
Abstract
Motivated by the recent claim of hot superconductivity with critical temperatures up to 550 K in La + x hydrides, we investigate the high-pressure phase diagram of compounds that may have formed in the experiment, using first-principles calculations for evolutionary crystal structure prediction and superconductivity. Starting from the hypothesis that the observed Tc may be realized by successive heating upon a pre-formed LaH10 phase, we examine plausible ternaries of lanthanum, hydrogen and other elements present in the diamond anvil cell: boron, nitrogen, carbon, platinum, gallium, gold. We find that only boron and, to a lesser extent, gallium form metastable superhydride-like structures that can host high-Tc superconductivity, but the predicted Tc’s are incompatible with the experimental reports. Our results indicate that, while the claims of hot superconductivity should be reconsidered, it is very likely that unknown H-rich ternary or multinary phases containing lanthanum, hydrogen, and possibly boron or gallium may have formed under the reported experimental conditions, and that these may exhibit superconducting properties comparable, or even superior, to those of currently known hydrides.
Similar content being viewed by others
Introduction
Since the discovery of high-temperature superconductivity (HTSC) in compressed sulfur hydride in 20141,1,2, the race for HTSC has dramatically accelerated, leading to a hydride rush fueled by ab initio predictions.
As of 2020, all binary hydrides have been computationally explored3,4,5, and many have been synthesized6,7,8,9,10,11,12. After LaH10 established the Tc record for binary hydrides in 2019, in 2020 Snider et al.13 reported a Tc of 288 K in a compressed mixture of carbon, sulfur, and hydrogen, effectively realizing a room temperature superconductor. Compared to binary hydrides, ternary (or, in general, multinary) hydrides exhibit an increased chemical versatility, which may be exploited to tune the superconducting properties. Since Migdal–Eliashberg theory does not pose a hard limit to Tc, it is possible that multinary hydrides may exhibit superconductivity at sensibly higher Tcs than the known binaries; for example, Tc’s largely exceeding room temperature have been predicted in a Li–Mg–H alloy14.
In the summer of 2020, Grockowiak et al. reported experimental evidence of superconductivity with onset temperatures growing from 294 to 550 K upon successive heating cycles of a mixture of lanthanum and ammonia borane at about 180 GPa15. This may have been an experimental observation of hot superconductivity in a multinary hydride; unfortunately, due to COVID restrictions, the authors were able to report only partial evidences, and did not provide information on the chemical composition and structure of the superconducting samples, which would be fundamental for reproducibility. Even if one is skeptical about the highest values of Tc reported, it is possible that one or more multinary phases may have formed, calling for further studies.
In the absence of conclusive experimental information, such a question can effectively be addressed by first-principles methods for crystal structure prediction and superconductivity, which have demonstrated an extraordinary accuracy for binary hydrides3,4,5,16.
In this paper, we perform an exploratory ab initio study of possible candidates for hot superconductivity, using evolutionary crystal structure prediction and linear-response calculations of the electron-phonon coupling. We explore all possible ternary combinations of lanthanum, hydrogen, and a third element present in the diamond anvil cell (DAC) in the experiment of ref. 15: boron and nitrogen (from the hydrogen source), carbon, from the epoxy, platinum, gallium, and gold, from the electrical contacts. One (or more) of these elements may react with lanthanum and hydrogen to form an unknown superhydride. Our aim is to sample the ternary phase diagrams with an accuracy sufficient to estimate the probability that stable or weakly metastable structures may form at high pressure, determine the characteristic structural motifs, and assess their potential for high-Tc conventional superconductivity. We choose to carry out all of our structural searches at 300 GPa, rather than 180 GPa, which is the highest pressure measured in the experiment, and examine both the thermodynamically stable and metastable structures when scanning for potential superconductors, to expand the landscape of possible candidates17. In fact, there is rather extensive evidence that static Density Functional Theory (DFT) calculations, which neglect quantum lattice effects, tend to overestimate the stabilization pressure of hydrides18,19 by as much as 100 GPa, and higher pressures typically yield better agreement with experiments also for superconducting properties. In addition, once we established that nitrogen and boron were the elements most likely to form a ternary superhydride, we performed additional searches for those elements at 150 GPa, to rule out the possibility of missing relevant phases.
We show that, among all elements present in the DAC, only nitrogen and boron form thermodynamically stable ternary structures with La and H, while the other elements do not form any stable ternary structure (Ga, Pt, C, Au); a few La–Ga–H hydrides are found very close to the convex hull, i.e. are likely metastable. Nitrogen forms stable and metastable structures that do not exhibit the typical characteristics of high-Tc superhydrides: high-symmetry, large hydrogen content, large fraction of H states at the Fermi level. On the other hand, some stable and metastable La–B–H structures are characterized by the same hydrogen cage-like motifs encountered in many record superhydrides20,21, and some metastable La–Ga–H structures present a similar dense hydrogen sublattice. In particular, within the limitations posed by the maximum cell size, our best superconducting phases are LaBH17 and LaGaH14, with a Tc of 180 and 137 K respectively, which are way too low to explain the hot superconductivity observed in ref. 15. Tuning of the electronic and vibrational properties through doping or impurities may increase this value up to a factor two, but it is extremely unlikely that this type of structures may reach Tc’s as high as 550 K.
This paper is organized as follows. The Results and Discussion section is further divided in the Phase diagrams subsection, where we discuss some general criteria leading to the formation of stable ternary phases, and present the predicted phase diagrams; In the Electronic properties subsection, where we analyze the electronic structure of the predicted phases; And in the Superconducting properties section, we discuss the superconducting properties of the metallic phases. The Methods section contains computational details on the structural searches and superconductivity calculations.
Results and discussion
Phase diagrams
Our structural searches were carried out using evolutionary algorithms as implemented in the Universal Structure Predictor: Evolutionary Xtallography (USPEX) code22,23. Further details on the structural searches can be found in the Computational Details section. Since full structural searches of ternary diagrams are extremely expensive computationally, before sampling in-depth the ternary compositions, we carried out a pre-screening process by first sampling roughly all possible combinations of elements, and then re-sampling those where stable compositions appeared.
The elements that we chose to analyze are based on a few considerations on the experimental report. According to the authors, the first onset of a superconducting transition occurred at 294 K (not far from the reported value for LaH10), and Tc gradually shifted towards higher temperatures, upon further heating. It seems reasonable to assume that LaH10 was formed first, and, with the subsequent heating cycles, the sample underwent further structural transitions into an unknown multinary hydride phase, incorporating one or more of the elements present in the diamond anvil cell during the experiment. The diamond anvil cell was loaded with pure lanthanum and ammonia borane (NH3BH3), which acts both as a hydrogen source and as pressure medium, and the authors mention as possible contaminants also platinum, gallium, and gold from the electrodes, and carbon from the epoxy binder. In principle, any combination of these elements may be responsible for the observed hot superconductivity phase.
We calculated the ternary convex hulls for La–X–H structures (X = B, N, Ga, Pt, C, Au), sampling 3000 structures for each diagram. This search resulted in no stable (or weakly metastable) compositions for La–C–H, La–Au–H, and La–Pt–H, while several stable/metastable ones were already found for La–N–H, and La–B–H, and a few metastable ones—but none stable—were found for La–Ga–H.
This led us to dismiss further investigation of La–C–H, La–Au–H, and La–Pt–H, and to devote our resources to a better sampling of the others. In La–Ga–H we re-sampled the (LaGa)xH1−x pseudo-binary, where the ternaries closest to the hull were present, to check whether any would become stable, but to no avail. Boron and nitrogen, on the other hand, were the most abundant elements other than hydrogen and lanthanum during the experiment, and thus are the most likely candidates to form ternary La–X–H hydrides. The existence of high-pressure compounds involving La, N, and B, as well as metal boron hydrides also supports this hypothesis24,25,26,27.
Having decided to focus on La–N–H, La–B–H, we improved our structural searches by performing a second structural search, sampling a total of 5000 structures, which add to the previously sampled 3000. In addition, we performed structural searches along several pseudo-binary phases, bringing the total number of structures sampled to about 15000. In these refined searches, we sampled unit cells as large as 48 atoms. See the Computational Details section for more information.
In Fig. 1 we report the convex hulls for La–N–H, La–B–H, and La–Ga–H. The convex hulls for La–Pt–H, La–C–H and La–Au–H, where no stable or metastable compounds are found are shown in Supplementary Fig. 1.

The convex hull shown are for La–N–H (a) and La–B-H (b), and La–Ga-H (c). Blue circles and red squares represent stable and metastable phases, respectively.
The La–N–H convex hull contains four stable ternary phases: LaN2H3, LaN3H10, La2N2H, and La4N4H, as well as several metastable phases along the (LaN)xH1−x line. We anticipate that none of the (meta)stable structures predicted in the La–N–H phase diagram is a likely candidate for HTCS since for most structures the hydrogen content is low, and H-rich weakly metastable structures are either low-symmetry or insulating.
In the La–B–H phase diagram we find three stable intermediate compositions: LaBH8, La2B6H5, and LaB8H. In addition, several H-rich phases are predicted to be metastable (within 50 meV/atom from the hull, shown as red squares), suggesting that there is a strong tendency for La and B to form H-rich phases. While this work was under review, Liang et al. published a preprint with a thorough investigation of the ternary La–B-H phase diagram at 100, 150, 200, and 300 GPa28. In their work, the authors construct the convex hull by sampling the LanBm (n = 1, m = 1–6; n = 2, m = 1) pseudo-binaries, and predict the formation of the same LaBH8 structure we report in our work.
In La–Ga–H phase diagram we could not find any stable ternary composition, but we do find a plethora of hydrogen-rich compositions close to the hull (i.e. within 50 meV/atom), indicating that metastable structures may in principle form. Among these, we analyzed three metastable structures with LaGaH6, LaGaH14, and LaGaH15 composition, which were characterized by a more or less symmetric Bravais lattice (at least orthorhombic), and promising structural characteristics: high hydrogen fraction, and intermediate H–H distance. Since the formation of any of these phases is unlikely, in the following we only included the LaGaH14 as representative of the La–Ga-H system. A discussion on the other phases can be found in the Supplemental Material.
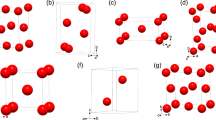
In Fig. 2, we show the crystal structures of the stable La–N–H and La–B–H phases (the structure with LaN3H10 composition is insulating, and is shown in Supplementary Fig. 3), together with two particularly significant metastable ones: LaBH17 and LaGaH14; additional information on the crystal structures with other elements, as well as all the structural data can be found in Supplementary Table 1 (The Supplemental Material is available at https://doi.org/10.1038/s41524-021-00691-6).

La, B, N, Ga and H atoms are shown as green, orange, gray, red, and blue spheres, respectively. Polyhedral surfaces match the color of the bonding atom. In LaGaH14 the 001 lattice plane is shown to highlight the planar nature of the hydrogen network.
The La4N4H and La2N2H phases are characterized by a La–N sublattice with a cubic CsCl arrangement, in which hydrogen occupies the interstitial sites, with a H–H distance (dH–H) of 3.6 and 2.6 Å, respectively. On the other hand, the LaN2H3 structure is characterized by the presence of La layers, alternated with a N–H network, and a H–H distance of 1.4 Å, while LaN3H10 exhibits a disordered mixture of H2 (dH–H = 0.74 Å), NH, NH2, and NH3 molecules scattered around a La atom. The few metastable phases at high hydrogen content that we predict, are also characterized by the presence of disordered H2 and NHx molecules.
The LaB8H phase exhibits a dense B–B network around each La atom, identical to the one predict for LaB8, with hydrogen occupying interstitial positions between the second-nearest La–La atoms, with a H–H distance of 3.7 Å. The La2B6H5 phase is characterized by the presence of two polymeric chains of BH2 and B2H2, connected by a shared hydrogen atom, resulting in a H–H distance of 1.4 Å, while the La atom acts as a spacer among the polymers. The LaBH8 phase exhibits a densely packed structure with two compenetrating face-centered cubic lattices for La and B, while H atoms form a rhombicuboctahedron around La and a cube around B. Nearest-neighboring hydrogen atoms form tetrahedra with a H–H distance of 1.33 Å. This phase is particularly interesting at lower pressures, as it remains stable down to 40 GPa, where it exhibits high-Tc superconductivity—further details have been discussed in ref. 26, while at 300 GPa the Tc is strongly suppressed by phonon hardening.
In the following we will focus on the crystal structure of LaBH17 which, as we will discuss later on, exhibits the highest Tc among the structures examined. Its crystal structure presents a slightly distorted orthorhombic structure (α-LaBH17), in which a cage of 32 H atoms surrounds a La atom. Neighboring cages are alternated with B2H10 molecules. The cages are stacked along the vertical axis, sharing a slightly distorted hydrogen hexagon, with a H–H distance of 0.95 Å. At 500 GPa this phase undergoes a structural transition into a tetragonal structure (β-LaBH17), which is essentially degenerate in energy with the orthorhombic one and only differs for a small rotation of the H cages with respect to the stacking axis and the regularization of the hydrogen hexagon. Most likely, the inclusion of quantum effects would remove the distortion at lower pressure, in the same way as reported for other superhydrides18,19. Both LaBH8 and LaBH17 have a relative hydrogen content above 50%, and are characterized by a symmetric lattice, with lanthanum and boron atoms encaged into hydrogen polyhedra, which form a sponge-like lattice, and hence are an example of a ternary hydride with a crystal structure that is reminiscent of sodalite clathrate hydrides.
The metastable LaGaH14 structure is characterized by a base-centered orthorhombic La sublattice, interpenetrated by a simple orthorhombic Ga one. A dense planar hydrogen network is sandwiched between La–Ga planes, with a H–H distance ranging between 0.9 and 1.2 Å, quite similarly to the structure of LaBH17. The structure of LaGaH15 (Fig. S3) is also very similar.
Electronic properties
In Fig. 3, we report the total and atom-projected electronic Density of States (DOS) for the stable La–N–H and La–B-H phases, and metastable LaBH17 and LaGaH14. The structures for La4N4H and La2N2H are metallic and the partial DOS in the valence region is dominated by lanthanum and nitrogen, with little contribution from interstitial hydrogen. In LaN2H3 hydrogen gives a rather small contribution to the DOS from −25 to −5 eV, while nitrogen strongly contributes in the −10 to 0 eV range, and makes up most of the states at the Fermi level. LaN3H10 (shown in Supplementary Fig. 6), on the other hand, is characterized by an insulating structure, with a band gap of 2.4 eV.

The total DOS and its projection onto La, N, B, Ga, and H are shown as black lines, and green, gray, orange, red, and blue filled lines, respectively. The Fermi energy is set as the zero.
Overall, none of the La–N–H structures shown exhibit the typical characteristics of superconducting hydrides: high-symmetry structures, with high hydrogen content, and a large DOS at the Fermi level which is mostly derived from hydrogen. In La–N–H, we observe that at low concentration, hydrogen plays no significant role in the band structure near the Fermi energy, while at higher concentration, the structures formed are low-symmetry molecular crystals, either insulating, or with negligible contribution of hydrogen to the states near the Fermi energy.
The stable La–B–H structures (and metastable LaBH17) are all metallic. In particular, LaB8H is characterized by a partial DOS with a predominant boron contribution, while hydrogen contributes only very little. The La2B6H5 structure exhibits a much larger, and rather constant, contribution of lanthanum to filled states. In both cases, however, hydrogen does not contribute significantly to the states at the Fermi level, nor does it play a significant role in the electronic structure. The structure for LaBH8 is characterized by a partial DOS character rather equally distributed among La, B, and H in the −25 to −5 eV range, which gives way to a hydrogen-dominated DOS in the −5 to 1 eV range, including states at the Fermi level. Last, LaBH17 exhibits a strong hydrogen character at all energies, as the structure is characterized by a weakly covalent hydrogen network, and a very high hydrogen concentration—The electronic band structures and Fermi surfaces of LaBH8 and LaBH17 are reported in Figs. S4 and S5. LaBH8 and LaBH17 are the only compositions for which the electronic structures are similar to the ones reported for sodalite-like superhydrides, i.e., a large hydrogen fraction of the states at the Fermi level NH/N(EF), as well as a large overall DOS at the Fermi level (N(EF)). Moreover, the structures of both LaBH8 and LaBH17 also satisfy a geometrical prerequisite: it was observed that the presence of weakly covalent hydrogen–hydrogen bonds positively correlates with Tc29. These bonds are associated with a H–H distance larger than 0.80 Å (i.e. no H2 molecules), but smaller than 2 Å (i.e., significant H–H interaction). When the H–H interatomic distance satisfies these constraints the H lattice is so dense that a quasi-free electron gas is realized30,31. A possible descriptor to characterize the nature of the H–H bond in hydrides, called connectivity, was recently proposed by Belli et al.29. The connectivity value ϕ represents the highest value of the Electron Localization Function (ELF) for which the isocontour spans the cell without discontinuities in all directions. In particular, isolated high ELF regions surrounding H–H bonds are associated with molecular hydrogen, while a ϕ between approximately 0.4 and 0.8 is considered an indication of weak covalent bonds. For LaBH8 and LaBH17 at 300 GPa we find a connectivity value ϕ of 0.33 and, 0.43, respectively and no molecular bonds, which classifies LaBH17, and possibly also LaBH8, as a weak covalent hydride. For LaGaH14 we find ϕ = 0.35, placing it in the same ballpark as LaBH8 and LaBH17. The ELF corresponding to the ϕ value for the two La–B–H structures is shown in Fig. S7. The electronic properties of the structures of other metastable hydrides are shown in Fig. S4.
The LaGaH14 structure is characterized by a large contribution of hydrogen to all occupied states. The occupied Ga-d states appear as a narrow peak around −18 eV, which however does not exhibit signs of hybridization with hydrogen or lanthanum, which instead occurs between −15 and 0 eV, as suggested by the three very similar projected DOS’s. The DOS at the Fermi level, however, is characterized by a strong hydrogen character, around 60%, indicating that the hydrogen covalent sublattice is metallic.
Superconducting properties
In order to asses whether there is a hot superconductor among the predicted structures, we calculated their vibrational and superconducting properties using Density Functional Perturbation Theory (DFPT). In addition to all stable La–N–H and La–B–H structures, we also included metastable ones from the other convex hulls if they respected the following criteria: (i) hydrogen content above 60%, (ii) symmetric lattice (at least orthorhombic), (iii) intermediate hydrogen–hydrogen distance (above 0.85 Å). Phonon frequencies were computed at the harmonic level, and the superconducting Tc due to e − ph interaction was calculated by numerically solving the T-dependent isotropic Migdal–Eliashberg equations, using a standard value of μ* of 0.1032,33. Our estimates do not take into account anharmonicity, whose effect should be to make our structures stable at lower pressures than the nominal harmonic instability pressure6,18,34.
A summary of the electronic and vibrational properties of the stable phases of La–B–H and La–N–H, and of selected La–Ga–H and La–Pt–H phases at 300 GPa is reported in Table 1. All structures are dynamically stable, and the predicted Tc’s are 8 K in LaB8H, 6 K in La2B6H5, 14 K in LaBH8 and 180 K in LaBH17, while La4N4H, La2N2H, and LaN2H3 exhibit a Tc below 1 K. Among the La–Ga–H and La–Pt–H metastable structures we find a few superconductors, and the highest Tc is found in LaGaH14, with 137 K. As expected, the highest Tcs are observed for the structures with the highest hydrogen content. The similarities between La–B and La–Ga hydrides, which are the only systems where high-Tc superconductivity are not surprising. In fact, gallium and boron both belong to the 13th group. This result suggests that, in combination with lanthanum, metalloids and other post-transition metals should favor the formation of ternaries with superconducting properties, whereas halogens and other nonmetals have the tendency to suppress it.
In Fig. 4, we report the total and the atom-projected Éliashberg functions for all stable structures (except LaN3H10, which is insulating), as well as for metastable LaGaH14 and LaBH17. As shown in the figure, in both La4N4H and La2N2H it is mostly nitrogen vibrations which contribute to the overall e-ph coupling, which is extremely small. In La4N4H, the H-derived modes with frequencies above 150 meV are absent, while they are present, albeit very weakly coupled, in La2N2H. It is clear that in these two structures hydrogen, which occupies the interstitial sites, does not play a significant role in the bonding or in the properties, and thus cannot contribute to the coupling. The spectrum of LaN2H3 is characterized by a few narrow peaks between 100 and 250 meV, with strong hydrogen character.

The atom projections on La, B, N, Ga, and H are shown in green, gray, orange, red, and blue, respectively. The Eliashberg function and ω-dependent e-ph coupling λ(ω) are defined in Supplemental Material. Note: due to the large differences in values, the y-axis scale is different for each subfigure.
In LaB8H the vibrational spectrum is dominated by boron, while in La2B6H5 about 30% of it is hydrogen. In both cases however, the integrated e-ph coupling λ is around 0.5, i.e. too low to lead to an appreciable Tc. In these cases the hydrogen content is rather low, therefore it is the B–B covalent bonding that dominates.
LaBH8 and LaBH17 are significantly different from LaB8H and La2B6H5: here essentially all of the coupling is concentrated into hydrogen modes. In particular, in LaBH8 the coupling largely comes from modes around 170 meV, yielding a high ωlog of 135 meV, and a relatively low λ = 0.5. LaBH17, on the other hand, exhibits a large value of the Eliashberg function at all energy ranges, concentrated on hydrogen modes. At 300 GPa, α-LaBH17 is on the verge of a structural instability, as witnessed by its small ωlog, and large electron-phonon coupling constant λ = 3.3. At higher pressures the phonon harden. Both LaBH8, LaBH17, and LaGaH14 exhibit the typical characteristic of high-Tc superhydrides, i.e., the presence of an interconnected, metallic hydrogen sublattice, which is manifested both in the large fraction of hydrogen states in the DOS at the Fermi level, and in a rather uniform distribution of the electron-phonon coupling over all phonon modes, as observed in binary high-Tc sodalite-like hydrides.
In another publication, we studied the superconducting behavior of LaBH8 as a function of pressure in greater detail35. This structure remains dynamically stable down to 40 GPa, where the phonon modes are softer, and the Tc reaches 126 K. At 300 GPa however, the pressure-induced hardening of the phonon modes is so strong that suppresses the high-Tc.
The Éliashberg function for LaGaH14, similarly to LaBH17, is completely dominated by hydrogen, which constitutes 99% of its spectral weight. The coupling is evenly spread over all modes from 30 to 300 meV, which all contribute significantly to the total coupling λ, supporting the idea that superconductivity originates from the hydrogen sublattice only, and not from a specific, strongly coupled mode.
All other La–N–H and La–B–H structures are undoubtedly not superhydrides, and given their small hydrogen fraction it is not surprising that they are not high-Tc superconductors.
To rule out that the discrepancy between predicted Tc and experiments may be due to the pressure shift introduced to empirically take into account the effect of quantum lattice fluctuations we recomputed the critical temperatures of LaBH8 and LaBH17 also at 150 GPa, i.e. below the maximum pressure reported in experiments. Here, LaBH8 is dynamically stable, with a Tc of 40 K, while LaBH17 has a weak dynamical instability; neglecting imaginary frequencies, the predicted Tc is 223 K, i.e. well below 550 K. We also performed a structural search at 150 GPa for the La–B–H and La–N–H systems, but could not find any other potential hot superconductor.
We can also estimate, albeit in a rather qualitative fashion, the effect of possible mechanisms that may positively influence the Tc of related ternary and multinary La–B–H phases. An obvious observation is that the Fermi energy in LaBH17 lies exactly in correspondence of a pseudogap. Carbon substitution at the boron site could sensibly enhance the Tc through charge doping; for example, a 50% replacement of boron with carbon would increase the DOS at the Fermi level by approximately a factor of two, and boost the Tc to about 290 K. If we assume that the Eliashberg function is rigidly multiplied by a factor equal to the increase in the DOS, in order to achieve 550 K one would need approximately an eight times larger DOS in LaBH8, and a four times larger DOS at the Fermi level in LaBH17. At most, a 100% substitution of boron with carbon would shift the Fermi energy enough to boost the DOS by a factor of two and a half, and the Tc to about 310 K, i.e. above room temperature, but well below 550 K.
Another possibility is to consider phases with a higher H content than LaBH17; the shape of the La–B-H ternary hull gave strong indications that they may form. In this case, one may speculate that average phonon frequencies may be increased compared to LaBH17, leading to an effective boost in Tc. However, even a doubling of all phonon frequencies of LaBH17, which is extremely unlikely, would be sufficient to bring the Tc only to 360 K. Similar arguments also apply for LaGaH14, as its Fermi energy is also situated in a pseudogap, and could be electron-doped via substitution of gallium with germanium, increasing the DOS at the Fermi level. However, even in the most optimistic scenario it would not be possible to reach the reported 550 K.
Outlook
In summary, following a recent experimental report of hot superconductivity at 550 K in a material with undetermined composition and structure15, we investigated from first-principles the high-pressure phase diagram of the most likely combinations of elements which could have formed, i.e. La–X–H ternary hydrides (X = B, N, Pt, Au, Ga, C), looking for a candidate to explain the experimental results. The choice of La-based ternary hydrides is motivated by the Tc measured after the first heating cycle, which is compatible with that of LaH10. As X element we considered all the elements that were reported to be present in the diamond anvil cell during the experiment: boron, nitrogen, hydrogen, and traces of platinum, gold, gallium and carbon.
In order to evaluate which ternary hydrides are thermodynamically favorable, we used variable-composition, evolutionary algorithms, at increasing levels of accuracy. According to our calculations, only La–N–H and La–B–H can form stable ternary phases, but formation of metastable La–Ga–H hydrides may be possible. Only in La–B–H and La–Ga-H do we predict the formation of H-rich, highly symmetric structures, which can host high-Tc superconductivity. In particular, we identified a high-Tc tetragonal LaBH17 phase, characterized by a dense hydrogen sublattice which is reminiscent of other high-Tc binary sodalite-like hydrides. For this structure we predicted a Tc of 180 K at 300 GPa by numerically solving the isotropic Migdal–Eliashberg equations. This result is way too far from the reported value of hot superconductivity to attribute the difference to numerical errors, and even within the most optimistic doping scenarios we could not increase Tc above 360 K. The discrepancy is too large for anharmonic lattice effects to affect our main conclusions.
While none of the binary or ternary phases of the elements considered in this work can explain the extreme Tcs reported, the La–B–H and La–Ga–H systems represent a very interesting starting point for further superconductivity studies. In fact, the extreme complexity of a ternary search limited our calculations in the maximum size of the unit cell, and the extent of the sampling for each composition, but our calculations suggest that the formation of high-H content La–B–H phases, or even quaternary phases involving lanthanum, hydrogen, and boron, nitrogen or gallium, with high Tc is definitely possible. Therefore, we urge the authors of ref. 15 to repeat their experiments under controlled conditions; it would be interesting, for example, to repeat the experiments employing diborane (B2H6), instead of ammonia borane as a hydrogen source, to discriminate between purely ternary La–B–H phases and quaternary La–B–N–H ones, or to intentionally include a larger amount of gallium in the cell.
We hope that a more precise determination of the critical temperature and a clearer indication of the elements and crystal structures will help elucidate the fascinating high-pressure physics of these systems.
Note: While this work was under review, we became aware of two other studies involving the La–B–H system28,36.
Methods
Structure search
To construct the phase diagrams, variable-composition structural searches were carried out using evolutionary algorithms as implemented in the USPEX software22,23. During the structural search, every structure was relaxed using a five-step process to minimize stress and forces, calculated within DFT. These calculations were performed using the Vienna ab initio Software Package37, using projector augmented waves pseudopotentials with Perdew–Burke–Ernzerhof exchange-correlation functional. We used a progressively tighter convergence, up to a cutoff on the plane waves expansion of 600 eV; a regular grid in \(\overrightarrow{k}\) space with a 0.04 spacing in units of \(\frac{2\pi }{\mathring{\rm A} }\), and a Methfessel–Paxton smearing with a width of 0.03 eV for reciprocal-space integration. The validity of the pseudopotentials was subject to an internal consistency check, as electronic structure and superconductivity calculations were carried out using Quantum ESPRESSO (QE) with Norm-Conserving pseudopotentials (see next Section). Therefore all the structures examined were re-relaxed using QE, with a different pseudopotential, and were always found to be already at the minimum.
The ternary hulls were constructed following a multi-step procedure similar to the one proposed in ref. 38:
-
1.
We performed a variable-composition evolutionary search with no restrictions, except for the maximum number of atoms in the unit cell, which was set to 20. For this search we considered 30 generations with 200 individuals in the first, and 60 individuals in each subsequent generation. While this level of accuracy is hardly enough to correctly identify the minima of the enthalpy for many compositions, it is sufficient to assess the possible formation of intermediate ternary phases.
-
2.
We extended our search to binary phases on the edges of the ternary hull, and for selected pseudo-binaries, the latter with a larger unit cell (up to 40 atoms). The pseudo-binary phases which were re-sampled are: (LaN)xH1−x, (LaN2)xH1−x, (LaN3)xH1−x, (LaH10)x(NH3)1−x, (LaH10)x(NH5)1−x, (LaH16)x(NH3)1−x, (LaH4)x(NH2)1−x, (LaB)xH1−x, (LaB3)xH1−x, (LaH2)x(BH)1−x, (LaH3)x(BH)1−x, (LaH4)x(BH)1−x, (LaH8)x(BH)1−x, (LaH10)x(BH)1−x, (LaH16)x(BH)1−x, (LaGa)xH1−x, and (LaPt)xH1−x
-
3.
Only for La–N–H and La–B–H. Motivated by the presence of stable ternary phases, we performed a final variable-composition evolutionary search, using all the stable structures found in the previous steps as seeds, with a maximum cell size of 40 atoms, a population size of 250 individuals, and 20 generations
-
4.
All enthalpies at the previous steps were collected, and the convex hulls in Fig. 1 were constructed using pymatgen39.
Where available, our convex hulls reproduce previous calculations for binary hydrides at comparable pressures8,21,40,41,42.
The figures of the crystal structures were generated using VESTA43.
Superconductivity
Calculations of electronic structure and electron-phonon properties were carried out in DFPT using QE33,44,45,46,47 using Optimized Norm-conserving Vanderbilt Pseudopotentials (ONCV)48. A cutoff of 80 Ry was used for the plane-wave expansion of the wave functions. The boron–carbon virtual crystal pseudopotentials were generated by mixing the two ONCV pseudopotentials, using the tools provided within QE.
The structures were re-relaxed in QE until each component of the forces acting on single atoms was less than 2 meV/Å. Calculations of the ground-state charge density were carried out using a 0.04 Ry smearing and a 6 × 6 × 6 grid in reciprocal space for \(\overrightarrow{k}\) space integration. Phonon calculations were performed on a 4 × 4 × 4 reciprocal-space grid for LaBH8, and a 2 × 2 × 2 grid for La4N4H, La2N2H, LaN2H3, LaB8H, La2B6H5, and a 3 × 3 × 3 grid for LaBH17, LaGaH6, LaGaH14, LaGaH15, and LaPtH6. The integration of the electron-phonon matrix elements on the Fermi surface was carried out using a 24 × 24 × 24 \(\overrightarrow{k}\) grid, and a gaussian smearing with a width of 200 meV to describe the zero-width limit of the electronic δ functions. The phonon DOS was obtained by performing Fourier interpolation on a 16 × 16 × 16 \(\overrightarrow{q}\) grid.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary information files. The authors also declare their availability to provide additional data and information upon request.
References
Einaga, M. et al. Crystal structure of the superconducting phase of sulfur hydride. Nat. Phys. 12, 835–838 (2016).
Duan, D. et al. Pressure-induced metallization of dense (S2H)2H2 with high-Tc superconductivity. Sci. Rep. 4, 6968 (2014).
Zurek, E. & Bi, T. High-temperature superconductivity in alkaline and rare-earth polyhydrides at high pressure: a theoretical perspective. J. Chem. Phys. 150, 050901 (2019).
Flores-Livas, J. A. et al. A perspective on conventional high-temperature superconductors at high pressure: Methods and materials. Phys. Rep. 856, 1–78 (2020).
Semenok, D. V., Kruglov, I. A., Savkin, I. A., Kvashin, A. G. & Oganov, A. R. On distribution of superconductivity in metal hydrides. Curr. Opin. Solid State Mater. Sci. 24, 100808 (2020).
Drodzov, A. P. et al. Superconductivity at 250 K in lanthanum hydride under high pressure. Nature 569, 528–531 (2019).
Somayazulu, M. et al. Evidence for superconductivity above 260 K in lanthanum superhydride at megabar pressures. Phys. Rev. Lett. 122, 027001 (2019).
Liu, H., Naumov, I. I., Hoffmann, R., Ashcroft, N. W. & Hemley, R. J. Potential high-Tc superconducting lanthanum and yttrium hydrides at high pressure. PNAS 114, 6990–6995 (2017).
Semenok, D. V. et al. Superconductivity at 161 K in thorium hydride thh10: synthesis and properties. Mat. Tod. 33, 36–44 (2020).
Troyan, I. A. et al. Anomalous high-temperature superconductivity in YH6. Adv. Mater. 33, 2006832 (2021).
Kong, P. P. et al. Superconductivity up to 243 K in yttrium hydrides under high pressure. Nat. Comm. 12, 5075 (2021).
Drodzov, A. P., Eremets, M. I. & Troyan, I. A. Superconductivity above 100 K in PH3 at high pressures. Preprint at https://arxiv.org/abs/1508.06224 (2015).
Snider, E. et al. Room-temperature superconductivity in a carbonaceous sulfur hydride. Nature 586, 373–377 (2020).
Sun, Y., Lv, J., Xie, Y., Liu, H. & Ma, Y. Route to a superconducting phase above room temperature in electron-doped hydride compounds under high pressure. Phys. Rev. Lett. 123, 097001 (2019).
Grockowiak, A. D. et al. Hot hydride superconductivity above 550 K. Preprint at https://arxiv.org/abs/2006.03004 (2020).
Boeri, L. & Bachelet, G. B. Viewpoint: the road to room-temperature conventional superconductivity. J. Phys.: Condens. Matter 31, 234002 (2019).
Shipley, A. M., Hutcheon, M. J., Needs, R. J. & Pickard, C. J. High-throughput discovery of high-temperature conventional superconductors. Phys. Rev. B 104, 054501 (2021).
Errea, I. et al. Quantum hydrogen-bond symmetrization in the superconducting hydrogen sulfide system. Nature 532, 81–84 (2016).
Errea, I. et al. Quantum crystal structure in the 250-Kelvin superconducting lanthanum hydride. Nature 578, 66–69 (2020).
Zurek, E., Hoffmann, R., Ashcroft, N. W., Oganov, A. R. & Lyakhov, A. O. A little bit of lithium does a lot for hydrogen. PNAS 106, 17640–17643 (2009).
Peng, F. et al. Hydrogen clathrate structures in rare earth hydrides at high pressures: possible route to room-temperature superconductivity. Phys. Rev. Lett. 119, 107001 (2017).
Glass, C. W., Oganov, A. R. & Hansen, N. USPEX–evolutionary crystal structure prediction. Comput. Phys. Commun. 175, 713–720 (2006).
Lyakhov, A. O., Oganov, A. R., Stokes, H. T. & Zhu, Q. New developments in evolutionary structure prediction algorithm uspex. Comput. Phys. Commun. 184, 1172–1182 (2013).
Teredesai, P. et al. High pressure phase transition in metallic LaB6: Raman and x-ray diffraction studies. Solid State Comm. 129 (2004).
Cava, R. J. et al. Superconductivity in lanthanum nickel boro-nitride. Nature 273, 245–247 (1994).
Cataldo, S. D., von der Linden, W. & Boeri, L. Phase diagram and superconductivity of calcium borohydrides at extreme pressures. Phys. Rev. B 102, 014516 (2020).
Kokail, C., von der Linden, W. & Boeri, L. Prediction of high-tc conventional superconductivity in the ternary lithium borohydride system. Phys. Rev. M 1, 074803 (2017).
Liang, X. et al. Prediction of high-Tc superconductivity in ternary lanthanum borohydrides. Phys. Rev. B, 134501 (2021).
Belli, F., Novoa, T., Contreras-Garcia, J. & Errea, I. Strong correlation between bonding network and critical temperature in hydrogen-based superconductors. Nat. Commun. 12, 1–11 (2021).
Borinaga, M., Errea, I., Calandra, M., Mauri, F. & Bergara, A. Anharmonic effects in atomic hydrogen: Superconductivity and lattice dynamical stability. Phys. Rev. B 93, 174308 (2016).
Azadi, S., Monserrat, B., Foulkes, W. M. C. & Needs, R. J. Dissociation of high-pressure solid molecular hydrogen: a quantum monte carlo and anharmonic vibrational study. Phys. Rev. Lett. 112, 165501 (2014).
Allen, P. B. & Dynes, R. C. Transition temperature of strong-coupled superconductors reanalyzed. Phys. Rev. B 12, 905 (1975).
Carbotte, J. P. Properties of boson-exchange superconductorsa. Rev. Mod. Phys. 62, 1027 (1990).
Heil, C., Cataldo, S. D., Bachelet, G. B. & Boeri, L. Superconductivity in sodalite-like yttrium hydrides. Phys. Rev. B 99, 220502(R) (2019).
Cataldo, S. D., Heil, C., von der Linden, W. & Boeri, L. LaBH8: towards high-tc low-pressure superconductivity in ternary superhydrides. Phys. Rev. B 104, L020511 (2021).
Zhang, Z. et al. Design principles for high temperature superconductors with hydrogen-based alloy backbone at moderate pressure. Preprint at https://arxiv.org/abs/2106.09879 (2021).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab-initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kvashin, A. G., Tantardini, C., Zakaryan, H. A., Kvashina, Y. A. & Oganov, A. R. Computational search for new w-mo-b compounds. Chem. Mater. 32, 7028–7035 (2020).
Ong, S. P. et al. Python materials genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Zhou, X.-F. et al. Superconducting high-pressure phase of platinum hydride from first principles. Phys. Rev. B 84, 054543 (2011).
Hu, C.-H. et al. Pressure-induced stabilization and insulator-superconductor transition of BH. Phys. Rev. Lett. 110, 165504 (2013).
Pickard, C. J. & Needs, R. J. Highly compressed ammonia forms an ionic crystal. Nat. Mater. 775–779 (2008).
Momma, K. & Izumi, F. Vesta: a three-dimensional visualization system for electronic and structural analysis. J. Appl. Cryst. 41, 653–658 (2008).
Giannozzi, P. et al. Quantum espresso: a modular and open-source software project for quantum simulation of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Baroni, S., de Gironcoli, S., Corso, A. D. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515 (2001).
Savrasov, S. Y. & Savrasov, D. Y. Electron-phonon interactions and related physical properties of metals from linear-response theory. Phys. Rev. B 54, 16487 (1990).
Giannozzi, P. et al. Advanced capabilities for materials modelling with quantum espresso. J. Phys. Condens. Matter 29, 465901 (2017).
Hamann, D. R. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 88, 085117 (2017).
Acknowledgements
We thank Antonio Sanna for kindly sharing with us the code for solving the isotropic Migdal–Eliashberg equations. The authors acknowledge computational resources from the dCluster of the Graz University of Technology and the VSC3 of the Vienna University of Technology, and support through the FWF, Austrian Science Fund, Project P30269-N36 (Superhydra). L.B. acknowledges funding through Progetto Ateneo Sapienza 2017-18-19 and computational Resources from CINECA, proj. Hi-TSEPH. S.D.C. acknowledges computational Resources from CINECA, proj. IsC90-HTS-TECH_C, and the dCluster of the Graz University of Technology.
Author information
Authors and Affiliations
Contributions
S.D.C performed the calculations. W.v.d.L. and L.B. supervised the project. All authors analyzed the results and contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Di Cataldo, S., von der Linden, W. & Boeri, L. First-principles search of hot superconductivity in La-X-H ternary hydrides. npj Comput Mater 8, 2 (2022). https://doi.org/10.1038/s41524-021-00691-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-021-00691-6
This article is cited by
-
Full-bandwidth anisotropic Migdal-Eliashberg theory and its application to superhydrides
Communications Physics (2024)
-
Quantum symmetrization transition in superconducting sulfur hydride from quantum Monte Carlo and path integral molecular dynamics
npj Computational Materials (2024)
-
Assessing the feasibility of near-ambient conditions superconductivity in the Lu-N-H system
Communications Materials (2024)
-
RETRACTED ARTICLE: Evidence of near-ambient superconductivity in a N-doped lutetium hydride
Nature (2023)
-
Quantum lattice dynamics and their importance in ternary superhydride clathrates
Communications Physics (2023)
