
Abstract
Thermoelectric materials have received much attention as energy harvesting devices and power generators. However, discovering novel high-performance thermoelectric materials is challenging due to the structural diversity and complexity of the thermoelectric materials containing alloys and dopants. For the efficient data-driven discovery of novel thermoelectric materials, we constructed a public dataset that contains experimentally synthesized thermoelectric materials and their experimental thermoelectric properties. For the collected dataset, we were able to construct prediction models that achieved R2-scores greater than 0.9 in the regression problems to predict the experimentally measured thermoelectric properties from the chemical compositions of the materials. Furthermore, we devised a material descriptor for the chemical compositions of the materials to improve the extrapolation capabilities of machine learning methods. Based on transfer learning with the proposed material descriptor, we significantly improved the R2-score from 0.13 to 0.71 in predicting experimental ZTs of the materials from completely unexplored material groups.
Similar content being viewed by others

Introduction
Thermoelectric material is a class of the materials that convert heat energy to electrical energy based on the Seebeck and Peltier effects1. These thermoelectric materials have been widely applied to scientific applications, such as energy harvesting2, thermoelectric cooling3, and thermopower generators4. Recently, thermoelectric materials have also received much attention as the materials for renewable energy5. To discover high-performance thermoelectric materials, various materials with the promising thermoelectric properties have been studied, such as such as tine selenide6, silicon-germanium7, and lead telluride8. In particular, various alloy and doped materials have been extensively studied around the promising host materials to improve the thermoelectric performances6,9,10.
In physical science, density functional theory (DFT)11 have been widely applied to estimate and interpret the relationships between the electronic structures and their physical properties of the materials, such as solar cell materials12, 2d materials13, and electrocatalysis14. However, although DFT achieved numerous successes as a generally applicable method to analyze the crystalline systems, the applicability of DFT is still limited to the materials of small unit cells due to exponential computational costs in huge unit cells15,16. For this reason, calculating the thermoelectric properties of the doped materials remains a challenging problem in physical science.
In a different direction from the conventional approach, machine learning has been studied to efficiently approximate the relationships between the materials and their physical properties17,18. Several machine learning methods outperformed the conventional calculation- and simulation-based methods in predicting the physical properties of the materials19,20. In particular, graph neural networks (GNNs)21 have shown remarkable prediction capabilities in various regression tasks on crystal structures19,22. The great successes of GNNs in materials science came from using a crystal graph representation that preserves structural information of the materials as well as their atomic attributes. However, the general applicability of GNNs in real-world applications is significantly limited because the crystal structures of the doped materials are not available in most cases.
In materials science, several machine learning methods that predict the physical properties of the materials from their chemical compositions have been proposed to extend the general applicability of machine learning to the applications where the crystal structures are not available. Representation learning from stoichiometry (Roost) tried to learn latent embeddings from the chemical compositions of the materials by representing the chemical compositions as an elemental graph23. In the experimental evaluations, Roost achieved state-of-the-art accuracies in predicting band gaps of inorganic crystals from their chemical compositions. However, its applicability is still limited to the pristine materials because the elemental graph is defined only for the pristine materials. DopNet was proposed to predict the physical properties of the alloy and doped materials from their chemical compositions based on a material space embedding approach18. By separately representing the host materials and the dopants, DopNet was able to learn more informative and latent features of the doped materials and consequently achieved state-of-the-art accuracies in predicting thermoelectric properties of the doped materials. However, the thermoelectric materials have different and complex thermodynamics for each material group24, i.e., a simple splitting of the host materials and the dopants cannot fully handle the complexities of the regression problems on the thermoelectric materials. In addition to DopNet, various machine learning approaches have been studied for thermoelectric materials. Neural networks were applied to determine the thermophysical properties of amino acid based ionic liquids25. Moreover, several ensemble methods were used to predict thermoelectric properties of the materials26. However, to the best of our knowledge, there is no public database and material descriptor for data-driven extrapolation to discover novel thermoelectric materials.
To accelerate the data-driven discovery of the thermoelectric materials, we constructed a public materials dataset containing 5205 experimental observations containing the chemical compositions of the experimentally synthesized thermoelectric materials (ESTM) and their experimental thermoelectric properties. We named the collected dataset as ESTM dataset. The ESTM dataset covers 880 unique thermoelectric materials and provides five experimentally measured thermoelectric properties: Seebeck coefficient, electrical conductivity, thermal conductivity, power factor, and figure of merit (ZT). In addition, we predicted the thermoelectric properties of the collected materials from their chemical compositions to validate the usefulness of the ESTM dataset in machine learning. In machine learning on the ESTM dataset, we achieved R2-scores27 greater than 0.9 in predicting five thermoelectric properties of the materials, and our prediction model showed a mean absolute error (MAE) less than 0.06 in predicting ZTs of the materials.
In addition to the public dataset, we devised a representation method for the alloy and doped materials, called system-identified material descriptor (SIMD), to accurately predict the target physical properties of the thermoelectric materials from their chemical compositions. SIMD makes a material cluster by collecting similar materials based on the chemical and physical attributes of the materials. Then, SIMD characterizes the relationships between the clustered materials and their target properties for each material cluster based on the least-square method28 to solve the system of equations. By SIMD, each material cluster is summarized as a vector that can be used to the input data of the machine learning algorithms.
Based on transfer learning with SIMD, we were able to improve R2-score from 0.13 to 0.71 in an extrapolation problem to predict ZTs of the materials from unexplored material groups, which is a key problem for the data-driven discovery of high-performance thermoelectric materials. We conducted a data-driven search in the materials space to evaluate the usefulness of the ESTM dataset and SIMD in real-world applications for material discovery. In the experiments of the materials discovery, a machine learning model based on SIMD showed a screening accuracy of 0.61 measured by F1-score29 as shown in Fig. 4 of the “Results and discussion” section, even though the high-throughput screening was conducted on the thermoelectric materials from completely unexplored material systems. Furthermore, we conducted a data-driven search based on SIMD to discover high-ZT materials under the temperature constraints, and the machine learning model with SIMD reduced the false positive by 50% for all search tasks, as shown in Fig. 5. The ESTM dataset and all resources of SIMD with the search results are publicly available at https://github.com/KRICT-DATA/SIMD. The contents of this paper can be summarized as:
-
We constructed a public dataset containing experimentally validated thermoelectric materials and their experimental thermoelectric properties.
-
For machine learning extrapolation, we devised a material descriptor that can incorporate information about similar materials beyond a single material.
-
The proposed descriptor significantly reduced the number of false positives in the high-throughput screening to discover novel thermoelectric materials.
Results and discussion
A public dataset of experimentally synthesized thermoelectric materials
For data-driven discovery of the thermoelectric materials, we performed the literature search to collected the chemical compositions and the experimentally measured thermoelectric properties of the materials. We collected 5205 experimental observations that are uniquely defined by a pair of the chemical composition and the measuring temperature. Each observation contains five target thermoelectric properties: Seebeck coefficient, electrical conductivity, thermal conductivity, power factor, and ZT. Table 1 describes the data row of the ESTM dataset. The first column of chemical composition presents the chemical composition of the thermolectric materials, and the second column is the measuring temperature of the thermoelectric properties. The first column presents the chemical composition of the collected thermoelectric materials. For machine learning, the chemical composition should be converted to the numerical data. The second column is the measuring temperature of the thermoelectric properties. The last column is DOI of the source literature of the collected experimental observation. The remaining columns are the thermoelectric properties that were experimentally collected or theoretically calculated from the experimental observations.
The ESTM covers 880 unique thermoelectric materials containing 65 elements from Li to Bi. The elemental distribution of the ESTM dataset is visualized in Supplementary Information (SI). The most common elements in the ESTM dataset were Se, Sb, and Te, which have been widely studied for high-performance thermoelectric materials30,31. In addition, the ESTM dataset contains popular and promising thermoelectric materials and their variants, such as lead tellurides (PbTe), bismuth tellurides (Bi2Te3), and tin selenide (SnSe). The maximum ZTs of the collected thermoelectric materials at high temperature (≥700 K) and near room temperature (≈300 K) were 2.16 and 1.17, respectively.
Machine learning interpolation for predicting thermoelectric properties
For the collected ETML dataset, we trained machine learning models to predict the thermoelectric properties from the chemical compositions of the materials. We predicted four experimentally measured thermoelectric properties: Seebeck coefficient, electrical conductivity, thermal conductivity, and ZT. In the experiment, we evaluated six different machine learning methods as follows:
-
Ridge regression (RidgeReg)32: it is a baseline linear regression model with weight regularization by the L2-norm.
-
K-nearest neighbor regression (KNNR)33: KNNR predicts the target value of the input data by interpolating the target values associated with K nearest neighbor data in the training dataset.
-
Support vector regression (SVR)34: SVR is a variant of support vector machine for the regression problems. It employs kernel methods to capture the nonlinear relationships between the input and the target data.
-
Gaussian process regression (GPR)35: GPR is a regression model of a Gaussian process. GPR assumes that the input variables are multivariate Gaussian random variables, and they are drawn from a multivariate normal distribution.
-
Fully-connected neural network (FCNN)36: FCNN is a feedforward neural network to approximate the relationships between the vector-shaped inputs and the scalar targets. We stacked two hidden layers between the input and the output layers of FCNN to extract latent and nonlinear information from the input data.
-
XGB37: XGB is an ensemble method that integrates multiple week prediction models to improve the prediction and generalization capabilities. XGB employs a gradient boosting method based on decision trees. XGB has shown state-of-the-art prediction accuracies in many scientific fields, such as organic chemistry and material science.
In the experiments, we used k-fold cross-validation method to train and evaluate the machine learning prediction models. We divided the entire ESTM dataset into three non-duplicated subsets (3-folds). In the training process, two folds of the dataset containing 3435 observations were used for training of the prediction models, and the remaining subset containing 1770 observations was used for evaluating the generalization capabilities of the trained prediction models. For each machine learning method, we repeated the training and evaluation processes until were subsets are used for the evaluation. We used the sparse encoding to convert the chemical compositions into machine-readable numerical vectors. The encoding process is described in Section 3 of Supplementary Information. For electrical and thermal conductivity, we applied the logarithm to adjust their high variances.
Table 2 presents the means and the standard deviations of the measured MAEs for the six machine learning methods in the interpolation problems that predict the thermoelectric properties on the ESTM dataset. For the comparison in a normalized metric, the mean and the standard deviations of the measured R2-scores are reported together in the table. In the evaluation, RidgeReg, SVR, and GPR failed to predict the thermoelectric properties for the input chemical compositions and measuring temperatures, and their R2-scores were less than 0.5 for all prediction tasks. Although KNNR and FCNN showed relatively high R2-scores over RidgeReg, SVR, and GPR, their prediction capabilities were still limited. By contrast, XGB achieved R2-scores greater than 0.9 for all prediction tasks. In addition, MAEs of XGB in predicting Seebeck coefficient, electrical conductivity, thermal conductivity, and ZT were 21.10 ± 0.48, 0.28 ± 0.02, 0.09 ± 0.01, and 0.06 ± 0.01, respectively. Figure 1 shows the prediction results of XGB, and the thermoelectric properties of the materials in the ESTM dataset were accurately predicted from the chemical compositions and the measuring temperature. The prediction results of XGB in predicting ZT show the availability of our ESTM dataset for rapid estimation of the experimentally measured thermoelectric performance of the materials in real-world applications.

Each abbreviation in the axis label means: S.C. Seebeck coefficient, E.C. electrical conductivity, and T.C. thermal conductivity.
System-identified material descriptor
Machine learning methods showed remarkable prediction capabilities in the interpolation problems17,19,22, and we were also able to observe the outstanding interpolation capabilities of machine learning methods on the ESTM dataset as shown in Fig. 1. However, it is not sufficient for the data-driven discovery of novel thermoelectric materials because our interest is in the extrapolation that predicts the thermoelectric properties of unexplored material systems. In other words, we should evaluate the prediction capabilities of machine learning methods before using them for the data-driven material discovery. For this reason, we evaluated the prediction capabilities of FCNN and XGB in the extrapolation problems on the ESTM dataset by randomly splitting the dataset based on the material groups rather than each material, i.e., the materials in the same material group were entirely removed in the training dataset. In the evaluation of the extrapolation, FCNN and XGB failed to predict ZTs of the materials from unknown material groups, and their R2-scores were just −0.15 and 0.13, respectively. This failure is natural because conventional machine learning methods are not effective in the extrapolation problems38,39.
To improve the extrapolation capabilities of the machine learning methods, we devised a material representation called SIMD that extracts system-level input features for each material group. The overall process of SIMD consists of three steps: (1) material cluster generation, (2) material cluster characterization, and (3) system-identified feature generation. Figure 2 illustrates the overall process of SIMD to generate the system-conditioned material representations for input tabular data of the materials. We will formally describe each step of SIMD to generate the system-conditioned material representations in the following subsections.

SIMD calculates the materials representations through the three steps. Step 1: the input materials are clustered based on their compositions. Step 2: the system vectors and target statistics are calculated for each material cluster. Step 3: the KNN method allocates the material cluster for each input material, and the representation generator concatenates the system vectors and the target statistics to the input features of the input material.
Material cluster generation
The purpose of material cluster generation is to construct the material clusters that cover alloy and doped materials derived from the same pristine materials. To this end, we define a cluster identifier that uniquely represent the material cluster. Formally, the cluster identifier for an input chemical composition s is defined as:
where e is the symbol of the element in s, re is the ratio of the element represented by e, and round() is the mathematical rounding operator to convert floating point values to integer values. For example, the input compositions of SnS1−xSnx (x = 0.09, 0.06, 0.12) and Ta1−xTixFeSb (x = 0.08, 0.12) generate the material clusters identified by SnS and TaFeSb, respectively. Note that the input engineering and measuring conditions of the material data are ignored in the process of the material clusters construction in order to cluster the input materials based on the chemical and physical attributes of the materials. After generating the material clusters, the input materials are clustered into the generated material clusters based on the compositions of the input materials. For example, the input materials of the compositions SnS0.91Se0.09, SnS0.94Se0.06, and SnS0.88Se0.12 are clustered into the material cluster of SnS regardless of their alloy and doping elements, as shown in Fig. 2. In the next step of the material cluster generation, the latent vector for each constructed material cluster is calculated to describe the material cluster as a machine-readable vector-shaped representation.
Material cluster characterization
In this step, we generate a vector representation of the constructed material clusters based on their cluster identifiers. Through this process, we can convert the material clusters described by chemical compositions into the vector-shaped representations that can be used for the input of the machine learning methods. Specifically, we extract two latent information called system vector and target statistics vector to generate the vector-shaped representations of the material clusters. The system vector for a material cluster represents a projection function from the input material space to the target space for the set of the materials in the material cluster, i.e., the system vector describes the relationships between the materials and the target properties in the local material space defined by the material cluster. The target statistics vector is defined by the mean, standard deviation, minimum, and maximum of the target properties of the materials in the material cluster. It briefly presents the distributions of the target properties in the material clusters, as shown in Fig. 2.
To calculate the system vector of the material cluster u, we represent the relationship between the input materials and the target properties in the material cluster as a mathematical system of material-wise equations as:
where xn = [xn1xn2, … , xnM] is a M-dimensional atomic feature vector calculated from the chemical composition of the nth material in the material cluster, cn = [cn1, cn2, … , cnL] is a L-dimensional condition vector of the nth material, yn is the target property of the nth material, and d = M + L is the dimensionality of the system vector w = [w1, w2, … , wd]. Note that ∣u∣ is the size of the material cluster which means the number of the materials in the material cluster u. The condition vector can be defined by the synthesis conditions and measurement factors such as temperature, pressure, and cooling time. In this equation system, the system vector w is the solution of the system, and it can be efficiently calculated by least-square method28 in linear algebra. In the implementation of SIMD, the atomic feature vector xn can be defined in various ways to transform the chemical composition of the material into the feature vector. We used a sparse encoding for the atomic feature vector xn, and the formal description of the atomic feature vector is given in the method section.
The target statistics vector v of the material cluster u is defined as a 4-dimensional vector of the mean, standard deviation, minimum, and maximum of the target properties of the materials in the material cluster. The target statistics vector is formally defined as:
where \(\bar{y}=\frac{1}{| u| }\mathop{\sum }\nolimits_{i = 1}^{| u| }{y}_{i}\) is the mean of target properties, and \({y}_{\sigma }=\sqrt{\mathop{\sum }\nolimits_{i = 1}^{| u| }{({y}_{i}-\bar{y})}^{2}/| u| }\) is the standard deviation of the target properties. Note that ymin and ymax mean the minimum and maximum values among the target values {y1, y2, … , y∣u∣}, respectively. As a result, the material clusters are represented as the (d + 4)-dimensional concatenated vector of the system and target statistics vectors.
System-identified feature generation
The purpose of system-identified feature generation is to convert the chemical compositions of the materials based on their physical attributes and the constructed material clusters. First, we determine the material clusters of the input chemical compositions. To this end, we define an anchor space where the material clusters are defined as anchor vectors corresponding to their cluster identifiers. The anchor vector is defined as an attribute vector based on the chemical attributes of the cluster identifiers and the chemical compositions. The implementation details of the anchor vector are provided in the method section. Then, the KNN method selects K nearest material clusters for the input chemical compositions in the anchor space. For the selected K nearest material clusters, the system and target statistics vectors are combined by a distance-weighted sum as:
where \({\phi }_{u,s}=q({a}_{u},{a}_{s})/{\sum }_{h\in {{{{\mathcal{N}}}}}_{s}}q({a}_{h},{a}_{s})\) is a distance-based weight for the input chemical composition s and a set of its nearest neighbor material cluster \({{{{\mathcal{N}}}}}_{s}\), and q(au, as) = 1/r(au, as) is the inverse distance for a distance function r. Finally, the system-identified material representation zs of the input chemical composition s is calculated as:
where ⊕ is the vector concatenation operator. Note that xs and cs are the atomic feature vector and the input conditions of the input chemical composition s. In machine learning with SIMD, we use the system-identified material representation zs rather than the original input xs and cs.
Transfer learning based on SIMD for machine learning extrapolation
The main feature of SIMD is to generate the vector-shaped representations of the material groups, and it can be used to summarize the large materials datasets. From this perspective, we applied SIMD to transfer learning that aims to transfer knowledge gained from source datasets in solving different but related problems. For the transfer learning on the thermoelectric materials, we used a large source dataset called Starry dataset40 that contains 215,683 observations of the thermoelectric materials and their ZT. However, although the Starry dataset covers extensive thermoelectric materials and their thermoelectric properties, it is not suitable for machine learning due to the following two reasons. (1) The experimentally collected and theoretically calculated thermoelectric materials and their properties are mixed without source labels in the Starry dataset, which makes the prediction models unreliable. (2) The parsing errors in the collected thermoelectric properties are inevitable in the Starry dataset because the data was automatically collected by a parsing algorithm. For this reason, we used Starry dataset as a source dataset for the transfer learning rather than the training dataset.
Figure 3 illustrates the overall process of SIMD to generate the system-identified features in the transfer learning environments. The transfer learning based on SIMD is performed through the following four steps.
-
(1)
SIMD constructs the material clusters and the system-identified features on the merged dataset of the source and training datasets.
-
(2)
The KNN method of SIMD determines K nearest material clusters of the input chemical compositions among the material clusters constructed on the source and training datasets.
-
(3)
SIMD transforms the original training dataset \({{{{\mathcal{D}}}}}_{{\rm{train}}}=\{({s}_{1},{{{{\bf{c}}}}}_{1},{y}_{1}),({s}_{2},{{{{\bf{c}}}}}_{2},{y}_{2}),\ldots ,({s}_{N},{{{{\bf{c}}}}}_{N},{y}_{N})\}\) into \({{{{\mathcal{Z}}}}}_{{\rm{train}}}=\{({{{{\bf{z}}}}}_{1},{y}_{1}),({{{{\bf{z}}}}}_{2},{y}_{2}),\ldots ,\left({{{{\bf{z}}}}}_{N},{y}_{N}\right.\}\) based on Eq. (6), where \(N=| {{{{\mathcal{D}}}}}_{{\rm{train}}}|\) is the number of observations in the training dataset.
-
(4)
The prediction model to predict the target property yn is trained on the transformed training dataset \({{{{\mathcal{Z}}}}}_{{\rm{train}}}\).

SIMD determines the nearest material systems of the input chemical composition in the anchor space and generates the material representation of the input chemical composition based on the selected material systems. In this example, Cu2SnS3, SnSe, and SnS are selected as the nearest material systems of the input SnS0.91Se0.09. Then, the material representation of SnS0.91Se0.09 is generated with the system vectors and target statistics of the selected material systems, which are stored in the material systems database of SIMD.
As a result, our transfer learning problem based on SIMD is formally defined as an optimization problem as:
where L is a loss function to measure the prediction errors, f( ⋅ ; θ) is a prediction model parameterized by θ, SIMD() is a function to generate zn for the input (sn, cn), and \({{{{\mathcal{D}}}}}_{s}\) is a source dataset of transfer learning. In the following subsections, we will evaluate the effectiveness of SIMD in transfer learning based extrapolation to predict thermoelectric efficiency of unknown materials.
SIMD-based transfer learning to extrapolate ZTs of unknown material groups
In this experiment, we conducted machine learning extrapolation to predict the target properties of the materials from unknown material groups. It is essential for material discovery based on machine learning, as we should explore unknown material groups to discover novel materials. To make an extrapolation problem on the ESTM dataset, we divided the entire dataset into the training and test datasets based on the material groups, i.e., none of the materials in the test material groups have never been included in the training dataset. For example, if a pristine material SnS is selected as a test material group, all alloy and doped materials derived from SnS (e.g., SnS0.91Se0.09 and SnS0.94Se0.06) are entirely removed in the training dataset. That is, the prediction models should predict the target properties of the materials that has never been seen in the training dataset, which is called extrapolation problem in machine learning.
To validate the effectiveness of SIMD, we generated four prediction models based on transfer learning approaches as:
-
FCNNf: FCNN is pretrained on the source Starry dataset. Then, FCNN is re-trained on the training dataset of the ESTM dataset.
-
FCNNd: FCNN is trained on the merged training dataset of the source Starry dataset and the training dataset of the ESTM dataset.
-
XGBd: XGB is trained on the merged training dataset.
-
SXGBd: XGB is trained on the merged training dataset transformed by SIMD.
After the training, the extrapolation capabilities of these four transfer learning methods were evaluated on the test dataset \({{{{\mathcal{D}}}}}_{{\rm{test}}}\) that contains completely unseen materials.
Table 3 summarizes the measured R2-scores of the four transfer learning methods in an extrapolation problem that predicts ZTs of the materials in unknown material groups. As shown in the results, the extrapolation capabilities of all machine learning methods were improved by employing the transfer learning approaches based on the Starry dataset. Specifically, FCNNd and XGBd showed R2-scores close to 0.5. However, SXGBd showed further improvement over the conventional XGBd and achieved a R2-score of 0.7.The improvement of SXGBd in R2-score was 0.58 and 0.19 compared to R2-scores of the baseline XGB model and the transfer learning based XGBd model, respectively. The R2-score of SXGBd around 0.70 in the extrapolation environment means that SXGBd roughly predicted the relationships between the test materials and their experimental ZTs, even for the test materials that have never been seen in the training dataset. In the following experiments, we will perform high-throughput screening based on SXGBd to demonstrate the effectiveness of SIMD in real-world applications for discovering novel thermoelectric materials.
High-throughput screening for discovering high-performance thermoelectric materials
The thermoelectric performance defined by ZT essentially determines the efficiency of power generation and energy harvesting in various real-world applications of the thermoelectric materials2,3,5. To discover novel materials of high ZTs, many experimental analyses and demonstrations have been conducted for various candidate material groups9,10,24. To validate the effectiveness of SIMD in material discovery, we conducted the high-throughput screening based on SXGBd for the thermoelectric materials that have never been provided in the training dataset of SXGBd.
We used SXGBd to predict the experimental ZTs of the materials from their chemical compositions and the given measuring temperatures. The high-throughput screening for discovering high-ZT materials can be defined as a binary classification problem determining whether ZTs of the given materials will actually be greater than the threshold ZT. In this classification problem, the true and false labels indicate whether ZTs of the materials are greater than the threshold ZT. For the high-throughput screening results, we calculated the screening accuracy using F1-score29 that can comprehensively evaluate the binary classification accuracy based on true positive, false positive, and false negative. Figure 4 shows the confusion matrices of the binary classification results of XGBd and SXGBd in the high-throughput screening for discovering the materials with ZTs of 1.5 or more. SXGBd achieved an F1-score of 0.61, and the improvement by SXGBd in F1-score was 0.12 compared to the F1-score of XGBd. In particular, SXGBd significantly reduced the number of false positives from 21 to 6. In the high-throughput screening for material discovery, a low false positive is crucial because it guarantees a high probability that the suggested materials will actually have desired properties. In other words, the low false positive can prevent the waste of time and labor to synthesize the materials incorrectly suggested by the prediction models. Quantitatively, the materials suggested by XGBd were actually the high-ZT materials with a probability of 50.00% (=100 × 21/42), whereas the materials suggested by SXGBd were actually the high-ZT materials with a probability of 78.57% (=100 × 22/28). These high-throughput screening results of SXGBd show the potential usability of SIMD in the data-driven discovery of high-performance novel thermoelectric materials.

The true label indicates whether the experimentally measured ZT of the material is actually 1.5 or more. On the other hand, the predicted label indicates whether ZT of the material is predicted to be 1.5 or more. Two abbreviations T and F mean the true and false labels, respectively.
High-throughput screening under temperature constraints
Since the applications of the thermoelectric materials are mainly categorized by the target temperatures, it is crucial to discover high-performance thermoelectric materials for a given temperature9,10,24. In this experiment, we evaluated the screening accuracies of XGBd and SXGBd in a high-throughput screening for discovering high-ZT materials under the given temperature ranges. We performed the high-throughput screening for three target ranges of the temperatures in kelvin: (1) near room temperature (290 ≤ T ≤ 310), (2) common thermoelectric temperature (300 ≤ T ≤ 600), and (3) high temperature (T ≥ 600). For the three target temperature ranges, we searched the materials of ZTs greater than or equal to 0.5, 0.8, and 1.5, respectively.
Figure 5 shows the confusion matrices of the classification results based on the predicted ZTs of XGBd and SXGBd in the high-throughput screening for discovering high-ZT materials under the temperature constraints. As shown in the confusion matrices, SXGBd showed higher F1-scores than XGBd for all high-throughput screening tasks. The false positive of XGBd was 65.22% in the high-throughput screening task in Fig. 5a. By contrast, the false positive of SXGBd was 25%. In addition, the false positive of XGBd was 41.04% in the tasks of Fig. 5b, but the false positive of SXGBd was 21.77%. Also, the false positives of XGBd and SGBd were 38.24% and 21.43% in the task of Fig. 5c, respectively. That is, we were able to reduce the number of false positive samples by more than 50% for all high-throughput screening tasks by applying the proposed SIMD. As we emphasized before, the low false positive is crucial in machine learning based high-throughput screening because it can prevent the waste of time and labor to synthesize the materials incorrectly suggested by the prediction models.

a Screening results at near-room temperature (290 ≤ T ≤ 310). b Screening results at common thermoelectric temperature (300 ≤ T ≤ 600). c Screening results at high temperature (600 ≤ T).
Exploration of virtual dopant spaces for discovering high-ZT materials
As shown in the experimental results, SXGBd achieved R2-score of 0.71 in the extrapolation problem of ZT prediction and showed reliable results in high-throughput screening of high-ZT materials. One of the most beneficial advantages of the extrapolation models is that we can efficiently explore unknown material spaces to discover novel materials without time-consuming experiments and simulations. In this section, we explored virtual dopant spaces using SXGBd to discover promising dopants for target given host materials. To this end, we generated the virtual dopant spaces for given host materials by concatenating the chemical compositions of the host materials and the candidate dopant elements. For example, we generated candidate materials Cu0.001SnSe, Cu0.002SnSe, … , Cu0.1SnSe for a given host material SnSe and a target dopant Cu and. Then, we predicted ZTs of the materials for the target measuring temperatures.
We conducted the virtual screening of the dopant space to discover novel dopants for a host material Bi0.5Sb1.5Te3 that was showed promising thermoelectric properties at low temperature. We generated virtual materials by concatenating Bi0.5Sb1.5Te3 and the elements from H to Fm with the doping concentrations in {0.001, 0.002, … , 0.1}. In other words, we predicted ZTs of 104 candidate virtual materials for the host material Bi0.5Sb1.5Te3. Then, we predicted ZTs of the generated materials at the temperatures in {300 K, 350 K, … , 800 K}. After that, we selected top 10% materials based on their predicted ZTs at 300 K.
In the exploration results, most selected materials contained the dopants of Ti, Fe, Ga, Se, and Ag, and we were able to crosscheck the improved thermoelectric performances by Ag and Ti in experiments41,42,43. We plotted ZTs of Ag- and Ti-doped Bi0.5Sb1.5Te3 for different measuring temperatures, as shown in Fig. 6. The red square and blue triangle lines present the predicted ZTs of the materials that were experimentally reported to have the highest and lowest ZTs in the Ag- and Ti-doped Bi0.5Sb1.5Te3, respectively. The gray dotted lines present the experimentally measured ZTs of the Ag- and Ti-doped Bi0.5Sb1.5Te3. As shown in the results, SXGBd accurately predicted the promising dopants Ag and Ti that were experimentally demonstrated to improve the thermoelectric performance of Bi0.5Sb1.5Te3. Moreover, SXGBd captured the tendency of ZTs of the doped materials for different measuring temperatures.

a Predicted ZTs of Ag-doped Bi0.5Sb1.5Te3. b Predicted ZTs of Ti-doped Bi0.5Sb1.5Te3. The gray dotted lines indicate the experimentally measured ZTs of the materials presented with the same marker.
Extrapolation accuracy for different number of nearest material clusters
The number of nearest material clusters (defined as K) is an important hyper-parameter of SIMD, as shown in Eqs. (4) and (5). In this section, we evaluated the extrapolation capabilities of SIMD for the changes in the values of K. We measured R2-scores of SXGBd in predicting ZTs in the extrapolation problems for different values of K in {1, 2, … , 2048}, as shown in Fig. 7a. Note that the number of material clusters in the training dataset was 2201 (~2048). As shown in the results, SXGBd showed the consistent R2 scores for the changes in the values of K. We also measured the effectiveness of K in the R2-scores of SXGBd for different data sizes. Figure 7b shows the measured R2-scores, and m means the number of material clusters in the training dataset. The small value of m indicates the small training dataset. In the experiment, SXGBd showed the highest R2-scores for K of 4 and 8 in the small training datasets containing 128 and 256 material systems (red and blue lines, respectively), which these small training dataset may not contain a material cluster that covers the input material. For these small training datasets, the values of K larger than 1 were helpful because SIMD was able to generate material descriptors by collecting the information from similar material clusters. By contrast, SXGBd achieved the best R2-score for K of 1 and 2 for more large dataset containing 512 material systems (indigo line). We can interpret this results that K = 1 was sufficient because various material systems covering the input materials were contained in the relatively large training dataset. Therefore, hyper-parameter selection of K is not a big problem in the implementation of SIMD if we have a large training dataset. However, when the training dataset is small, a sufficiently large K will help to improve the extrapolation ability of SIMD.

a Prediction accuracies of SXGBd for different values of K in the original source dataset. b Prediction accuracies of SXGBd for different values of K in subsets of the source dataset. K is the number of nearest material clusters in Eqs. (4) and (5). m is the number of material clusters in the training dataset. For the quantitative comparisons, we present the R2-score of the baseline XGBd using the black dotted line.
SIMD for system-conditioned prediction
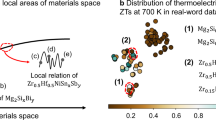
In the high-throughput screening to discover novel thermoelectric materials, SIMD reduced the false positive of the prediction models by about 50% compared to the conventional methods. To reveal physical or chemical insights from the high-throughput screening results, we conducted a case study for the Mg1−xLixGe0.9Si0.1 system that generated most false positive samples in the baseline XGBd. Figure 8 illustrates the distribution of the thermoelectric materials around the Mg1−xLixGe0.9Si0.1 system in the chemical space based on the sparse encoding of the chemical compositions. In this chemical space, three high-ZT materials denoted by (a), (c), and (e) in Fig. 8 are distributed near the materials from the Mg1−xLixGe0.9Si0.1 system because they commonly contain two elements Ge and Mg. Basically, the similar feature representations yields the similar target values in machine learning. Hence, although the thermoelectric materials from the Mg1−xLixGe0.9Si0.1 system actually have the low-ZT (<0.4), XGBd incorrectly predicted them as the high-ZT (≥1.5) materials because the similarly-encoded materials (a), (c), and (e) in the training dataset have the high-ZT (>1.4).

Distribution of the thermoelectric materials around the Mg1−xLixGe0.9Si0.1 system in the chemical space based on the sparse encoding of the chemical compositions.
By contrast, SXGBd with SIMD greatly reduced the false positive from the Mg1−xLixGe0.9Si0.1 system in the high-throughput screening. As shown in Fig. 8, the three materials (a), (c), and (e) belong to the material clusters (b), (d), and (f) in SIMD. The materials from the Mg1−xLixGe0.9Si0.1 system have different sparse encodings to the materials (b), (d), and (f) because they contain only one element Mg in common or no element in common. Therefore, SIMD can generate the representations of the Mg1−xLixGe0.9Si0.1 system that are different to high-ZT materials (b), (d), and (f). As a result, the prediction models based on SIMD were able to correctly learn that the thermoelectric materials from the Mg1−xLixGe0.9Si0.1 system are low-ZT materials.
Material space exploration based on global search method with SIMD
In the result section, SIMD was successfully applied to discover high-ZT thermoelectric materials based on high-throughput screening. However, we can further extend SIMD to an automated global search method for discovering novel materials in unexplored material space by integrating SIMD with randomized iterative search algorithms called metaheuristic algorithms44. In chemical science, metaheuristics have been successfully applied to discover novel molecules and materials45,46,47. Various metaheuristic optimization algorithms have been proposed based on evolutionary method48, swarm intelligence strategy49, and physics-inspired approach50. Although existing metaheuristic algorithms employ different optimization schemes, we can integrate SIMD without algorithmic modifications because we can identically define a problem to discover novel materials based on SIMD as a constrained optimization problem as:
where x is the sparse encoding of the elements in the chemical composition, f( ⋅ ; θ*) is a trained extrapolation model to predict target material properties from the input x, and g(x) is a penalty term for checking violation of user-defined constraints on discovered materials. In this formulation, f( ⋅ ; θ*) can be implemented by SXGBd to extrapolate ZTs of the unexplored materials. Furthermore, g(x) can be defined as domain-specific constraints regardless of whether it is differentiable or not. For example, if we focus on the materials containing maximum three elements, we can define the penalty term as:
In addition to the example constraint, we can impose various domain-specific constraints on the chemical characteristics of discovered materials, such as target elements, ranges of target properties, and the number of dopants. Note that if we want to discover the materials maximizing the target properties, the optimization problem in Eq. (8) can be defined as a maximization problem with a negative penalty term. In this study, we conducted the global search using equilibrium optimizer50 based on SXGBd to discover novel high-ZT materials, and the search results are provided in SI.
Methods
Sparse encoding of chemical compositions
In the implementation of SIMD in Eqs. (6) and (8), we used the sparse encoding x to represent the input chemical compositions as the vector-shaped data. Formally, the sparse encoding of the input chemical composition s is defined as:
where e is an element in the input chemical composition s, re is the ratio of e, and ne is the atomic number of e. In the implementation, we considered the elements from H to Fm, i.e., the dimensionality of the sparse encoding was 100.
Anchor space for material clusters allocation
To determine the material clusters for the input chemical compositions based on the KNN method, the cluster identifiers defined by the chemical compositions and the input chemical composition should be converted into compact low-dimensional vectors. To this end, we defined an anchor space where the chemical compositions are represented as compact 12-dimensional vectors. Formally, the chemical compositions of the material clusters and the input materials are defined as an anchor vector as:
where qmean is a three-dimensional vector of the average atomic numbers, atomic volumes, and atomic weights of the elements in the input chemical composition. Similarly, qstd, qmin, and qmax are calculated as the standard deviations, minimum values, and the maximum values of the three atomic attributes of the elements in the input chemical composition, respectively. Based on the anchor vectors of the material clusters and the input materials, we determined the material clusters of the input materials by calculating the Euclidean distances between the anchor vectors of the material clusters and the input materials.
Algorithmic description of SIMD
We present a Python-based algorithmic description of SIMD for reproducibility of SIMD. Algorithm 1 describes the overall process to generate the system-identified material features based on SIMD. For given inputs \({{{\mathcal{D}}}}\) and K, the algorithm returns the transformed dataset \({{{\mathcal{Z}}}}\) and the material clusters \({{{{\mathcal{C}}}}}_{m}\), where \({{{\mathcal{Z}}}}\) contains the system-identified features of the materials and \({{{{\mathcal{C}}}}}_{m}\) is a dictionary storing the identifiers of the generated material clusters.
Algorithm 1
The overall process to generate the system-identified material features. 
Data availability
The collected dataset and all resources of the proposed method are publicly available at https://github.com/KRICT-DATA/SIMD.
Code availability
All experimental scripts and source codes of SGL are publicly available at https://github.com/KRICT-DATA/SIMD.
References
Rademann, K., Raghuwanshi, V. & Hoell, A. Chapter 3—crystallization and growth mechanisms of nanostructures in silicate glass: from complete characterization toward applications. In Glass Nanocomposites, (eds Karmakar, B., Rademann, K. & Stepanov, A. L.) 89–114 (William Andrew Publishing, 2016).
Nozariasbmarz, A. et al. Review of wearable thermoelectric energy harvesting: from body temperature to electronic systems. Appl. Energy 258, 114069 (2020).
Zhao, D. & Tan, G. A review of thermoelectric cooling: materials, modeling and applications. Appl. Therm. Eng. 66, 15–24 (2014).
Tan, G., Ohta, M. & Kanatzidis, M. G. Thermoelectric power generation: from new materials to devices. Philos. Trans. R. Soc. A 377, 20180450 (2019).
Elsheikh, M. H. et al. A review on thermoelectric renewable energy: principle parameters that affect their performance. Renew. Sustain. Energy Rev. 30, 337–355 (2014).
Zhao, L.-D. et al. Ultralow thermal conductivity and high thermoelectric figure of merit in snse crystals. Nature 508, 373–377 (2014).
Joshi, G. et al. Enhanced thermoelectric figure-of-merit in nanostructured p-type silicon germanium bulk alloys. Nano Lett. 8, 4670–4674 (2008).
Sharma, P. K., Senguttuvan, T., Sharma, V. K. & Chaudhary, S. Revisiting the thermoelectric properties of lead telluride. Mater. Today Energy 21, 100713 (2021).
Yang, L., Chen, Z.-G., Dargusch, M. S. & Zou, J. High performance thermoelectric materials: progress and their applications. Adv. Energy Mater. 8, 1701797 (2018).
Tan, G., Zhao, L.-D. & Kanatzidis, M. G. Rationally designing high-performance bulk thermoelectric materials. Chem. Rev. 116, 12123–12149 (2016).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Jacobs, R., Booske, J. & Morgan, D. Understanding and controlling the work function of perovskite oxides using density functional theory. Adv. Funct. Mater. 26, 5471–5482 (2016).
Patra, A. et al. Efficient band structure calculation of two-dimensional materials from semilocal density functionals. J. Phys. Chem. C 125, 11206–11215 (2021).
Liao, X. et al. Density functional theory for electrocatalysis. Energy Environ. Mater. 5, 157–185 (2022).
Schuch, N. & Verstraete, F. Computational complexity of interacting electrons and fundamental limitations of density functional theory. Nat. Phys. 5, 732–735 (2009).
Cohen, A. J., Mori-Sánchez, P. & Yang, W. Insights into current limitations of density functional theory. Science 321, 792–794 (2008).
Zhuo, Y., Mansouri Tehrani, A. & Brgoch, J. Predicting the band gaps of inorganic solids by machine learning. J. Phys. Chem. Lett. 9, 1668–1673 (2018).
Na, G. S., Jang, S. & Chang, H. Predicting thermoelectric properties from chemical formula with explicitly identifying dopant effects. Npj Comput. Mater. 7, 106 (2021).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Schleder, G. R., Padilha, A. C., Acosta, C. M., Costa, M. & Fazzio, A. From dft to machine learning: recent approaches to materials science–a review. J. Phys. Mater. 2, 032001 (2019).
Kipf, T. N. & Welling, M. Semi-supervised classification with graph convolutional networks. ICLR (2017).
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mater. 31, 3564–3572 (2019).
Goodall, R. E. & Lee, A. A. Predicting materials properties without crystal structure: deep representation learning from stoichiometry. Nat. Commun. 11, 1–9 (2020).
Alam, H. & Ramakrishna, S. A review on the enhancement of figure of merit from bulk to nano-thermoelectric materials. Nano Energy 2, 190–212 (2013).
Cancilla, J. C., Perez, A., Wierzchoś, K. & Torrecilla, J. S. Neural networks applied to determine the thermophysical properties of amino acid based ionic liquids. Phys. Chem. Chem. Phys. 18, 7435–7441 (2016).
Wang, T., Zhang, C., Snoussi, H. & Zhang, G. Machine learning approaches for thermoelectric materials research. Adv. Funct. Mater. 30, 1906041 (2020).
Draper, N. R. & Smith, H. Applied Regression Analysis 3rd edn (Wiley-Interscience, 1998).
Moré, J. J. The Levenberg-Marquardt algorithm: implementation and theory. In Numerical Analysis, 105–116 (Springer, 1978) https://link.springer.com/chapter/10.1007/BFb0067700.
Powers, D. M. W. Evaluation: from precision, recall and f-measure to roc., informedness, markedness and correlation. J. Mach. Learn. Technol. 2, 37–63 (2011).
Chen, Z.-G., Shi, X., Zhao, L.-D. & Zou, J. High-performance snse thermoelectric materials: progress and future challenge. Prog. Mater. Sci. 97, 283–346 (2018).
Wei, J. et al. Review of current high-zt thermoelectric materials. J. Mater. Sci. 55, 12642–12704 (2020).
McDonald, G. C. Ridge regression. Wiley Interdiscip. Rev. Comput. Stat. 1, 93–100 (2009).
Taunk, K., De, S., Verma, S. & Swetapadma, A. A brief review of nearest neighbor algorithm for learning and classification. In ICCS, 1255–1260 (IEEE, 2019).
Smola, A. J. & Schölkopf, B. A tutorial on support vector regression. Stat. Comput. 14, 199–222 (2004).
Schulz, E., Speekenbrink, M. & Krause, A. A tutorial on gaussian process regression: modelling, exploring, and exploiting functions. J. Math. Psychol. 85, 1–16 (2018).
Rosenblatt, F. The perceptron: a probabilistic model for information storage and organization in the brain. Psychol. Rev. 65, 386 (1958).
Chen, T. & Guestrin, C. Xgboost: a scalable tree boosting system. In SIGKDD, 785–794 (Association for Computing Machinery, 2016).
Xu, K. et al. How neural networks extrapolate: from feedforward to graph neural networks. In ICLR (2021).
Na, G. S., Jang, S. & Chang, H. Nonlinearity encoding to improve extrapolation capabilities for unobserved physical states. Phys. Chem. Chem. Phys. 24, 1300–1304 (2022).
Katsura, Y. et al. Data driven analysis of electron relaxation times in PbTe-type thermoelectric materials. Sci. Technol. Adv. Mater. 20, 511–520 (2019).
Lee, J. et al. Control of thermoelectric properties through the addition of ag in the bi0. 5sb1. 5te3alloy. Electron. Mater. Lett. 6, 201–207 (2010).
Cao, S. et al. Enhanced thermoelectric properties of ag-modified bi0. 5sb1. 5te3 composites by a facile electroless plating method. ACS Appl. Mater. Interfaces 9, 36478–36482 (2017).
Feng, B., Tang, Y. & Lei, J. The influential mechanism of ti doping on thermoelectric properties of bi0. 5sb1. 5te3 alloy. J. Mater. Sci. Mater. Electron. 32, 28534–28541 (2021).
Gogna, A. & Tayal, A. Metaheuristics: review and application. J. Exp. Theor. Artif. Intell. 25, 503–526 (2013).
Leguy, J., Cauchy, T., Glavatskikh, M., Duval, B. & Da Mota, B. Evomol: a flexible and interpretable evolutionary algorithm for unbiased de novo molecular generation. J. Cheminform. 12, 1–19 (2020).
Pirgazi, J., Alimoradi, M., Esmaeili Abharian, T. & Olyaee, M. H. An efficient hybrid filter-wrapper metaheuristic-based gene selection method for high dimensional datasets. Sci. Rep. 9, 1–15 (2019).
Mlinar, V. Utilization of inverse approach in the design of materials over nano-to macro-scale. Ann. Phys. 527, 187–204 (2015).
Katoch, S., Chauhan, S. S. & Kumar, V. A review on genetic algorithm: past, present, and future. Multimed. Tools. Appl. 80, 8091–8126 (2021).
Poli, R., Kennedy, J. & Blackwell, T. Particle swarm optimization. Swarm Intell. 1, 33–57 (2007).
Faramarzi, A., Heidarinejad, M., Stephens, B. & Mirjalili, S. Equilibrium optimizer: a novel optimization algorithm. Knowl. Based Syst. 191, 105190 (2020).
Acknowledgements
This study was supported by a project from the Korea Research Institute of Chemical Technology (KRICT) [grant number: SI2151-10]. We thank Jian Jeong for assistance with dataset construction.
Author information
Authors and Affiliations
Contributions
G.S.N. contributed to design of experiments and conducted experiments. G.S.N. and H.C. wrote the original manuscript and analyzed the results. All the authors were involved in writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Na, G.S., Chang, H. A public database of thermoelectric materials and system-identified material representation for data-driven discovery. npj Comput Mater 8, 214 (2022). https://doi.org/10.1038/s41524-022-00897-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-022-00897-2
This article is cited by
-
Leveraging language representation for materials exploration and discovery
npj Computational Materials (2024)
-
High-throughput deformation potential and electrical transport calculations
npj Computational Materials (2023)
