evaluate our MoE framework’s ability to handle data scarcity, we assessed the framework on nineteen materials property regression tasks from Matminer38 with dataset sizes ranging from 120 to 8043 examples. The task datasets span thermodynamic, electronic, mechanical, and dielectric properties; intrinsic and extrinsic properties at fixed temperature and doping concentration; as well as data generated with various DFT exchange-correlation functionals and physical experiments. We set the sparsity hyperparameter k in Eq. (3) to 18, allowing the model to utilize all source tasks. Parameter vector θ (Eq. (2)) was initialized to a vector of ones, corresponding to a uniform distribution of probability scores assigned to each pre-trained model. Hyperparameters were held fixed across all tasks.
Source tasks used to pre-train the MoE extractors are described in Supplementary Table 2. The tasks consisted of eighteen scalar property regression tasks with dataset sizes ranging from 10,855 to 132,752 examples and included properties like formation energies, bandgaps, average conduction band effective masses, dielectric constants, and bulk moduli from the Materials Project and JARVIS.
Mean absolute errors for MoE, single-task learning without pre-training (STL), and TL are reported in Table 3. To avoid expensive, brute-force trial and error of every source task for every downstream task during TL, we used the common heuristic of transferring from the largest available source task, Materials Project (MP) formation energies1. This TL strategy has been employed by several works suggesting TL performance for property prediction improves when the model is pre-trained with more data7,11,15.
Across the 19 downstream tasks, MoE achieved the best performance in 14 of 19 target properties and comparable results on four of the remaining five properties. Notably, TL performed worse than single-task training from random initialization (STL) on 6 of the 19 tasks. These include predicting piezoelectric moduli, Poisson ratios, and other dielectric properties. A possible explanation is that representations trained on formation energies of 3D bulk crystals do not transfer well to dissimilar properties. In contrast, MoE outperformed STL on all 19 tasks, highlighting MoE’s ability to avoid negative transfer without any task-specific hyperparameter tuning.
For one task, predicting phonon mode peak positions (PhonDOS peak), MoE strongly outperformed STL but performed worse than TL from MP formation energies. A plausible explanation is that the MP formation energy dataset is significantly more useful for predicting phonon mode peak positions than any other source task used by the MoE model. Indeed, we found that the MoE model assigned an extremely large probability score of 0.808 to the extractor pre-trained on MP formation energies (recall scores for all eighteen extractors are non-negative and sum to 1). Thus, possible avenues for future improvement include task-specific hyperparameter tuning (e.g., decreasing k or increasing λ to encourage focusing on the most useful task), the inclusion of more generalizable source tasks, and/or development of ML methods which transfer information from more relevant datasets (e.g., other vibrational properties).
Figure 3 depicts a scatter plot of TL and MoE improvement of MAEs over STL as a function of downstream dataset size. Unlike TL, scatter points for MoE lie entirely above zero improvement, highlighting MoE’s ability to yield positive transfer over the entire range of downstream dataset sizes. Interestingly, Fig. 3 also shows no correlation between improvement over STL and downstream dataset size. This result is likely because improvement over STL depends not only on downstream dataset size, but also on the similarity between the source and downstream tasks. In addition, the dependence on downstream dataset size is not necessarily monotonic. Small downstream dataset sizes can lead to overfitting, while large downstream dataset sizes may have less to gain from information transfer. We discuss these factors affecting transferability next.

Percent improvement on MAE across 19 materials property regression tasks of transfer learning from Materials Project formation energies (TL) and our mixture of experts approach (MoE) over single-task learning with random initialization (STL). For clarity, the y axis above and below the break have different scales. Examples of positive and negative transfer are indicated by points above and below the gray dotted line, respectively.
Understanding transferability
Negative transfer is a pervasive phenomenon in ML wherein transferring information from a source task(s) to a downstream task exhibits worse performance than training on the downstream task from random initialization. Wang et al.21 and Gong et al.47 discussed the key factors from which negative transfer arises: divergence between the source and downstream tasks’ joint distributions over the domain and label spaces as well as the size of the labeled downstream task data. Formally, we denote PS(X, Y) and PT(X, Y) as the joint distribution of the source and downstream tasks, respectively. Random variable X corresponds to the input (e.g., materials), and Y is the label (e.g., corresponding values for a specific property). Negative transfer can result from the divergence d(PS(X, Y), PT(X, Y)), where d( ⋅ , ⋅ ) is a divergence metric over distributions. Wang et al. also argued that the downstream dataset size has a mixed effect on negative transfer; if the downstream task is too small, then it becomes difficult for the learning algorithm to properly learn the similarity between the source and target tasks. Yet, if the downstream task is too large, then transferring from a source task with even a slightly different joint distribution could harm generalization and perform worse than STL.
To understand performance variations of MoE across different downstream tasks, we examined the divergence in feature and label space between the source and downstream tasks. In general, atomic structures X and their associated materials properties Y are not independent; the connection between structure and properties is central to materials science. A data-driven comparison of two materials property prediction tasks S and T should thus compare their full joint distributions, PS(X, Y) and PT(X, Y). Unfortunately, computing PS(X, Y), \(\left.{P}_{T}(X,Y)\right)\), and subsequently \(d\left.({P}_{S}(X,Y),{P}_{T}(X,Y))\right)\) is difficult in practice. Instead, we decoupled the feature and label spaces, separately measuring empirical domain and label shifts between source and downstream tasks, d(PS(X), PT(X)), and d(PS(Y), PT(Y)). To compute these shifts, we used central moment discrepancy (CMD)48, a distance metric for probability distributions on compact space. Intuitively, CMD compares distribution means and arbitrarily high central moments to capture differences in distribution positions and shapes. Up to 50th-order central moments were included in our experiments. To measure domain shift, CMD was computed in the learned feature space of the last frozen convolutional layer of each extractor. We found that CMD computed in this feature space showed a strong positive trend with CMD computed in the space of features procedurally generated from local atomic structure order parameters49,50 (see Supplementary Fig. 1). To measure label shift, each task’s label distribution was first normalized by subtracting the mean and dividing by the standard deviation. Since CMD requires each distribution to be on compact space, a sigmoid function was applied on each dimension of the feature and normalized label spaces to maintain compact support between 0 and 1.
For each downstream task, we plotted the average CMD in label and feature space to the top-n source tasks (i.e., the “closest” n source tasks) for n = 4 (Fig. 4). Other values of n were explored without yielding ostensibly significant differences. The size of each plotted data point indicates the size of the downstream task data, and the color indicates MoE’s improvement over STL on that task.

Scatter plot of the average central moment discrepancy (CMD) in label and feature space for each downstream task across the top-n (i.e., “closest” n) extractors for n = 4. Other values of n were also explored without ostensibly different results. Marker sizes are scaled with the downstream dataset size and colored by MoE’s improvement on MAE over STL.
Figure 4 reveals no discernable trends, highlighting the difficulty of predicting transferability with simple proxies. Instead, machine-learned models, like MoE, are needed to determine which source tasks are most useful for a downstream task. Plausible explanations for the lack of emergent patterns are (1) uncertainty in the improvement over STL obfuscates any trends, (2) 19 downstream tasks is too small of a sample size, (3) CMD is not the optimal distance metric for capturing divergences in task distributions, and (4) decoupling the domain and feature spaces X and Y catastrophically ignores the connection between structure and properties.
Model interpretability
A natural consequence of our MoE gating function (Eq. (2)) is that for each downstream task, the model automatically learns to associate a probability score to each pre-trained extractor. By analyzing these scores, we can readily interpret which pre-trained extractors and source tasks were most relevant to learning each downstream task. In Fig. 5, we visualize a heatmap of these learned scores for all downstream tasks.

Source tasks are ordered left to right from smallest to largest dataset size. Contrary to the heuristic of transferring from the largest source task, our MoE framework did not usually assign the largest score to the largest source task. Instead, the model often assigned scores which were physically intuitive.
Despite initializing the model with uniform probability scores assigned to each extractor, we make two notable observations. (1) For a given downstream task, learned scores are relatively robust across different random seeds. (2) The model often learns scores which are physically intuitive rather than simply assigning large scores to extractors pre-trained with more data. For example, MP bandgaps and dielectric constants computed with the OptB88vDW DFT functional were assigned the highest scores for predicting the electronic component of MP dielectric tensors (possibly because larger bandgaps result in fewer free electrons and lower electronic polarizability). When predicting experimental formation energies, the model heavily concentrated probability mass onto the JARVIS and MP formation energy extractors. MP bandgaps were also assigned the highest score when predicting 2D materials’ bandgaps and experimental bandgaps. Such correspondences suggest our MoE framework’s strong performance results in part from learning physically meaningful relationships between source and downstream tasks.
However, there were some instances of counterintuitive task relationships being emitted by the MoE model. For example, while the JARVIS and MP shear moduli extractors were assigned large scores for predicting Poisson ratios as expected, the JARVIS and MP bulk modulus extractors were not. Similarly, when predicting piezoelectric modulus, the MoE model automatically assigned the highest scores to electronic properties like n- and p-type electronic conductivities, electronic thermal conductivities, and Seebeck coefficients, as well as MP bandgaps (perhaps because a nonzero band gap is required to maintain electronic polarizations). However, the model did not assign large scores to any mechanical properties. These unexpected results are perhaps better explained by divergences in the datasets’ joint distributions in domain and label space rather than by domain knowledge.
Scaling to an arbitrary number of extractors
We anticipate that the number of large materials task datasets, and hence the number of potential source tasks, will increase as growing compute resources, new DFT functionals, high-throughput experimental methods, and novel pre-training tasks emerge from the community. To handle many source tasks, we experimented with sparse gating by allowing the sparsity hyperparameter, k, from Eq. (3) to be less than the number of extractors. During inference, only k extractors would be activated, and thus gradients would only be computed for k extractors in each training iteration. Utilizing sparsity consequently decouples the speed per training iteration from the number of extractors, enabling the MoE framework to scale to an arbitrary number of extractors without concomitant increases in computing cost. In anticipation of the community leveraging performance boosts from model scale51, we note that extractors can be distributed across multiple GPUs.
We compared k values of 2, 4, 6, and 10 for three downstream tasks: predicting piezoelectric modulus, 2D exfoliation energies, and experimental formation energies. Surprisingly, we observed no significant detriment to performance compared to k = 18, even when setting k as small as 2 (Table 4). This result suggests practitioners can supply our MoE framework with as many pre-trained extractors as desired without fear of increasing compute cost or harming predictive performance.
In conclusion, we presented a mixture of experts framework combining complementary materials datasets and ML models to achieve consistent state-of-the-art performance on a suite of data-scarce property prediction tasks. We demonstrated the interpretability of our framework, which readily emits automatically learned relationships between a downstream task and all source tasks in a single training run. We often found these relationships to be physically intuitive. By introducing a sparsity hyperparameter, we also showed that MoE is scalable to an arbitrary number of source tasks and extractors without performance detriment. The MoE framework is general, allowing any model architecture or hand-crafted featurizer to act as extractors and any dataset to act as a source task. We invite the community to engineer new source tasks to train generalizable extractors; explore mixtures of different extractor model classes such as hand-crafted descriptors or equivariant neural networks which predict non-scalar properties; and share materials datasets spanning diverse properties, dataset sizes, and fidelities.
Methods
Crystal graph convolutional neural networks
For a full treatment of CGCNNs, see ref. 8. Briefly, CGCNNs operate on graph representations of crystals. A crystal structure with N atoms is represented as a graph \(G=({\{{{{{\bf{v}}}}}_{i}^{0}\}}_{i = 1}^{N},{\{{{{{\bf{u}}}}}_{(i,j)}\}}_{i,j = 1}^{N})\) with initial node features \({{{{\bf{v}}}}}_{i}^{0}\) representing atom i and edge features u(i, j) representing bond(s) features between atoms i and j. In the original implementation, \({{{{\bf{v}}}}}_{i}^{0}\) is a trainable linear transformation of vectorized elemental features like group number and electronegativity.
Node/atom features are sequentially updated with graph convolutional layers, passing information from node features and shared edges of locally neighboring atoms. CGCNN implements their graph convolutional layer as
where \({{{{\bf{v}}}}}_{i}^{(t)}\) is the node feature of atom i after t graph convolutions, σ(⋅) is a sigmoid function, g(⋅) is a softplus, k represents the kth bond between atoms i and j, ⊙ is elementwise multiplication, ⊕ is concatenation, and \({{{{\bf{W}}}}}_{{{{\rm{f}}}}}^{(t)}\), \({{{{\bf{W}}}}}_{{{{\rm{s}}}}}^{(t)}\), \({{{{\bf{b}}}}}_{{{{\rm{f}}}}}^{(t)}\), and \({{{{\bf{b}}}}}_{{{{\rm{s}}}}}^{(t)}\) are trainable parameters for the tth graph convolutional layer. After T convolutional layers, all node features are averaged to produce a feature vector vc representing the entire crystal. Finally, vc is passed as input to a multilayer perceptron to yield a prediction.
Model training
Datasets were split into 70% training, 15% validation, and 15% testing data for five random seeds. For each dataset, the same five random splits were re-used across STL, TL, and MoE experiments for consistency.
All random splits were sampled uniformly except for the experimental formation energy dataset. The MP fits to experimental formation energies to obtain energy corrections for certain anions (O, S, F, Cl, Br, I, N, H, Se, Si, Sb, and Te) and +U corrections for GGA+U calculations on oxides and fluorides with certain transition metals (V, Cr, Mn, Fe, Co, Ni, W, and Mo)52,53. Thus, to avoid information leakage from MP formation energy pre-training data to the experimental formation energy test data, we excluded compounds with O, S, F, Cl, Br, I, N, H, Se, Si, Sb, Te, V, Cr, Mn, Fe, Co, Ni, W, and Mo from all experimental formation energy test splits.
Models were trained for 1000 epochs unless validation error did not improve for 500 epochs, in which case early stopping was applied. Models were optimized with Adam, mean-squared error loss, a Cosine Annealing scheduler, and a batch size of 250 (or the entire training split - whichever was smaller) on NVIDIA Tesla V100 and A100 GPUs. Each dataset’s regression labels were normalized by subtracting the mean and dividing by the standard deviation of labels in the training and validation sets.
During STL and extractor training, all layers were updated with an initial learning rate of 1e-2. During TL and MoE training, all extractor layers were frozen except for the last convolutional layer, which was updated with an initial learning rate of 5e-3. Head layers were updated with an initial learning rate of 1e-2.
Batches were always sampled with uniform random sampling, except when pre-training extractors on the n- and p-type electronic thermal and electronic conductivity source tasks, which had heavily skewed label distributions. For those tasks, batches were sampled with weighted sampling. Specifically, label distributions were split into 30 bins, and the sampling weight for bin i was computed as
where Ib represents the set of bin indices with at least one example. Bins with no examples were reassigned a sampling weight of 0.
While some hyperparameter tuning was conducted for pairwise TL (see Supplementary Figs. 2 and 3), we did not do any hyperparameter tuning for MoE, instead drawing the same hyperparameters from TL or from literature. Thus the strong performance of MoE is likely robust and can possibly be further improved with hyperparameter tuning.
Data availability
All data are available from Matminer (see https://hackingmaterials.lbl.gov/matminer/)38. Downstream task datasets, training, validation, and test splits are available at https://github.com/rees-c/MoE.
Code availability
All code is openly accessible at https://github.com/rees-c/MoE.
References
Jain, A. et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater 1, 11002 (2013).
Curtarolo, S. et al. AFLOWLIB.ORG: a distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235 (2012).
Choudhary, K. et al. The joint automated repository for various integrated simulations (JARVIS) for data-driven materials design. npj Comput. Mater. 6, 1–13 (2020).
Saal, J. E., Oliynyk, A. O. & Meredig, B. Machine learning in materials discovery: confirmed predictions and their underlying approaches keywords. Annu. Rev. Mater. Res. 11, 45 (2020).
Sendek, A. D. et al. Holistic computational structure screening of more than 12,000 candidates for solid lithium-ion conductor materials. Energy Environ. Sci 10, 306 (2017).
Dunn, A., Wang, Q., Ganose, A., Dopp, D. & Jain, A. Benchmarking materials property prediction methods: the matbench test set and automatminer reference algorithm. npj Comput. Mater. 6, 138 (2020).
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mater 31, 3564–3572 (2019).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Bartel, C. J. et al. A critical examination of compound stability predictions from machine-learned formation energies. npj Comput. Mater. 6, 97 (2020).
Das, K. et al. CrysXPP: an explainable property predictor for crystalline materials. npj Comput. Mater. 8, 1–11 (2022).
Chen, C. & Ong, S. P. AtomSets as a hierarchical transfer learning framework for small and large materials datasets. npj Comput. Mater. 7, 1–9 (2021).
Wang, Z. et al. Deep learning for ultra-fast and high precision screening of energy materials. Energy Storage Mater. 39, 45–53 (2021).
Chen, C., Zuo, Y., Ye, W., Li, X. & Ong, S. P. Learning properties of ordered and disordered materials from multi-fidelity data. Nat. Comput. Sci. 1, 46–53 (2021).
Jha, D. et al. Enhancing materials property prediction by leveraging computational and experimental data using deep transfer learning. Nat. Commun. 10, 1–12 (2019).
Lee, J. & Asahi, R. Transfer learning for materials informatics using crystal graph convolutional neural network. Comput. Mater. Sci 190, 110314 (2021).
Hutchinson, M. L. et al. Overcoming data scarcity with transfer learning. Preprint at https://arxiv.org/abs/1711.05099 (2017).
Chen, X., Wang, S., Fu, B., Long, M. & Wang, J. Catastrophic forgetting meets negative transfer: batch spectral shrinkage for safe transfer learning. in Adv. Neural Inf. Process. Syst. (eds. Wallach, H., Larochelle, H., Beygelzimer, A., Alché-Buch, F., Fox, E., & Garnett, R.) 32 (Curran Associates, Inc., 2019). https://proceedings.neurips.cc/paper/2019/hash/c6bff625bdb0393992c9d4db0c6bbe45-Abstract.html.
Kirkpatrick, J. et al. Overcoming catastrophic forgetting in neural networks. Proc. Natl. Acad. Sci. USA 114, 3521–3526 (2017).
Zamir, A. R. et al. Taskonomy: disentangling task transfer learning. in IEEE/CVF Conference on Computer Vision and Pattern Recognition, 3712–3722 (IEEE, 2018).
Vu, T. et al. Exploring and predicting transferability across NLP tasks. in Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing, 7882–7926 (Association for Computational Linguisitcs, 2020).
Wang, Z., Dai, Z., Póczos, B. & Carbonell, J. Characterizing and avoiding negative transfer. in Proc. IEEE/CVF International Conference on Computer Vision and Pattern Recognition, 11285–11294 (IEEE, 2019).
Yamada, H. et al. Predicting materials properties with little data using shotgun transfer learning. ACS Cent. Sci. 5, 1717–1730 (2019).
Magar, R., Wang, Y. & Farimani, A. Crystal twins: self-supervised learning for crystalline material property prediction. npj Comput. Mater. 8, 231 (2022).
Sanyal, S. et al. MT-CGCNN: integrating crystal graph convolutional neural network with multitask learning for material property prediction. Preprint at https://arxiv.org/abs/1811.05660 (2018).
Zamir, A. R. et al. Robust learning through cross-task consistency. in IEEE/CVF International Conference on Computer Vision and Pattern Recognition, 11194–11203 (IEEE, 2020).
Standley, T. et al. Which tasks should be learned together in multi-task learning? in Proc. 37th International Conference on Machine Learning, Vol. 119, 9120–9132 (PMLR, 2020).
Fifty, C. et al. Efficiently identifying task groupings for multi-task learning. in Adv. Neural Inf. Process. Syst. (eds. Ranzato, M., Beygelzimer, A., Dapuhin, Y., Liang, P.S., & Vaughan, J.W.) 34, 27503–27516 (Curran Associates, Inc., 2021).
Chen, Z. et al. Just pick a sign: optimizing deep multitask models with gradient sign dropout. in Adv. Neural Inf. Process. Syst. (eds. Larochelle, H., Ranzato, M., Hadsell, R., Balcan, M.F., & Lin, H.) 33, 2039–2050 (Curran Associates, Inc., 2020).
Chen, Z., Badrinarayanan, V., Lee, C.-Y. & Rabinovich, A. Gradnorm: gradient normalization for adaptive loss balancing in deep multitask networks. in Proc. 35th International Conference on Machine Learning, Vol. 80, 794–803 (PMLR, 2018).
Javaloy, A. & Valera, I. RotoGrad: gradient homogenization in multitask learning. in Proc. International Conference on Learning Representations (ICLR, 2022). https://openreview.net/forum?id=T8wHz4rnuGL.
Xie, T. et al. Atomistic graph networks for experimental materials property prediction. Preprint at https://arxiv.org/abs/2103.13795 (2021).
Jacobs, R. A., Jordan, M. I., Nowlan, S. J. & Hinton, G. E. Adaptive mixtures of local experts. Neural Comput. 3, 79–87 (1991).
Shazeer, N. et al. Outrageously large neural networks: the sparsely-gated mixture-of-experts layer. in Proc. International Conference on Learning Representations (ICLR, 2017).
De Jong, M., Chen, W., Geerlings, H., Asta, M. & Persson, K. A. A database to enable discovery and design of piezoelectric materials. Sci. Data 2, 1–13 (2015).
Choudhary, K., Kalish, I., Beams, R. & Tavazza, F. High-throughput identification and characterization of two-dimensional materials using density functional theory. Sci. Rep. 7, 1–16 (2017).
Wang, A. et al. A framework for quantifying uncertainty in DFT energy corrections. Sci. Rep. 11, 15496 (2021).
Kim, G., Meschel, S. V., Nash, P. & Chen, W. Experimental formation enthalpies for intermetallic phases and other inorganic compounds. Sci. Data 4, 1–11 (2017).
Ward, L. et al. Matminer: an open source toolkit for materials data mining. Comput. Mater. Sci. 152, 60–69 (2018).
Lin, Z. et al. A Structured self-attentive sentence embedding. in Proc. International Conference on Learning Representations (ICLR, 2017). https://openreview.net/forum?id=BJC_jUqxe.
Chen, Z. et al. Direct prediction of phonon density of states with Euclidean neural network. Adv. Sci. 8, 2004214 (2020).
Tshitoyan, V. et al. Unsupervised word embeddings capture latent knowledge from materials science literature. Nature 571, 95–98 (2019).
Xie, T., Fu, X., Ganea, O.-E., Barzilay, R. & Jaakkola, T. Crystal diffusion variational autoencoder for periodic material generation. in Proc. International Conference on Learning Representations (ICLR, 2022). https://openreview.net/forum?id=03RLpj-tc_.
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B. & Wolverton, C. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM 65, 1501–1509 (2013).
De Jong, M. et al. Charting the complete elastic properties of inorganic crystalline compounds. Sci. Data 2, 1–13 (2015).
Choudhary, K., Cheon, G., Reed, E. & Tavazza, F. Elastic properties of bulk and low-dimensional materials using van der Waals density functional. Phys. Rev. B 98, 14107 (2018).
Castelli, I. E. et al. New cubic perovskites for one- and two-photon water splitting using the computational materials repository. Energy Environ. Sci. 5, 9034–9043 (2012).
Gong, M. et al. Domain adaptation with conditional transferable components. in Proc. 33rd International Conference on Machine Learning, Vol. 48, 2839–2848 (PMLR, 2016).
Zellinger, W., Lughofer, E., Saminger-Platz, S., Grubinger, T. & Natschläger, T. Central moment discrepancy (CMD) for domain-invariant representation learning. in Proc. International Conference on Learning Representations (ICLR, 2017). https://openreview.net/forum?id=SkB-_mcel.
Pan, H. et al. Benchmarking coordination number prediction algorithms on inorganic crystal structures. Inorg. Chem 60, 1590–1603 (2021).
Zimmermann, N. E. & Jain, A. Local structure order parameters and site fingerprints for quantification of coordination environment and crystal structure similarity. RSC Adv. 10, 6063–6081 (2020).
Frey, N. C. et al. Neural scaling of deep chemical models. Preprint at https://doi.org/10.26434/chemrxiv-2022-3s512 (2022).
Jain, A. et al. Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 84, 045115 (2011).
Jain, A. et al. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 50, 2295–2310 (2011).
Acknowledgements
We acknowledge Dr. Alex Ganose for helpful discussion on the Materials Project’s formation energy calculation methodology. This material is based upon work supported by the National Science Foundation under Grant Nos. 1922758, 2118201, and 2106825. It utilizes computational resources supported by the National Science Foundation’s Major Research Instrumentation program (Grant No. 1725729) and the Delta research computing project (Grant No. 2005572).
Author information
Authors and Affiliations
Contributions
R.C., Y.-X.W., and E.E. conceived the idea. R.C. wrote the code, carried out the experiments, and performed the analysis. Y.-X.W. and E.E. supervised and guided the project. The manuscript was prepared by R.C. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, R., Wang, YX. & Ertekin, E. Towards overcoming data scarcity in materials science: unifying models and datasets with a mixture of experts framework. npj Comput Mater 8, 242 (2022). https://doi.org/10.1038/s41524-022-00929-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-022-00929-x
This article is cited by
-
Leveraging language representation for materials exploration and discovery
npj Computational Materials (2024)
-
Advances of machine learning in materials science: Ideas and techniques
Frontiers of Physics (2024)
