
Abstract
Databases for two-dimensional materials host numerous ferromagnetic materials without the vital information of Curie temperature since its calculation involves a manually intensive complex process. In this work, we develop a fully automated, hardware-accelerated, dynamic-translation based computer code, which performs first principles-based computations followed by Heisenberg model-based Monte Carlo simulations to estimate the Curie temperature from the crystal structure. We employ this code to conduct a high-throughput scan of 786 materials from a database to discover 26 materials with a Curie point beyond 400 K. For rapid data mining, we further use these results to develop an end-to-end machine learning model with generalized chemical features through an exhaustive search of the model space as well as the hyperparameters. We discover a few more high Curie point materials from different sources using this data-driven model. Such material informatics, which agrees well with recent experiments, is expected to foster practical applications of two-dimensional magnetism.
Similar content being viewed by others

Introduction
The recent experimental demonstration of ferromagnetism in two-dimensional (2D) materials: CrI31 and Cr2Ge2Te62 at low temperatures, has opened a new horizon of nanotechnology research since these materials inherit the potential to revolutionize engineering fields like spintronics3, valleytronics4, sensing and memory technologies5. In their classical work, Mermin and Wanger6 showed that under an isotropic Heisenberg model, long-range magnetic order must be absent in 2D. However, the more recent discovery of even room-temperature ferromagnetism in monolayer VSe27 and MnSe28 has been possible as the strong magnetocrystalline anisotropy of these 2D materials lifts the Mermin–Wanger restriction. So far, a plethora of 2D ferromagnetic (FM) materials9,10,11,12,13,14 have been computationally predicted, including a few general-purpose 2D materials databases15,16,17 containing hundreds to thousands of entries. However, none of these databases contain the most crucial parameter for 2DFM materials relevant for practical applications: the transition temperature or Curie point (TC). This is due to the fact that the computational determination of TC is a highly complex process, which involves a manual heuristics-based search for the ground-state and low-energy spin configurations. Identification of different magnetic exchanges (direct, super or double) within the neighbouring atoms and mapping them appropriately in a Monte Carlo based spin-flipping simulator has also been a manually intensive exercise. The choice of the model Hamiltonian (Ising instead of Heisenberg) used to simulate the spin-flipping with temperature, also raises a question on the reliability of the Curie temperatures of 2D materials predicted so far in the literature10,14.
Recently, an algorithm18 has been proposed which can search and predict the collinear, experimentally verified ground and low-energy spin states for bulk materials, almost optimally and exhaustively. Building on this, we develop a code, which performs first principles-based computations followed by Heisenberg model-based Monte Carlo simulations to predict the Curie point accurately from any magnetic 2D material crystal structure. Software engineering on this code makes it capable to execute such rigorous calculations in a high-throughput manner, even on a workstation-grade computer with GPU (graphical processing unit) acceleration. We use this code to determine the Curie points of materials from a suitable database16. To our surprise, almost 47% of the 786 materials classified as FM, turned out to be antiferromagnetic (AFM) upon close inspection by our code. The TC and other magnetic properties could be successfully determined for 157 materials, among which 26 materials reveal beyond 400 K Curie point. Close agreement with experimentally measured TC for a few materials validates our high-throughput methodology. In pursuit of faster discovery of high-TC materials, we further develop a machine-learning (ML) pipeline using these 157 data points. Using this ML model, we identify a few high TC 2DFM materials from the literature and other databases.
The informatics, which optimally balances the rigorousness and efficiency, gives us unprecedented opportunity to compare the magnetic properties of a very large number of materials with diverse structures, which may lead to many new insights on 2D magnetism. For example, we understand why the inclusion of the higher-order neighbours is important for TC calculation for certain materials and why it is not for the others. We observe several violations of the Goodenough–Kanamori19,20 rules for superexchange, the origin of which is open for further exploration. We also demonstrate that a machine-learning model can capture the complex process of temperature-dependent spin-flipping with exceptional accuracy. Our work thus significantly upgrades the computational materials toolbox to foster practical applications of two-dimensional magnetism.
Results
High-throughput computational framework
We first explain the workflow of our automated code as illustrated in Fig. 1. The unit cell of the material is first fed to a recently developed module18 of the open-source python library pymatgen21, which generates different FM and AFM spin configurations of the material based on symmetry analysis. Eliminating the heuristics-based approach, pymatgen not only helps to automate the process of the TC calculation, but also makes it more rigorous and thus reliable [see Methods]. The code-generated spin configurations for experimentally synthesized materials CrI31 and Cr2Ge2Te62 and newly predicted material Cr3Te410 are shown in Supplementary Figs. 1–3 as examples. These structures are then relaxed using collinear density functional theory with Hubbard correction (DFT + U) and their energies are calculated. At this stage, if the ground state is found to be AFM, the material is discarded. Here we also calculate the magnetic moment (µ) of each atom of the structure. Since the magnetocrystalline anisotropic energy (MAE) is essential for the existence of long-range magnetic order in 2D materials, it is calculated using non-collinear DFT including the effects of spin–orbit coupling (SOC) in the next step. These calculations also reveal the easy magnetization axis (EMA) of the 2DFM material, which can be important for specific applications. All these calculations provide us enough information to fit the DFT energy values to the following Heisenberg Hamiltonian:
Here, J1, J2, J3 and J4 are the nearest-neighbour (N1), 2nd nearest-neighbour (N2), 3rd nearest-neighbour (N3) and 4th nearest-neighbour (N4) exchange coupling constants and Si, Sj, Sl, Sm and Sn are the spins at sites i, j, l, m, and n, respectively. kx, ky, and kz are the magnetic anisotropy constants in the x, y and z directions. S is computed as µ/(2µB), where µ is the local magnetic moment of the magnetic ions.

The different building blocks of our end-to-end code for determining properties of 2DFM materials along with an estimation of CPU and GPU usage of each block.
For 2D materials, the classical Monte Carlo (MC) based solution of the Heisenberg Model is known to accurately predict the transition temperature22. First, the 2DFM unit cell is multiplied to make a supercell large enough to eliminate size effects, and using a GPU accelerated search, all the neighbours of all the sites are mapped into an 1D array structure. This is then used to perform a Heisenberg model-based classical MC simulation using a semi-compiled dynamic-translation based module, from which the TC of the material can be obtained. For a few materials, no in-plane anisotropy is observed which are classified as XY magnets. Mermin–Wagner theorem6 prohibits spontaneous symmetry breaking in these kinds of systems where the spin degree of freedom is ≤2. Instead, XY magnets exhibit a Berezinskii−Kosterlitz−Thouless (BKT) transition to a quasi-long-range ordered low-temperature phase. For these materials, TC is calculated from the following equation obtained from Monte Carlo simulations of the XY model23:
where kB is the Boltzmann constant and EFM and EAFM are energies of the FM and the most stable AFM solutions normalized by the number of atoms.
Though the Ising model can provide good results for materials with extremely high anisotropy, significant overestimation of TC may happen for materials with moderate to low anisotropy10,11,14. The anisotropic Heisenberg model takes care of this problem and effectively balances the contributions between exchange and anisotropy. However, it requires further MAE calculations and the MC simulation becomes computationally much more expensive. Although this is a high-throughput study, leveraging software engineering [see Methods], we decide to use the rigorous Heisenberg model without compromising the accuracy.
Database search
We identify the database C2DB16 as the ideal database to conduct our study based on the following reasons. (1) The authors have performed a preliminary classification between FM and AFM materials and a large number (786) of materials are classified as FM. (2) These materials have been explored by a “systematic combinatorial approach” where almost all known layered exfoliable materials are covered15, and by substituting the atoms, the authors have predicted a lot of new materials. This kind of variation is ideal to train machine-learning models, which is one of our primary goals. Also, recent synthesis of janus24 and other species substituted25 2D materials with no bulk analogues have made practical application of this kind of “synthetic” 2D materials possible. (3)The authors have calculated several properties of interest including thermodynamic stability and energy above the convex hull which helps us to estimate the chance of potential synthesis of these materials. The electronic structures have also been computed, which tells us about the presence of important properties like half-metallicity.
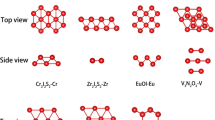
Interestingly, after a close examination by our code, 368 of the 786 materials classified as FM in the database turns out to be actually AFM. The pymatgen magnetism module explores the symmetry allowed spin-configuration space almost exhaustively, and in the process also explores large AFM supercells, which probably the authors of C2DB could not afford to do in their general-purpose study. A few discrepancies can also arise from the difference in DFT settings between the studies. Analyses of a considerable amount of materials have failed and thus have been discarded due to various computational limitations [see Methods]. Sheet 1 of Supplementary Spreadsheet 1 lists all the examined materials with FM/AFM classification as found by our method. In the end, the TC of 157 2DFM materials could be successfully computed, among which 12 materials are found to be XY magnets. These materials belong to more than 20 different prototype structures, which are shown in Fig. 2.

Top view of prototypes (according to C2DB) of all the 2DFM materials for which TC could be determined using our method. Blue balls represent the magnetic metal ions and the red and brown balls signify non-magnetic ions. Note that the grouped prototypes, e.g. FeOCl, FeSe, and NiO3 show the same structure cation-wise, but have different numbers of anions. However, all prototypes in such a group look similar from the top.
Discovery of high-T c materials
Almost all experimentally synthesized 2D materials are there in C2DB, except Cr2Ge2Te6, which too we have included manually in this set. We have not included those 2DFM materials whose ferromagnetism cannot be accurately modelled by the Heisenberg or the XY model, such as known itinerant material Fe3GeTe226,27. Sheet 1 of Supplementary Spreadsheet 2 lists all the calculated properties, as well as the computed Curie temperatures of these 2DFM materials. To include interactions of N neighbours in the Hamiltonian, N + 1 spin configurations are required, and the number of configurations generated by pymatgen is also listed in Supplementary Spreadsheet 2. Given enough configurations, we have included up to the 4th nearest-neighbour interaction in this work. The TCs are calculated using two methodologies: (1) commonly used nearest-neighbour approach: including only the N1 interaction and fitting the energies of the FM and the most stable AFM configurations, (denoted as ‘TC’ in the spreadsheet) and (2) multi-neighbour approach: interactions including up to N4 (listed under the ‘TC_exact’ column). Apparently, the TC_exact should provide a better estimate of the real TC of the material. However, to our surprise, we observe that TC, which is computationally much economic, is close to TC_exact in most of the cases, except a few as described below. (1) Materials with more than one distinct metal layer (prototypes CH, GaSe, CdI2-MXene, Ti2CO2, and Ti2CH2O2). For prototypes CH and GaSe, there is no layer containing anions between the metal layers, which gives rise to strong inter-layer direct exchange in addition to the intra-layer superexchange. In prototypes CdI2, Ti2CO2 and Ti2CH2O2, which are all MXenes, the distinct metal layers are connected by anionic layers, where both the inter-layer as well as intra-layer exchange interactions play a pivotal role in deciding the TC. Clearly, considering only the N1 interactions in these materials is not accurate enough, as reflected in the huge differences between TC and TC_exact. In passing, we note that, for a few MXenes, pymatgen could only generate two configurations, thus, the TC value has been repeated as TC_exact. (2) Materials with square or rectangular lattice (prototype FeOCl, FeSe, GeS2 and NiSe). Here, the N1s or N2s are the atoms situated in the diagonally opposite corners of the square or rectangle, where superexchange is expected to be feeble at best and only strong direct exchange could persist. The difference in distance between N1s and N2s are also very small in these materials, which again makes the inclusion of the higher neighbour interactions necessary [see Supplementary Figs. 4 and 5]. In a few materials (Janus) the effect of higher-order neighbours is not possible to take into account due to moderate distortion in the lattice and TC value has been repeated as TC_exact.
The calculated TC (TC_exact) by our automated code for the experimentally synthesized materials CrI31, Cr2Ge2Te62, and MnSe28 matches the experimental reports very closely without any manual tinkering of parameters, validating the generalization and accuracy of our method. For T-phase VSe2 our predicted TC is only 114.33 K, whereas room temperature ferromagnetism has been reported7. However, it must be noted that the authors have reported strong substrate dependence of the magnetism and TC in this study which explains this apparent discrepancy. Also, our code confirms the magnetism to be in-plane in this material which matches the experimental report. With Supplementary Table 1, Supplementary Note 1 and Supplementary Figs 1, 2, and 6, we explain in detail how our code works for 2 FM (CrI31, CrGeTe32) and 2 AFM (FePS328, NiPS329) experimentally synthesized 2D materials.
We finally discover a total of 26 materials with TC > 400 K and 32 materials with TC ≥ 300 K, making these materials suitable for practical device applications. Interestingly, many of these materials are known to show a “low” amount of magnetism in bulk forms, such as materials containing Rh, Ru, Mo, W, Sc, Ti, and Zr, which were ignored in previous heuristics-based searches10. However, our study suggests that the materials containing the above-mentioned metals can indeed show a “decent” (0.59–3.96 µB/atom) magnetic moment in 2D crystal form along with high-TC, possibly because of the enhanced electron localization. Also, for some of these materials the anisotropy is not great, but the difference in energy between the FM and AFM states, which ultimately translates to exchange parameters, helps to lift the TC beyond the room-temperature.
Since the magnetic properties of these materials can greatly depend on the value of U (Hubbard Correction), we also calculate the TC of the 26 promising high-TC materials with much more accurate material-specific U valuess30 [see Methods] and present the results in Sheet 3 of Supplementary Spreadsheet 2. Apart from a single material (MoIN_Pmmn), we observe that the TC of the rest of the materials remains either close to 400 K or becomes much higher than that. We also noticed in some cases the TC value has been significantly enhanced with the application of these tailor-made U values. Thus, we expect that some of the materials whose TC values fall in the 250–400 K range might exhibit much higher TC if it is calculated using material-specific U.
Machine-learning model
Due to an increasing amount of available data, machine-learning has recently found many applications in the field of solid-state materials science31. Very recently, training on about 2500 experimentally reported Curie temperature of bulk materials, accurate ML models to predict TC of bulk materials have been developed32. 2D magnetism is fundamentally different from the magnetism of the bulk materials as most of the time anisotropy does not play such a significant role there. Therefore, In this emerging field, one doesn’t have the luxury of a sufficient amount of data points to train on. Based on the 157 data points obtained from our database-search we develop a machine-learning model to predict the TC from the crystal structures. To decide the best model and features, we use the autoML library automatminer (https://github.com/hackingmaterials/automatminer). This tool takes structures as input and decorates the dataset with easily computable and chemically and physically meaningful features33. Then the dataset is cleaned and reduced and is sent to the autoML library TPOT34, which stochastically searches the model and the hyperparameter space using a genetic algorithm and finds the best model for the given dataset. After this extensive search [see Methods], we find an excellently fitted pipeline with average cross-validation (CV) score 94.57 K2, which is reported as the mean square error (MSE) on the training set. For the FM/AFM classification problem, we also try to find a suitable pipeline using the same method, with our examined 525 FM + AFM data points. The fitted pipeline reports an average CV score of a lowly 72.89% (accuracy) on the training set, which is understandable considering the complexity of the problem and the size and skewness of the data.
To test the generalization and predictive power of these ML pipelines, we construct a test set from reported 2DFM materials. Also, quite a few selected materials are included in the test set from a separate database17 with completely new structures and complex compositions. After inspecting these materials using our code, we find a few materials to be AFM which have been claimed as FM. This discrepancy can originate from the use of different DFT settings. This, along with fitting with a large number of spin configurations also causes a difference in fitted J values as observed for few materials (see Supplementary Fig. 3). Details of 22 materials identified as FM are provided in Sheet 2 of Supplementary Spreadsheet 2. The new prototypes encountered in this set are illustrated in Fig. 3.

Figure 4a illustrates the accuracy of the ML pipeline prediction against the DFT-MC calculated TC for all the train and test data. The ML predicted TC is also listed in the ‘TC_exact predicted’ column of Supplementary Spreadsheet 2. The distribution of absolute errors in train and test data have been plotted in Fig. 4b. Although the pipeline has fitted to train data with high accuracy, generalization to test data does not seem so well. The MSE value also turns out to be 30335.46 K2. Although the small train data size could be partially responsible for this, the main reason is possibly the introduction of unseen crystal structures. For instance, although we have not trained with even a single material containing La, the prediction for LaBr2 is exceptionally close probably because we had a lot of crystals with similar structures in our train data. The same argument can be applied to Mn2H2NO2 and Cr3Te4. The ML classifier pipeline has also been tried on the test data containing a total of 123 FM + AFM samples, which yields a decent 73.17% accuracy. Sheet 2 of Supplementary Spreadsheet 1 tabulates all the materials tried and their ultimate fate as well as the classification prediction.

a plot of DFT-MC calculated TC and machine learning predicted TC for all the train as well as test samples. The green line represents a perfectly accurate prediction line. b distribution (histogram plot) of the absolute error of the machine learning prediction for test and train data combined.
During this exercise, we identify CrO2 _P4/mmm and ZnNi2O5 as high-TC materials, while the claim of Cr3Te4 possessing high-TC has also been verified, albeit the Curie temperature turns out to be much lower than the reported Ising model predicted value10, but higher than the experimentally reported value for bulk Cr3Te4.
Violation of Goodenough-Kanamori rule
The classical Goodenough-Kanamori semi-empirical rules19,20 for magnetic materials essentially states that when the magnetic-ion-anion-magnetic-ion bond angle is close to 180°, a strong AFM structure due to superexchange should prevail, whereas if this angle is close to 90°, the material should show ferromagnetism. However, in this work, we notice quite a few violations of this rule. We have tabulated these bond angles for all the train and test materials in Supplementary Spreadsheet 2 and the apparent violation cases are marked in red. The most significant violation of the Goodenough-Kanamori criterion can be seen in the strong high-TC FM material CrO2 _P4/mmm, where the cation–anion–cation bond angle is precisely 180°. Although a full investigation is out of the scope of this work, it appears that these postulates were developed for bulk materials and are failing here because of highly covalent bonds of 2DFM materials. Especially CrO2 _P4/mmm exhibits a highly planar structure, and thus should manifest highly covalent bonds. However, this material seems to be dynamically unstable and possesses a much higher total energy compared to the experimentally available bulk phase which might make it unlikely to be experimentally synthesized9.
High-temperature structural stability
According to the thermodynamic stability classification in C2DB, we choose three highly stable high-TC materials, namely CrIN_Pmmm, RhCl2_C2/m, and Mn2H2CO2_P-3m1 along with newfound ZnNi2O5_Pmmn for further structural stability evaluation at high temperatures. An ab-initio molecular dynamics (AIMD) run at 400 K for a total of 6.5 ps is conducted for this. Although CrIN, RhCl2, and ZnNi2O5 retain their crystal structure during this, albeit with less crystallinity, the MXene Mn2H2CO2 starts to melt away just after 3 ps, rendering it unsuitable for practical applications. Supplementary Figs 7–10 show the structural differences resulted from the MD runs.
Discussion
In this study, we predict a total of 26 2D materials to have TC beyond 400 K. Many of these could be easily synthesizable, either by straight exfoliation from their bulk counterparts or by bottom-up chemical methods24,25. It is worth noting that, low thermodynamic stability does not necessarily mean the material would be unusable for practical applications. For instance, according to C2DB, Silicene is classified as a material with low thermodynamic stability. However, Silicene transistors have been demonstrated to work in room temperature35. Some of these materials screened by us even show dynamic instability. Again, many commonly used 2D materials, such as T-phase MoS2 showing dynamic instability36 in free-standing form stabilizes themselves on substrates through possible substrate interaction and even finds application in room-temperature devices37. Also, these materials can show charge density wave (CDW) characteristics and stabilize in a larger supercell with slight structural distortion7. However, CDW distortions can sometimes impact the magnetic order adversely38.
To summarize, using high-throughput automated codes and data-driven models, we thoroughly screen 2D materials databases and predict a host of 2DFM materials with high Curie point. With the emergence of novel synthesis techniques, these materials could indeed be of interest to experimentalists and engineers in terms of practical application in various devices. The ML model and the automated code developed in this work could find use in the community for rapid magnetic property prediction. The model complements the rigorous DFT-MC based code and if trained with sufficiently large datasets the model could eventually replace the code32. State-of-the-art software engineering enables us to achieve an optimal balance between rigor and computational efficiency, which is very important for reliable high-throughput material screening. As a result, we discover many important magnetic materials involving metals like Mo, W and Ti which have so far been ignored by heuristic-based formula-screening10.
Methods
Spin configurations generation
As mentioned before, the python library pymatgen21 has been extensively used in this study for generating spin configurations, managing, and parsing input-output files and performing the neighbour mappings. The python module ASE39 has also been used to parse the input structure files. To ensure the reliability and coverage of the pymatgen generated spin configurations, we manually verify that the code-generated spin configuration set almost always includes all heuristics-based configurations reported in the literature9,10,11,12,13,14. Often, the code generates even more unexplored but symmetrically valid configurations that we leverage to include a higher number of neighbour interactions for a more accurate prediction of the TC. At the same time, we ignore ferrimagnetic configurations since these are usually energetically highly unstable as well as asymmetric. For instance, in contrast to the previous report10, our TC prediction of Cr3Te4 is based on a larger number of symmetric FM and AFM configurations and thus seems to be much more accurate.
It is worth mentioning that since we cover such a huge variety of materials using an automated workflow, we use such values of various DFT and numerical parameters [see Supplementary Readme (readme file at OSF repository)], which would yield reasonable results for all materials. For instance, in case of CrI3, the default value of the parameter enum_prec = 0.001 (enum_precision_parameter in pymatgen) generates 3 configurations, where the Néel AFM configuration gets excluded (Supplementary Fig 1). But with enum_prec = 1e-7, all 4 configurations can be generated. For the first case, we obtain J1 = 2.78 and J2 = 0.43 meV/link, while the second set gives us almost identical values of J1 = 2.82, J2 = 0.41 and J3 = 0.009 meV/link and in both cases, we obtain the same TC. The Néel AFM solution turns out to be the most energetically unfavourable state and gets truncated18 by pymatgen-enumlib with default settings. For this high-throughput study, we find the default values to be accurate enough for our one-fits-all scheme. However, for focused studies on some specific material one might want to obtain extremely accurate results and thus might need to tune the parameters a little.
DFT parameters
Because the energy differences between various configurations for magnetism calculations could be as low as ≈µeV, the computations need to be performed with high accuracy. We use heavily modified versions of predefined configurations ‘MPRelaxSet’, ‘MPStaticSet’ and ‘MPSOCSet’ available in pymatgen for relaxations, static runs and MAE calculations. These modifications, as well as other details of DFT, are highlighted below.
Spin-polarized DFT calculations are carried out using generalized gradient approximation (GGA) as implemented in the code VASP40 with projector augmented-wave (PAW)41 method using the Perdew–Burke–Ernzenhof (PBE)42 exchange-correlation functional. Along with the CPU version, The GPU port43 of VASP has been used extensively. For all calculations, a correction on the strongly correlated d-shell electrons (GGA + U) is applied using the Dudarev44 formulation. The default value of the cut-off energy (520 eV) is used which proved to be sufficiently large. For relaxations, the default reciprocal density of 64 Å−3 is employed whereas for all collinear and non-collinear static runs a much denser reciprocal density of 300 Å−3 has been used. Electronic convergence is set to be attained when the difference in energy of successive electronic steps becomes less than 10−6 eV, whereas the structural geometry is optimized until the maximum Hellmann–Feynman force on every atom falls below 0.01 eV/Å. For the high-precision MAE calculations, a stricter electronic convergence criterion of 10−8 eV is imposed. A large vacuum space of >25 Å in the direction of c is applied to avoid any spurious interaction between periodically repeated layers. The Bader charge and magnetization analysis are performed using the code developed by the Henkelman group45, where charge densities generated from DFT static runs are used as inputs. These Bader partitioned magnetic moments have been used as the local magnetic moments of the magnetic elements. All crystal structure images are generated using the tool VESTA46.
For metals, Co, Cr, Fe, Mn, Mo, Ni, V and W, the effective U values have been taken from the Materials Project (https://wiki.materialsproject.org/GGA%2BU_calculations#Calibration_of_U_values) where these effective U values have been calibrated by performing a fitting to experimental binary formation enthalpies47. This is an established practice and has been used in similar high-throughput screening studies before10. We also find that the application of an effective U is essential to perform accurate DFT calculations on materials with “low” magnetization (materials containing Nb, Sc, Ru, Rh, Pd, Cu, Os, Ti, Zr, Re, Hf, Pt, and La) as the AFM solutions become difficult to obtain for these materials without a proper effective U. In these cases, for a specific metal, first the effective U values are obtained using the linear-response approach30 using 3 × 3 supercells for a few materials containing the element and then the average value of these effective U is taken as the final effective U. A complete list of effective U values (in eV) used in this study, as well as the DFT parameters imposed can be found in Supplementary Code 1 (e2e.py at OSF repository).
It is worth noting that, these effective U values depend on the element type, charge state and coordination mode of magnetic species in a certain material, which implies that the value of U is quite material-specific and should be determined carefully for accurate predictions. Thus, we also calculate the material-specific U values for the most promising 26 materials with predicted high-TC using the linear-response approach30. We observe that the U values obtained from the linear-response method can be quite different from the high-throughput U values we have used so far (see Sheet 3 of Supplementary Spreadsheet 2). However, the computational budget of the linear-response method is excessively high to be adopted for high-throughput material screening.
Hamiltonian fitting
The coupling constant (J) values are fitted using the collinear energy values of different FM and AFM spin configurations. However, for a lot of cases, when the energies of all configurations are taken, the determinant of the system of equations becomes zero which makes the set of equations unsolvable. As an automated remedy for these problems, again a fitting is tried omitting the most unstable AFM configuration and using one less neighbour than before. This process is repeated until a set of physically meaningful solutions is found, or the code runs out of configurations to fit. The anisotropic constant (k) values are fitted using the non-collinear energy values with spins oriented in different directions ([100], [010], [110], [001]).
Despite our best efforts, calculations for 261 materials (out of the 786 materials classified as FM in C2DB) had to be cancelled because of the following reasons: (1) pymatgen could not recognize the symmetry and could generate only one configuration, (2) material turned out to be non-magnetic after DFT calculations, (3) severe convergence issues occurred during DFT calculations, (4) AFM configurations could not be retained even after application of proper U and manual tuning of parameters, (5) the Hamiltonian could not be fitted properly, (6) phase change of crystal structure after relaxation.
Monte Carlo simulation
To study ferromagnetic (FM) to paramagnetic (PM) transition in these monolayer materials, Monte Carlo (MC) simulations of the Heisenberg model have been performed using the Metropolis algorithm with single-spin update scheme48. To eliminate the size effects, a 50 × 50 supercell containing 2500-8000 sites has been used to simulate the system. Total 105 Monte Carlo steps have been performed for each temperature, while the results from the first 104 steps have been discarded, as the system is allowed to equilibrate (thermalize) during this time. The final values of magnetization and susceptibility are calculated as the average over the last 9 × 104 MC steps for each temperature.
Software engineering
We develop the complete end-to-end code in python to take advantage of pymatgen. However, python being an interpreted language, the MC simulations turned out to be excessively slow, especially with high coordination numbers and inclusion of higher neighbours, which made the code unsuitable for the high-throughput study. As a remedy to this problem, we decide to use python-based just-in-time (JIT) compiler numba49 which compiles specific decorated python modules at the first encounter to low-level instructions, and when these modules are repeatedly called, the compiled version is used which makes the code extremely fast. However, the trade-off is, a lot of powerful functions and the coding flexibility offered by python (like heterogeneous data structures, appending to a list) cannot be successfully compiled and significant software engineering, as well as timing and cost-benefit analyses, are required to achieve an optimal code. GPU acceleration for neighbour-mapping of large lattices (2500–8000 sites) has also been implemented, which on a CPU must be done serially and takes a lot more time.
The engineered code was optimized to such an extent that the whole study could be performed using even a workstation-grade machine, albeit with GPU acceleration. A video of real-time execution of the code, where the TC of four materials (CrI3, Cr2Ge2Te6, MnSe2, and MoC3) are being calculated in parallel in a single-CPU (18 cores), three-GPU enabled workstation within ≈10 hour, can be found at https://youtu.be/HJkR-03OzBI. At the same time excellent scalability is observed, when the code is executed on a high performance computing node (https://youtu.be/GQaFfm29LR4).
Machine learning
The python libraries automatminer and matminer33 have been used to featurize the datasets and search for optimal ML pipeline for the FM/AFM classification problem as well as the TC predicting regression problem. Dataset cleaning and feature reduction are handled by automatminer. Then, various pre-processing algorithms along with a host of commonly used ML models employed for materials science problems31 for small to moderate datasets have been searched by the autoML library TPOT34, such as naive Bayesian, decision tree, extra trees, random forest, gradient boosting, k-neighbors, linear SVC and logistic regression for classification and elastic net CV, decision tree, extra trees, random forest, gradient boosting, k-neighbors, lasso lars CV, ridge CV and linear SVR for regression. Also, the hyperparameters are tuned at the same time. A full list of pre-processing algorithms, ML models and hyperparameters searched can be found at https://github.com/hackingmaterials/automatminer/blob/master/automatminer/automl/config/tpot_configs.py. It is worth noting that TPOT uses the ML library scikit-learn50 for the ML as well as the feature-engineering models. For each autoML search, more than 60,000 pipelines have been explored.
The generic python codes using scikit-learn for the best pipelines for both FM/AFM classification and TC regression have been given in Supplementary Codes 2 and 3 (TPOT_*.py at OSF repository). For the former, a combination of SelectPercentile, MaxAbsScaler, and ExtraTreesClassifier turns out to be the best pipeline, whereas for the latter it is a combination of SelectPercentile, ZeroCount, and GradientBoostingRegressor. The list of features used for these problems can be found in Supplementary Logs 1 and 2 (*_digest at OSF repository). Moreover, the pickled pipelines have also been provided in Supplementary ML Pipes 1 and 2 (*.pipe at OSF repository) which can be loaded into automatminer to make predictions on any dataset. The Supplementary Readme file provides detailed instructions on how to use the Supplementary codes and pipes.
AIMD simulations
For the chosen materials, to minimize the temperature oscillations a large supercell containing ≥144 atoms has been constructed to run the AIMD on. Because of severe convergence issues, non-spin-polarized DFT calculations are performed with Gamma-point only sampling which can be sufficient to determine structural stability. A canonical ensemble (NVT) is used and a Nosé-Hoover thermostat51,52 at 400 K is employed. The simulations run for 6.5 ps with a 2 fs time step. The crystal structures of the tested materials after the simulation can be seen in Supplementary Figs 7–10.
Data availability
The authors declare that the main data supporting the findings of this study are available within the paper and its Supplementary files, the OSF repository (https://osf.io/6ebjp/) and other open online resources. Other relevant data are available from the corresponding author upon reasonable request.
Code availability
All relevant python codes and pickled ML pipelines are provided in the OSF Repository (https://osf.io/6ebjp/). Also, the end-to-end code for Curie point determination (e2e.py) is available at GitHub (https://github.com/NSDRLIISc/e2e), which we plan to update periodically.
References
Huang, B. et al. Layer-dependent ferromagnetism in a van der Waals crystal down to the monolayer limit. Nature 546, 270–273 (2017).
Gong, C. et al. Discovery of intrinsic ferromagnetism in two-dimensional van der Waals crystals. Nature 546, 265 (2017).
Feng, Y. P. et al. Prospects of spintronics based on 2D materials. Wiley Interdiscip. Rev. Comput. Mol. Sci. 7, e1313 (2017).
Farooq, M. U. & Hong, J. Switchable valley splitting by external electric field effect in graphene/CrI3 heterostructures. npj 2D Mater. Appl. 3, 3 (2019).
Soumyanarayanan, A., Reyren, N., Fert, A. & Panagopoulos, C. Emergent phenomena induced by spin-orbit coupling at surfaces and interfaces. Nature 539, 509–517 (2016).
Mermin, N. D. & Wagner, H. Absence of ferromagnetism or antiferromagnetism in one- or two-dimensional isotropic Heisenberg models. Phys. Rev. Lett. 17, 1133–1136 (1966).
Bonilla, M. et al. Strong room-temperature ferromagnetism in VSe2 monolayers on van der Waals substrates. Nat. Nanotechnol. 13, 289–293 (2018).
O’Hara, D. J. et al. Room temperature intrinsic ferromagnetism in epitaxial manganese selenide films in the monolayer limit. Nano Lett. 18, 3125–3131 (2018).
van Gog, H. et al. Thermal stability and electronic and magnetic properties of atomically thin 2D transition metal oxides. npj 2D Mater. Appl. 3, 18 (2019).
Zhu, Y., Kong, X., Rhone, T. D. & Guo, H. Systematic search for two-dimensional ferromagnetic materials. Phys. Rev. Mater. 2, 81001 (2018).
Kumar, H. et al. Tunable magnetism and transport properties in nitride MXenes. ACS Nano 11, 7648–7655 (2017).
Jiang, Z., Wang, P., Xing, J., Jiang, X. & Zhao, J. Screening and design of Novel 2D ferromagnetic materials with high curie temperature above room temperature. ACS Appl. Mater. Interfaces 10, 39032–39039 (2018).
Zhuang, H. L., Xie, Y., Kent, P. R. C. & Ganesh, P. Computational discovery of ferromagnetic semiconducting single-layer CrSnTe3. Phys. Rev. B 92, 35407 (2015).
Miao, N., Xu, B., Zhu, L., Zhou, J. & Sun, Z. 2D intrinsic ferromagnets from van der Waals antiferromagnets. J. Am. Chem. Soc. 140, 2417–2420 (2018).
Mounet, N. et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Nat. Nanotechnol. 13, 246–252 (2018).
Haastrup, S. et al. The Computational 2D Materials Database: high-throughput modeling and discovery of atomically thin crystals. 2D Mater. 5, 42002 (2018).
Zhou, J. et al. 2DMatPedia, an open computational database of two-dimensional materials from top-down and bottom-up approaches. Sci. Data 6, 86 (2019).
Horton, M. K., Montoya, J. H., Liu, M. & Persson, K. A. High-throughput prediction of the ground-state collinear magnetic order of inorganic materials using density functional theory. npj Comput. Mater. 5, 64 (2019).
Goodenough, J. B. An interpretation of the magnetic properties of the perovskite-type mixed crystals La1−xSrxCoO3−λ. J. Phys. Chem. Solids 6, 287–297 (1958).
Kanamori, J. Superexchange interaction and symmetry properties of electron orbitals. J. Phys. Chem. Solids 10, 87–98 (1959).
Ong, S. P. et al. Python Materials Genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Torelli, D. & Olsen, T. Calculating critical temperatures for ferromagnetic order in two-dimensional materials. 2D Mater. 6, 15028 (2018).
Ashton, M. et al. Two-dimensional intrinsic half-metals with large spin gaps. Nano Lett. 17, 5251–5257 (2017).
Zhang, J. et al. Janus monolayer transition-metal dichalcogenides. ACS Nano 11, 8192–8198 (2017).
Sreepal, V. et al. Two-dimensional covalent crystals by chemical conversion of thin van der Waals materials. Nano Lett. 19, 6475–6481 (2019).
Fei, Z. et al. Two-dimensional itinerant ferromagnetism in atomically thin Fe3GeTe2. Nat. Mater. 17, 778–782 (2018).
Zhuang, H. L., Kent, P. R. C. & Hennig, R. G. Strong anisotropy and magnetostriction in the two-dimensional Stoner ferromagnet Fe3GeTe2. Phys. Rev. B 93, 134407 (2016).
Wang, X. et al. Raman spectroscopy of atomically thin two-dimensional magnetic iron phosphorus trisulfide (FePS3). Cryst. 2D Mater. 3, 31009 (2016).
Kim, K. et al. Suppression of magnetic ordering in XXZ-type antiferromagnetic monolayer NiPS3. Nat. Commun. 10, 345 (2019).
Cococcioni, M. & de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA+U method. Phys. Rev. B 71, 35105 (2005).
Schmidt, J., Marques, M. R. G., Botti, S. & Marques, M. A. L. Recent advances and applications of machine learning in solid-state materials science. npj Comput. Mater. 5, 83 (2019).
Nelson, J. & Sanvito, S. Predicting the Curie temperature of ferromagnets using machine learning. Phys. Rev. Mater. 3, 104405 (2019).
Ward, L. et al. Matminer: an open source toolkit for materials data mining. Comput. Mater. Sci. 152, 60–69 (2018).
Olson, R. S., Bartley, N., Urbanowicz, R. J. & Moore, J. H. Evaluation of a tree-based pipeline optimization tool for automating data science. In Proc Genetic and Evolutionary Computation Conference 2016 (ed. Friedrich, T.) 485–492 (ACM, 2016).
Tao, L. et al. Silicene field-effect transistors operating at room temperature. Nat. Nanotechnol. 10, 227–231 (2015).
Saha, D. & Mahapatra, S. Atomistic modeling of the metallic-to-semiconducting phase boundaries in monolayer MoS2. Appl. Phys. Lett. 108, 253106 (2016).
Kappera, R. et al. Phase-engineered low-resistance contacts for ultrathin MoS2 transistors. Nat. Mater. 13, 1128 (2014).
Zheng, F., Zhou, Z., Liu, X. & Feng, J. First-principles study of charge and magnetic ordering in monolayer NbSe2. Phys. Rev. B 97, 81101 (2018).
Hjorth Larsen, A. et al. The atomic simulation environment—a Python library for working with atoms. J. Phys. Condens. Matter 29, 273002 (2017).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Hacene, M. et al. Accelerating VASP electronic structure calculations using graphic processing units. J. Comput. Chem. 33, 2581–2589 (2012).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Hu, L. et al. Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds. Phys. Rev. B 6, 1–7 (2017).
Momma, K. & Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr 44, 1272–1276 (2011).
Wang, L., Maxisch, T. & Ceder, G. Oxidation energies of transition metal oxides within the GGA+U framework. Phys. Rev. B 73, 195107 (2006).
Vatansever, E. et al. Strain effects on electronic and magnetic properties of the monolayer α - RuCl3: A first-principles and Monte Carlo study. J. Appl. Phys. 125, 83903 (2019).
Lam, S. K., Pitrou, A. & Seibert, S. Numba: a LLVM-based Python JIT Compiler. In Proc. 2nd Workshop on the LLVM Compiler Infrastructure in HPC (ed. Finkel, H.) 7:1–7:6 (ACM, 2015).
Pedregosa, F. et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985).
Acknowledgements
The authors are grateful to Dr. Naihua Miao, Beihang University and Prof. Ümit Akıncı, Dokuz Eylül University for several insightful discussions. Authors also thank Abdul Samadh, Manipal University, and Kota Hemanth, IIT Delhi, for their help with software engineering and machine learning. The authors thank Supercomputer Education and Research Centre (SERC), Indian Institute of Science (IISc) Bangalore, for CPU based computations. The GPU computing nodes used in this study were procured through the research funding from the Department of Science and Technology (DST), Government of India, under grant number DST/TMD/MES/2K18/28.
Author information
Authors and Affiliations
Contributions
A.K. developed the automated end-to-end code, implemented it on CPU and GPU based architectures, developed the ML models and analysed the data. M.K. scanned the materials using the automated code, collected and verified the results. S.M. conceived the problem statement and overall supervised the work. All authors contributed to the writing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kabiraj, A., Kumar, M. & Mahapatra, S. High-throughput discovery of high Curie point two-dimensional ferromagnetic materials. npj Comput Mater 6, 35 (2020). https://doi.org/10.1038/s41524-020-0300-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-020-0300-2
This article is cited by
-
Realizing unipolar and bipolar intrinsic skyrmions in MXenes from high-fidelity first-principles calculations
npj Computational Materials (2023)
-
High-throughput computation and structure prototype analysis for two-dimensional ferromagnetic materials
npj Computational Materials (2022)
-
High-throughput design of functional-engineered MXene transistors with low-resistive contacts
npj Computational Materials (2022)
-
High-throughput inverse design and Bayesian optimization of functionalities: spin splitting in two-dimensional compounds
Scientific Data (2022)
-
Describing chain-like assembly of ethoxygroup-functionalized organic molecules on Au(111) using high-throughput simulations
Scientific Reports (2021)
