
Abstract
The accelerated growth rate of repository entries in crystallographic databases makes it arduous to identify and classify their prototype structures. The open-source AFLOW-XtalFinder package was developed to solve this problem. It symbolically maps structures into standard designations following the AFLOW Prototype Encyclopedia and calculates the internal degrees of freedom consistent with the International Tables for Crystallography. To ensure uniqueness, structures are analyzed and compared via symmetry, local atomic geometries, and crystal mapping techniques, simultaneously grouping them by similarity. The software (i) distinguishes distinct crystal prototypes and atom decorations, (ii) determines equivalent spin configurations, (iii) reveals compounds with similar properties, and (iv) guides the discovery of unexplored materials. The operations are accessible through a Python module ready for workflows, and through command line syntax. All the 4+ million compounds in the AFLOW.org repositories are mapped to their ideal prototype, allowing users to search database entries via symbolic structure-type. Furthermore, 15,000 unique structures — sorted by prevalence — are extracted from the AFLOW-ICSD catalog to serve as future prototypes in the Encyclopedia.
Similar content being viewed by others
Introduction
Scientists have been struggling for decades to identify prototypes (e.g. Strukturbericht series1 and Pearson’s Handbook2) and duplicates in crystallographic databases; and to label structures in a concise way to recognize (and enable searching by) structure-types. The recent rapid growth of online repositories has worsened the problem3. Distinguishing distinct crystalline compounds is becoming increasingly difficult, leading to the repetition of previously studied materials, hindering database variety — biasing data-driven analyses and machine learning methods4,5,6 — and wasting valuable computational and experimental resources. The multitude of crystal geometries make by-hand detection of prototypes and repeated entries intractable. A major complication for finding structure-types is the non-standard representation of crystals. Determination of unique crystallographic structures is obfuscated by (i) unit cell representations and (ii) origin choices. While standard forms exist — such as Niggli7 and Minkowski8 unit cells — the conversion procedures are highly sensitive to numerical tolerance values and can cast similar structures into differing descriptions9,10. Additionally, lattice standardization techniques do not address differences in origin choices. The lack of commensurate representations impedes the search for prototypes and inhibits mappings between similar crystals and their corresponding properties. To overcome non-standard descriptions, crystal comparison tools have been developed to identify similar structures. Programs such as Structure Matcher11, XTALCOMP10, SPAP12, CMPZ13, CRYCOM9, STRUCTURE-TIDY14, and COMPSTRU15 are available with varying objectives related to structure comparison. For instance, XTALCOMP is coupled with the XTALOPT infrastructure for identifying distinct materials generated via their evolutionary algorithm16. Despite the considerable number of platforms, none are suitable for autonomous prototype detection. Crystallographic symmetry is neglected in Structure Matcher, XTALCOMP, and SPAP; while STRUCTURE-TIDY, CRYCOM, and COMPSTRU rely on external symmetry packages. Additionally, most tools only feature single pairwise comparisons (with the exception of Structure Matcher) and others require additional inputs (e.g. space group, Wyckoff positions, and unit cell choice). Aside from technical functionality, the codes do not offer built-in methods to compare structures to existing crystallographic libraries and material repositories. To promote materials discovery, routines must analyze compounds with respect to established prototypes to identify new structure-types. This would enable the expansion of prototype libraries — such as the AFLOW Prototype Encyclopedia (or Prototype Encyclopedia for brevity)17,18 — fueling the generation of unique compounds via prototype decoration. Comparing compounds to those in materials databases can prevent duplication. Moreover, the properties of database entries can be used to estimate those of similar uncalculated compounds, exploiting the structure-property relationship of materials. Clearly, an automatic and reliable large-scale method for discerning unique crystallographic structures is therefore crucial for the materials science community.
AFLOW-XtalFinder (AFLOW crystal finder, XtalFinder for brevity) addresses many of the previously mentioned issues in a high-throughput fashion. The primary objective of XtalFinder is to identify/classify the prototypes of materials and relate them via structural similarity metrics. To accomplish this, XtalFinder determines the ideal prototype designation of crystal structures, consistent with the International Tables for Crystallography (ITC)19. Any structure in this representation can be automatically generated via a symbolic prototype generator. Similarity between structures is analyzed on multiple fronts. Crystallographic structures are first compared by symmetry (isopointal analysis), leveraging a robust software implementation, AFLOW-SYM, which calculates self-consistent symmetry descriptions freeing the user from tolerance adjustments20. Local atomic geometries are also computed to match neighborhoods of atoms in crystals (isoconfigurational snapshots). Finally, crystal similarity is resolved by rigorous structure mapping procedures (complete isoconfigurational analysis) and quantified via a misfit criterion21. The prototype finder accommodates automatic workflows, with the functionality to analyze multiple materials/structures simultaneously via multithreading. Features are provided to identify crystallographic structures, distinct materials, atom decorations, and spin configurations. Methods are also included to compare compounds/prototypes to the AFLOW.org repository and AFLOW prototype libraries. Every entry in the AFLOW.org repository has been mapped to its prototype label, enabling users to search the database by structure-type. The XtalFinder code — written in C++ — is part of the AFLOW (Automatic flow) framework22,23,24,25 and is open-source under the GNU-GPL license. For seamless integration into different work environments, this functionality is accessible via the command-line and a Python module.
Results
Problem of the ideal prototype
Prototype structures are generally classified in terms of their symmetry characteristics. For example, the rocksalt prototype has a face-centered cubic lattice and 8 atoms in the conventional cell (i.e. Pearson symbol of cF8), space group \(Fm\bar{3}m\) (#225), and Wyckoff positions \(4\ \ a\ \ m\bar{3}m\) and \(4\ \ b\ \ m\bar{3}m\). Determining this information for any arbitrary structure is often a challenge: numerical noise in the atomic positions inhibits the detection of crystal isometries, requiring by-hand modification of tolerance thresholds. Furthermore, consistency between real- and reciprocal-space symmetries is often overlooked, and yet it is imperative for reliable ab initio simulations. Thus, accurate prototype detection relies on robust symmetry analyses.
XtalFinder employs a self-consistent mechanism to find the ideal prototype of a given structure. The space group, Pearson symbol, and occupied Wyckoff positions are calculated via the AFLOW-SYM routines20. The prototype classification is sensitive to the symmetry tolerance (ϵsym). For example, the AlCl structure (ICSD #56541, DFT-relaxed) in Fig. 1(a) can be classified as one of six different prototypes as a function of ϵsym: (i) mP4, SG #11 (0 < ϵsym≤0.25 Å); (ii) mC4, SG #12 (0.26≤ϵsym≤0.27 Å); (iii) oI4, SG #71 (0.34≤ϵsym≤0.36 Å); (iv) tI4, SG #139 (0.37≤ϵsym≤0.44 Å); (v) hR6, SG #166 (0.45≤ϵsym≤0.49 Å); and (vi) cF8, SG #225 (0.51≤ϵsym≤1.0 Å). For certain tolerance values — e.g. 0.27 < ϵsym < 0.34 Å and 0.49 < ϵsym < 0.51 Å — incommensurate symmetry descriptions are calculated. To overcome this, the symmetry tolerance is automatically changed, scanning tighter and looser tolerances around the initial value, to find consistent symmetry descriptions at a new ϵsym. This autonomous approach ensures prototype classifications are correct and compatible against all symmetry descriptors (e.g. space group, Wyckoff position, lattice type, Brillouin zone, etc.).

a The prototype for an input structure, e.g. AlCl (ICSD #56541), is identified by analyzing its symmetry. Classification of the prototype may change depending on the symmetry tolerance (ϵsym): mP4, SG #11 (0 < ϵsym ≤ 0.25 Å); mC4, SG #12 (0.26 ≤ ϵsym ≤ 0.27 Å); oI4, SG #71 (0.34 ≤ ϵsym ≤ 0.36 Å); tI4, SG #139 (0.37 ≤ ϵsym ≤ 0.44 Å); hR6, SG #166 (0.45 ≤ ϵsym ≤ 0.49 Å); and cF8, SG #225 (0.51 ≤ ϵsym ≤ 1.0 Å). An adaptive routine is employed for tolerance regions with incommensurate symmetry descriptions (gray arrows for 0.27 < ϵsym < 0.34 Å and 0.49 < ϵsym < 0.51 Å), ensuring self-consistent prototype/symmetry designations. b The structure is then mapped into its prototype label and symbolic and numeric internal degrees of freedom (dof), consistent with the International Tables for Crystallography (ITC). Structures in this representation can be generated with the symbolic prototype generator.
The default symmetry tolerance value for classifying prototypes in XtalFinder is proportional to the minimum interatomic distance (\({d}_{{\rm{nn}}}^{\min }\)/100). The tolerance is thus system-specific, and it has been shown to be consistent with experimentally resolved symmetries20. Nevertheless, the tolerance can also be adjusted by the user, and is guaranteed to return a commensurate designation due to the adaptive prototype protocol shown in Fig. 1a.
Once the symmetry attributes of the crystal are calculated, XtalFinder automatically maps the structure to its AFLOW prototype label and symmetry-based degrees of freedom (Fig. 1b), i.e. lattice parameters/angles and non-fixed Wyckoff coordinates17,18. These designations are commensurate with the ITC cell choices and Wyckoff positions; the de facto standard for crystallography. The label specifies the stoichiometry and symmetry of the structure in underscore-separated fields. The fields indicate the following (example system: esseneite structure, ABC6D2_mC40_15_e_e_3f_f17)
-
first field: the reduced stoichiometry based on alphabetic ordering of the compound, e.g. a quaternary with stoichiometry ABC6D2,
-
second field: the Pearson symbol, e.g. mC40,
-
third field: the space group number, e.g. space group #15,
-
fourth field: the Wyckoff letter(s) of the first atomic site, e.g. site A: one Wyckoff position with letter e,
-
fifth field: the Wyckoff letter(s) of the second atomic site, e.g. site B: one Wyckoff position with letter e,
-
sixth field: the Wyckoff letter(s) of the third atomic site, e.g. site C: three Wyckoff positions with letters f, and
-
seventh field: the Wyckoff letter(s) of the fourth atomic site, e.g. site D: one Wyckoff position with letter f.
The prototype parameters specify the degrees of freedom allowed by the symmetry of the structure. For the esseneite structure, there are 18 parameters: a, b/a, c/a, β, y1, y2, x3, y3, z3, x4, y4, z4, x5, y5, z5, x6, y6, and z6. The first three variables are the lattice parameters — with b and c represented in relation to a — the fourth variable is the lattice angle β, and the subsequent variables are the Wyckoff coordinates (fractional) that are not fixed by symmetry. The sequence of the Wyckoff parameters is based on the alphabetic ordering of the Wyckoff letters, followed by alphabetic ordering of the species. Additional information regarding the label and parameters are discussed in the refs. 17,18.
Mapping structures into this format characterizes prototypes in a concise and descriptive manner. The representation also easily distinguishes isopointal and isoconfigurational prototypes. Two compounds with similar labels are isopointal (i.e. same symmetry), and are isoconfigurational if their parameters are the same (i.e. equivalent geometric configurations). However, a strict parameter comparison does not distinguish isoconfigurational structures, e.g. parameters may differ by an origin shift. Moreover, the representation reveals the degrees of freedom that can be altered, while preserving the underlying symmetry. This is useful for showing continuous structure transitions within the same symmetry-type and performing symmetry-constrained structure relaxations26. Lastly, with this format, structures are now easily regenerated with the AFLOW software.
Symbolic prototype generator
Structures represented in the ideal prototype designation can be created and decorated with any atomic elements via a symbolic prototype generator, enabling automatic materials design. The procedure introduced in refs. 17,18 has been extended to create all possible prototype structures, going beyond those previously described in the Prototype Encyclopedia. Given a crystal’s composition, Pearson symbol, space group, and occupied Wyckoff positions, the generator determines the degrees of freedom in symbolic notation (i.e. a, b/a, c/a, α, β, γ, x, y, and z) that must be specified, based on the ITC conventions19. Feeding in the ideal prototype label and degrees of freedom to the symbolic generator will produce the corresponding geometry file, substituting the appropriate degrees of freedom with the input values. Prototypes, including those in the Prototype Encyclopedia, no longer need to be tabulated (hard-coded) in the AFLOW software, and are now created on-the-fly. With this prototype generator, AFLOW is capable of creating structures to span all regions of crystallographic space.
Structures are generated with the following prototype command syntax: --proto=label --params=parameter_1,parameter_2,.... Here, the label is the ideal prototype label, e.g. AB_mP4_11_e_e as shown in Fig. 1(b), and parameter_1,parameter_2,... are the comma-separated values for the prototype’s degrees of freedom, e.g. 5.586, 0.719, 0.698, 91.992, 0.252, 0.234, 0.751, and 0.261 as shown in Fig. 1b. By default, structures are generated with fictitious species in alphabetical order (i.e. A, B, C, D, etc.). Users can override this order by specifying other permutations after the prototype label (separated by a period), i.e. --proto=label.BAC...; a useful feature for controlling the atomic site decorations. Specific elements can be decorated onto the prototype by appending the element abbreviations to the command in colon-separated alphabetical order, e.g. --proto=label:Ag:Cu:Zr. The generator checks for any inconsistencies with the provided label and/or parameter values, terminating prematurely with a message listing possible fixes to the command. The generator supports multiple geometry file formats, including VASP (POSCAR)27, FHI-AIMS28, QUANTUM ESPRESSO29, ABINIT30, ELK31, and CIF. Swapping the command --proto=label with --aflow_proto=label, will build an aflow.in file, AFLOW’s input file (using a standard set of DFT parameters by default32), automating ab initio simulations of these compounds.
The generator can also print the symbolic representation of the lattice and Wyckoff positions. Adding the option --add_equations to the prototype command returns both a numerical and symbolic version of the geometry file, and the option --equations_only only prints the symbolic version. Symbolic geometry files can be printed with respect to the conventional cell (ITC) or symbolically transformed into the primitive cell (using the SymbolicC++ open-source software33). By default, AFLOW provides the primitive cell, since fewer-atom unit cells are more computationally efficient.
With a robust prototype classifier and generator in place, comparison of prototypes is required to (i) identify unique structure-types and (ii) group similar ones together. The prototype label and parameters alone cannot establish structural similarity due to variations in the choice of lattice and origin, potentially affecting both the label (e.g. Wyckoff letters) and the parameters (e.g. lattice and non-fixed Wyckoff parameters). Therefore, XtalFinder offers three levels of comparison: symmetry, local atomic geometry, and complete crystal geometry. They are described in the following three subsections.
Isopointal structures: compare symmetry
Symmetry analyses of crystals are required to identify structures of the same symmetry-type. The isometries of crystals (e.g. rotations, roto-inversions, screw axes, and glide planes) are calculated via the routines of AFLOW-SYM20 to determine the space group and occupied Wyckoff positions (Fig. 2a). Results from AFLOW-SYM are robust against numerical tolerance issues and are consistent with experimentally determined symmetries in comparison to other symmetry software20.

a Crystal isometries are calculated internally with AFLOW-SYM. The space groups and occupied Wyckoff positions are compared, revealing isopointal structures. b The local least-frequently occurring atom (LFA) geometries are computed and compared between structures. An example local LFA geometry (2-D projection) is shown for the quaternary Heusler structure (ABCD_cF16_216_c_d_b_a)17,18, highlighting the closest neighbors (via solid lines) for each LFA type to the central Mg atom (purple). Shaded concentric circles indicate the tolerance threshold for capturing atoms in the coordination shell with a thickness of 10% of the distance from the central and connected atom. Local geometry vectors are compared against local geometries in other structures to determine mapping potential. c Two structures (\({{\mathbb{X}}}_{{\rm{ref}}}\) and \({{\mathbb{X}}}_{{\rm{test}}}\)) are mapped onto one another by expanding \({{\mathbb{X}}}_{{\rm{test}}}\) into a supercell and exploring commensurate lattice and origin choices with respect to \({{\mathbb{X}}}_{{\rm{ref}}}\). The yellow lattice (highlighted by the green box) is a potential match with \({{\mathbb{X}}}_{{\rm{ref}}}\). \({{\mathbb{X}}}_{{\rm{test}}}\) is transformed into the new representation (\({\widetilde{{\mathbb{X}}}}_{{\rm{test}}}\)), and the structures are quantitatively compared via the misfit criteria. The structures are evaluated via their lattice deviation (ϵlatt), coordinate displacement (ϵcoord), and figure of failure (ϵfail). Distances between mapped atoms (dmap) that are less than half the atom’s nearest neighbor (dnn/2) are accounted for in the coordinate displacement (green dashed lines and arrows), while larger distances are described in the figure of failure (red dashed lines and arrows).
Crystals are isopointal if they have commensurate space groups (equivalent or enantiomorphic pairs) and Wyckoff positions34. Wyckoff positions are compatible if they have the same multiplicity and similar site symmetry designations. Due to different setting and origin choices for the conventional cell, a strict site symmetry match is insufficient. For instance, the Wyckoff positions with multiplicity 2 in space group #47 (Pmmm) — four 2mm (letters i-l), four m2m (letters m-p), and four mm2 (letters q-t) — form a Wyckoff set and are related via an automorphism of the space group operations19,35,36. Depending on the assignment of the lattice parameters (a, b, and c) and origin choice, different — and potentially equivalent — Wyckoff decorations are possible. Consequently, XtalFinder tests permutations of the site symmetry symbol to expose positions that may be within the same Wyckoff set. Permuting the site symmetry symbol does not always reveal Wyckoff positions belonging to the same set since the site symmetry may originate from higher point symmetries (see example of space group #66 (Cccm) and Wyckoff positions i and k in ref. 36). Nevertheless, Wyckoff positions belonging to different sets cannot be matched, which will be revealed via the geometric structure comparison.
The symmetry calculation is performed automatically, i.e. it does not require input from the user. Options are available to ignore symmetry and force geometric comparison of structures, which can identify crystals associated via symmetry subgroups.
Isoconfigurational snapshots: compare local geometry
Beyond isopointal analyses, structures are further compared by inspecting arrangements of atoms, i.e. local atomic geometries. Local geometry analyses have been fruitful in providing structural descriptors and similarity metrics between crystals of different types37,38. However, the positions of these environments are often neglected, precluding the determination of one-to-one mappings between similar crystals. Nevertheless, the analysis quickly identifies local geometries and is employed here to analyze structures beyond symmetry considerations (i.e. isoconfigurational versus isopointal34).
Rather than determine the complete local atomic geometry for each atom, XtalFinder builds a reduced representation: neighborhoods comprised of only the least frequently occurring atom (LFA) type(s). The local LFA geometry analysis provides the connectivity for a subset of atoms (i.e. LFA-type) to discern if patterns are present in both structures, regardless of cell choice and crystal orientation. This description is preferred over the full local geometry because it is i. computationally less expensive to calculate and ii. generally less sensitive to coordination cutoff tolerances. The latter is attributed to the fact that LFA geometries are more sparse.
An example of a local LFA geometry is shown for the quaternary Heusler structure (ABCD_cF16_216_c_d_b_a)17,18 in Fig. 2b. A local LFA atomic geometry (AG) is a set of vectors connecting a central atom (c) to its closest neighbors:
where \({{\bf{d}}}_{ic}^{\min }\) is the minimum distance vector to the i atom — restricted to LFA-type(s) only — and is calculated via the method of images for periodic systems39:
Here, na, nb, and nc are the lattice dimensions along the lattice vectors a, b, and c; and xi and xc are the Cartesian coordinates of the i and c (center) atoms, respectively. A coordination shell with a thickness of \({d}_{ic}^{\min }/10\) captures other atoms of the same type to control numerical noise in the atomic coordinates (a similar tolerance metric is defined in AFLOW-SYM, i.e. loose preset tolerance value20). This cutoff value yields expected coordination numbers for well-known systems and is comparable to results provided by other atom environment calculators37,38. If there is only one LFA type — e.g. Si in α-cristobalite (SiO2, A2B_tP12_92_b_a)17,18 — then the distance to the closest neighbor of that LFA type is calculated. If there are multiple LFA types — e.g. four for the quaternary Heusler (as illustrated in Fig. 2b) — then the minimum distances to each LFA type are computed. The local atomic geometry is calculated for each atom of the LFA type(s) in the unit cell, resulting in a list of atomic geometries ({AGc}). Therefore, α-cristobalite has a set of four Si LFA geometries (one for each Si in the unit cell: {AGSi,1, AGSi,2, AGSi,3, AGSi,4}) and the quaternary Heusler has a set of four LFA geometries (one for each element type: {AGAu, AGLi, AGMg, \(A{G}_{{\rm{Sn}}}\)}, respectively).
To investigate structural compatibility, local atomic geometry lists for compounds are compared. In general, the local geometry comparisons err on the side of caution. For instance, comparing the cardinality of the coordination is often too strict. Despite a more sparse geometry space, slight deviations in position can move atoms outside the coordination shell threshold, changing the atom cardinality and neglecting potential matches. Local atomic geometries are thus compatible if (i) the central atoms are comparable types (i.e. same element and/or stoichiometric ratio in the crystal), (ii) the neighborhood of surrounding atoms have distances that match within 20% after normalizing with respect to \(\max (A{G}_{c})\) (i.e. the largest distance in the local geometry cluster), and (iii) the angles formed by two atoms and the center atom match within 10°. To further alleviate the coordination problem, an exact geometry match is not required, i.e. some distances and angles are permitted to be missing. Grouping local atomic geometries as compatible is favored to mitigate false negatives for equivalent structures.
Isoconfigurational structures: compare geometric structure
To resolve a commensurate representation between two structures for geometric comparison, one structure — the reference \({{\mathbb{X}}}_{{\rm{ref}}}\) — remains fixed and the other structure — the potential duplicate \({{\mathbb{X}}}_{{\rm{test}}}\) — is expanded into a supercell. Lattice vectors are identified within the supercell and compared against the reference structure. For any similar lattices to \({{\mathbb{X}}}_{{\rm{ref}}}\), \({{\mathbb{X}}}_{{\rm{test}}}\) is transformed into the new lattice representation (\({\widetilde{{\mathbb{X}}}}_{{\rm{test}}}\)). Origin shifts for this cell are then explored in an attempt to match atoms. If one-to-one atom mappings exist between the two structures, then the similarity is quantified with the crystal misfit method (see "Quantitative similarity measure” subsection)21. Misfit values below a given threshold indicate equivalent structures and the search terminates. Alternatively, misfit values larger than the threshold are disregarded and the search continues until all lattices and origin shifts are exhausted. The procedure is detailed below and an illustration of the process is depicted in Fig. 2c.
The lattice search algorithm begins by scaling the volumes of the unit cells to compare structures with different volumes (an option is available to quantify the similarity between structures at fixed volumes). Once scaled, the routine searches for translation vectors by generating a lattice grid of \({{\mathbb{X}}}_{{\rm{test}}}\). The size of the grid is defined to encompass a sphere with a radius (rgrid) equal to the maximum lattice vector length of \({{\mathbb{X}}}_{{\rm{ref}}}\), i.e.
Similar to a procedure described in ref. 20, the necessary grid dimensions are given by the set of vectors perpendicular to each pair of \({{\mathbb{X}}}_{{\rm{ref}}}\) lattice vectors scaled by the grid radius (e.g.\({{\bf{n}}}_{1}={r}_{{\rm{grid}}}\left({\bf{b}}\times {\bf{c}}/| | {\bf{b}}\times {\bf{c}}| | \right)\)). The scaled vectors are then transformed into the lattice basis (L), via \({\bf{n}}^{\prime} ={{\bf{L}}}^{-1}{\bf{n}}\), and the ceiling of the \({\bf{n}}^{\prime}\) components indicate the grid dimensions: \({n}_{a,b,c}={\rm{ceil}}({\bf{n}}^{\prime} )\). The grid dimensions span between − na,b,c → na,b,c to account for different orientations/rotations between the structures. To optimize the lattice search, translation vectors are explored in a grid comprised of only the LFA-type in \({{\mathbb{X}}}_{{\rm{test}}}\) (since they are the minimal set of atoms exhibiting crystal periodicity). In addition to verifying crystal periodicity, candidate lattice vectors must be similar to those in the \({{\mathbb{X}}}_{{\rm{ref}}}\) lattice based on (i) lattice vector moduli (Δl), (ii) angles formed between pairs of lattice vectors (Δθ) and (iii) volumes enclosed by three lattice vectors (ΔV). The tolerances values (Δl, Δθ, ΔV) are chosen based on how much the lattices are allowed to differ. If the lattices are significantly different, then the lattice is ignored (see the "lattice deviation” in the "Quantitative similarity measure” subsection). Additionally, as a speed increase, commensurate lattices are sorted by minimum lattice deviation to find matches more quickly. Upon finding a similar cell to \({{\mathbb{X}}}_{{\rm{ref}}}\), \({{\mathbb{X}}}_{{\rm{test}}}\) is transformed into the new lattice representation \({\widetilde{{\mathbb{X}}}}_{{\rm{test}}}\) and is stored if the representations have the same number of atoms (and types).
For each prospective unit cell, possible origin choices are explored. The origin of \({{\mathbb{X}}}_{{\rm{ref}}}\) is placed on one of the LFA-type atoms, and the origin of \({\widetilde{{\mathbb{X}}}}_{{\rm{test}}}\) is cycled through all atoms of its LFA-type. Given an origin choice, a mapping procedure is attempted for all atoms in the unit cell. The minimum Cartesian distance — via the method of images for periodic systems39 — is determined for every atom i in \({{\mathbb{X}}}_{{\rm{ref}}}\) to each atom j in \({\widetilde{{\mathbb{X}}}}_{{\rm{test}}}\)
where na, nb, and nc are the lattice dimensions along the lattice vectors a, b, and c; and xi and xj are the Cartesian coordinates of the i and j atoms, respectively. Given the set of distances {dij}, the minimum distance over all j atoms is identified as the mapping distance, i.e.
regardless of the element type. Once \({d}_{i}^{{\rm{map}}}\) is computed for all i, the following conditions are verified: i. one-to-one mappings (i.e. no duplicate j indices between i indices), and ii. no cross-matching between element types (i.e. cannot map a single element type to multiple types in \({\widetilde{{\mathbb{X}}}}_{{\rm{test}}}\)). If either condition is violated, the mappings are ignored and the search continues.
Given a successful mapping, the similarity of the two crystals in the corresponding representations are quantified, indicating equivalent or unique structures. If no mapping is found for any lattice and origin choice, then the structures are considered distinct and are not assigned a similarity value.
Quantitative similarity measure
To compare two crystals in a given representation, a method proposed by Burzlaff and Malinovsky is employed21. The similarity between structures is quantified by a misfit value, ϵ, which incorporates differences between lattice vectors and atomic coordinates via21:
The misfit quantity is bound between zero and one: structures with a value close to zero match and those with a value close to one do not match. Special misfit ranges defined by Burzlaff and Malinovsky are adopted here21
The "same family” designation generally corresponds to crystals with common symmetry subgroups. Burzlaff and Malinovsky recommend ϵmatch = 0.1 and ϵfamily = 0.2 based on definitions from Pearson40 and Parthé41. In the "Finding ϵmatch: structural misfit versus enthalpy” section, heuristic misfit thresholds are identified based on the allowed maximum enthalpy differences between similar structures.
The deviation of the lattices, ϵlatt, captures the difference between the lattice face diagonals of \({\widetilde{{\mathbb{X}}}}_{{\rm{test}}}\) and \({{\mathbb{X}}}_{{\rm{ref}}}\)21
where fkl and dkl denote the diagonals by adding and subtracting, respectively, the k and l lattice vectors. In the lattice search algorithm, Δl, Δθ, and ΔV tolerances are coupled to ϵlatt, and are tuned to ensure ϵlatt ≤ ϵfamily.
The coordinate deviation — measuring the disparity between atomic positions in the two structures — is based on the mapped atom distances (\({d}_{i}^{{\rm{map}}}\) or \({d}_{j}^{{\rm{map}}}\) as computed with Equations (4) and (5)) and the atoms’ nearest neighbor distances in the respective structures, dnn21
\({\widetilde{N}}^{{\rm{test}}}\) and Nref are the number of atoms in the two crystals. If dmap < dnn/2, then a "switch” variable n is set to zero and the mapped atom distance is included in ϵcoord. Otherwise, n is set to one, signifying the mapped atoms are far apart and not considered in ϵcoord. These atoms are represented in the figure of failure, ϵfail21
Other metrics can be used to assess structural similarity, including the root mean square (rms) of the atom positions11 and coordination characterization functions12. XtalFinder employs the crystal misfit criteria to incorporate structural differences between both the lattice and atom positions. Differences between common similarity metrics — and their software implementations — are discussed in more detail in the “Comparison Accuracy” subsection.
Super-type comparisons
To explore new areas of materials space, the XtalFinder module (i) identifies equivalent and unique materials, (ii) uncovers common structure-types across different compounds (i.e. prototypes), (iii) determines inequivalent atom decorations for a given crystal structure, and (iv) discerns distinct magnetic structure configurations. The corresponding comparison modes are denoted as material-, structure-, decoration-, and magnetic-type, respectively (Fig. 3). Each variant uses the underlying procedures discussed in the “Results” section (i.e. symmetry, local atomic geometry, and geometric structure comparisons) with different restrictions on mapping atom types.

a Material-type: maps same element types, revealing duplicate compounds. b Structure-type: maps structures regardless of the element types, identifying crystallographic prototypes. c Decoration-type: creates and compares all atom decorations for a given structure, determining unique and equivalent decorations (in this case, atom decorations AB and BA match). d Magnetic-type: maps compounds by element types and magnetic moments, discerning distinct spin configurations.
Material-type
Material-type comparisons map atoms of the same atomic species (Fig. 3a). For example, given two ZnS zincblende compounds (AB_cF8_216_c_a)17,18, a material-type comparison maps Zn → Zn and S → S in the two structures. Therefore, the method reveals duplicate compounds within a data set.
Structure-type
Conversely, structure-type comparisons ignore atomic species and map any atom-type with compatible stoichiometric ratios (Fig. 3b). In the case of zincblende structures ZnS and SiC, a structure-type comparison attempts to map Zn → Si and S → C, or Zn → C and S → Si, since the compounds are equicompositional. This mode exposes unique backbone structures and is practical for crystallographic prototyping. Identifying prototypes is also useful for modeling solid solutions and disordered materials42,43.
Decoration-type
The decoration-type (or permutation-type) mode determines unique atom decorations for a given crystal structure, i.e. inequivalent colorings of a structure, where each element is denoted by a different color (Fig. 3c). Continuing with the zincblende example, the A and B atomic sites are equivalent: swapping elements on the sites results in a duplicate compound compared to the original decoration. Thus, only one site decoration choice is necessary to create a distinct compound. Given a compound with n species, there are n! possible atom permutations. XtalFinder automatically (i) generates compounds with the different atom decorations for a crystal, (ii) compares the decorations (via a material-type comparison), and (iii) identifies the unique configurations. Atom decorations are only compared if atomic types have the same Wyckoff multiplicity and similar site symmetries (see subsection "Isopointal structures: compare symmetry").
Equivalent decoration groups need to obey Lagrange’s theorem44: the order h of subgroup H divides the group G with order g (i.e. \({\rm{mod}}(g,h)=0\)). Accordingly, the numbers of unique and equivalent decorations must divide the total number of decorations, i.e. satisfy divisor theory. The possible equivalent decoration groups — out of n! — are dictated by its divisors, and are enumerated below for 2 < n < 5 (elemental compounds, n = 1, are excluded):
2! = 2 : 2, 1
3! = 6 : 6, 3, 2, 1
4! = 24 : 24, 12, 8, 6, 4, 3, 2, 1
5! = 120 : 120, 60, 40, 30, 24, 20, 15, 12, 10, 8, 6, 5, 4, 3, 2, 1
For example, the possible groupings for a ternary compound (n = 3) are: 6, 3, 2, and 1 unique sets with 1, 2, 3, and 6 decorations per set, respectively.
Depending on the matching (misfit) tolerance and the choice of the reference decoration, calculated equivalency groups can violate divisor theory. For instance, two decorations can match with a certain misfit; however, a better match with a smaller misfit can exist with another decoration. To combat incorrect groupings, XtalFinder executes a consistency check, verifying the groupings are commensurate with the possible divisors. If they are not, XtalFinder searches for better matches and regroups the compatible decorations.
For example, ICSD entry BiITe #10500 (original geometry) has six possible atom decorations: ABC, BAC, CBA, ACB, CAB, and BCA. Since the three equicompositional sites are comprised of the same Wyckoff multiplicity and site symmetry (multiplicity 1 and site symmetry 3m. in space group #156), all structures are placed in the same initial comparison group, with ABC chosen as the reference decoration (since it is the first in the set). After comparing, the equivalent groups and their misfit values are:
-
ABC = BAC (ϵ = 0.0889) = CBA (ϵ = 0.0144),
-
CAB = ACB (ϵ = 0.0889), and
-
BCA (no equivalent decorations).
However, the number of equivalent decorations in each set are not the same, violating Lagrange’s theorem44. Furthermore, all misfits values should be the same, since the underlying structure is unchanged. The incommensurate groupings are a symptom of only comparing to the reference decoration, as opposed to cross-comparing with other decorations.
To remedy incorrect groupings, XtalFinder checks for better matches (i.e. potential equivalent decorations with lower misfit values). Therefore, the “duplicate” decorations are compared to the other reference decorations and regrouped to minimize the misfit value. In this case, the subsequent cross-comparisons are performed:
-
BAC with CAB and BCA,
-
CBA with CAB and BCA, and
-
ACB with BCA (not compared with ABC; performed previously).
Consequently, the final equivalent decorations are
-
ABC = CBA (ϵ = 0.0144),
-
CAB = BAC (ϵ = 0.0144), and
-
BCA = ACB (ϵ = 0.0144).
The groupings above satisfy Lagrange’s theorem, and the equivalent structures in each group have the same misfit value with respect to their reference decoration.
Magnetic-type
Magnetic-type comparisons map atoms of the same atomic species and similar magnetic moments, i.e. analyzes spin configurations (Fig. 3d). For instance, given two body-centered cubic chromium compounds with antiferromagnetic ordering, the routine attempts to map Cr↑ → Cr↑ and Cr↓ → Cr↓. A magnetic moment tolerance threshold denotes equivalent spin sites; where the default tolerance is 0.1μB. The analysis can be performed for both collinear and non-collinear systems. The magnetic-type comparison can be joined with a magnetic structure generator to create distinct spin configurations for high-throughput simulation.
Multiple comparisons
With the plethora of compounds generated by computational frameworks — such as AFLOW22,45, NoMaD46, Materials Project47, High-Throughput Toolkit48, Materials Cloud/AiiDA49, and OQMD50 — automatically comparing structures is necessary for high-throughput classification of unique/duplicate compounds and structure-types. For this purpose, we developed an automatic comparison procedure for multiple crystals (Fig. 4). Compounds are first grouped into isopointal sets by analyzing and comparing the symmetries of the structures, aggregating them by stoichiometry, space groups, and Wyckoff sets (calculated via AFLOW-SYM20). Next, compounds are further partitioned into near isoconfigurational sets by determining and comparing the local LFA geometries in each structure. Within each near isoconfigurational group, one representative structure — generally the first in the set — is compared to the other structures via geometric comparisons and the misfit values are stored. Once the comparisons finish, any unmatched structures (i.e. misfit values greater than ϵmatch) are reorganized into new comparison sets. The process repeats until all structures have been assembled into matching groups or all comparison pairs are exhausted. The three comparison analyses are performed in this order for two reasons: (i) to categorize structural similarity to varying degrees (isopointal, near isoconfigurational, and isoconfigurational) and (ii) to efficiently group compounds to reduce the computational cost of the geometric structure comparison (see "Speed and scaling considerations” in the Results). This procedure is the same for material-, structure-, decoration-, and magnetic-type comparisons; however, different atom mapping restrictions are applied depending on the comparison mode.

Compounds are compared in the following sequence: symmetry, local LFA geometry, and geometric structure. The algorithms determine isopointal, near isoconfigurational, and isoconfigurational structures, respectively, and aggregate them into similar sets (enclosed in black solid-lined boxes). Unmatched structures (i.e. ϵ > ϵmatch) after the initial geometric structure comparison are put into new groups and re-compared until all equivalent structures are grouped. This sequence is the same for material-, structure-, decoration-, and magnetic-type comparisons; however, the criteria for atom mappings differ (see subsection ”Super-type comparisons” for details). The symmetry, local LFA geometry, and geometric structure comparisons (blue boxes) are multithreaded for parallel computation.
Multithreading
To enhance calculation speed, multithreading capabilities can be employed. The three computationally intensive procedures — calculating the symmetry, constructing the local LFA geometry, and performing geometric comparisons — are partitioned onto allocated threads, offering significant speed increases for large collections of structures.
Automatic comparisons
There are three built-in functions to compare multiple structures automatically: (i) compare structures provided by a user, (ii) compare an input structure to prototypes in AFLOW17,18, and (iii) compare an input structure to entries in the AFLOW.org repository. An overview of each high-throughput method is discussed below and usage is detailed in the Methods section.
Compare user datasets
Users can load crystal geometries and compare them automatically with XtalFinder. Options to perform both material-type and structure-type comparisons are available to identify unique/duplicate compounds or prototypes, respectively. For structure-type comparisons, the unique atom decorations for each representative structure are determined. Once the analysis is complete, XtalFinder groups compatible structures together and returns the corresponding misfit values.
Compare to AFLOW prototypes libraries
Given an input structure, this routine returns similar AFLOW prototype(s) along with their misfit value(s) (Fig. 5a). AFLOW contains structural prototypes that can be rapidly decorated for high-throughput materials discovery: 590 in the Prototype Encyclopedia17,18 and 1,492 in the High-throughput Quantum Computing library25. In this method, AFLOW prototypes are extracted — based on similar stoichiometry, space group, and Wyckoff positions to the input — and compared to the user’s structure. Since only matches to the input are relevant, the procedure terminates before regrouping any unmatched prototypes. The attributes of matched prototypes are also returned, including the prototype label, mineral name, Strukturbericht designation, and links to the corresponding Prototype Encyclopedia webpage. The scheme identifies common structure-types with the AFLOW libraries or — if no matches are found — reveals new prototypes. Absent prototypes can be characterized automatically in the AFLOW standard designation with XtalFinder’s prototyping tool (discussed in subsection “Problem of the ideal prototype”).

An input Al2O3 (corundum) compound is compared to entries in a AFLOW Prototype Encyclopedia and b the AFLOW.org repository. Potential equivalent entries are retrieved automatically from the respective catalog and compared with XtalFinder. Matching entries and their level of similarity (misfit) are returned.
Compare to AFLOW.org repository
Compounds are compared to entries in the AFLOW.org repository using the AFLOW REST- and AFLUX Search-APIs51,52 (Fig. 5b). An AFLUX query (i.e. matchbook and directives) is generated internally and returns database compounds similar to the input structure based on species, stoichiometry, space group, and Wyckoff positions. With the AURL from the AFLUX response, structures for the entry are retrieved via the REST-API. The most relaxed structure is extracted by default; however, options are available to obtain structures at different ab-initio relaxation steps. The set of entries from the database are then compared to the input structure. Similar to the AFLOW prototype comparisons, candidate entries are only compared against the input structure, i.e. the procedure terminates without regrouping unmatched entries.
With the underlying AFLUX functionality, material properties can also be extracted, highlighting the structure-property relationship amongst similar materials. For instance, the enthalpy per atom (Hatom) for matching database entries are printed by including the enthalpy_atom API keyword in the query. Any number or combination of properties can be queried; available API keywords are located in refs. 51,52. Table 1 shows the comparison results between a rocksalt NaCl compound and matching DFT-relaxed structures in the AFLOW.org repository along with their misfits and enthalpies per atom.
This routine reveals equivalent AFLOW.org compounds, if similar materials exists in the database. As such, it can estimate structural properties a priori; before performing any calculations. The estimation is based on the following assumptions: i. the matching AFLOW material resides at a local minimum in the energy landscape and ii. the input structure relaxes to the same geometry as that AFLOW compound, given comparable calculation parameters. The functionality can explore properties that are not calculated for a given entry, but are calculated for an equivalent entry. For example, compounds in AFLOW’s prototype catalogs (LIB1, LIB2, LIB3, etc.) do not usually have band structure data; however, corresponding ICSD entries can be found which do provide band structure information. Finally, the method can identify compounds that are absent from the database and prioritize them for future calculation, enhancing the diversity of the AFLOW.org repositories.
Using AFLOW-XtalFinder
For ease-of-use, the XtalFinder routines are accessible via a command-line interface and a Python environment (see Methods for details).
Ideal prototype analysis in AFLOW.org
The ideal prototype designations — for both the original and relaxed geometries — have been successfully determined for all 4+ million entries in the AFLOW.org repository. The prototype label, parameter variables, and parameter values are incorporated into the AFLOW REST- and Search-APIs51,52. The corresponding API keywords for the original geometries are
-
aflow_prototype_label_orig,
-
aflow_prototype_params_list_orig, and
-
aflow_prototype_params_values_orig.
For the DFT-relaxed geometries, the keywords are
-
aflow_prototype_label_relax,
-
aflow_prototype_params_list_relax, and
-
aflow_prototype_params_values_relax.
The prototype keywords enable researchers to search for materials by structure. This feature is useful for identifying possible crystal structures given experimental data. For example, with composition, space group, and occupied Wyckoff information (characteristics often known to experimentalists); users can construct the corresponding prototype label(s) and extract all compounds based on the provided structure-type. The keywords are also used to identify the frequency of certain prototypes in the AFLOW.org repository. For example, all compounds that are isopointal to the corundum prototype (labels: A2B3_hR10_167_c_e and A3B2_hR10_167_e_c) can be retrieved for both the original and relaxed geometries. Moreover, this search capability is used to discern if a structure-type is novel or has been reported previously.
The ideal prototype keywords also reveal whether a compound retains the same prototype designation before and after relaxation. For structures that retain the same prototype label, the parameter values show the continuous structure transition during relaxation. For structures that transform into different prototypes, the symmetry-based designations highlight the symmetries that were broken. This can indicate that certain element combinations/arrangements are averse to certain prototype structures. More advanced relaxation techniques, e.g. symmetry-constrained relaxations26, would be required to restrict the relaxation to a given prototype structure.
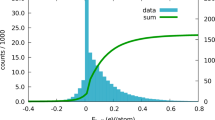
Finding ϵ match: structural misfit versus enthalpy
To identify a suitable threshold for matching similar structures (ϵmatch), Fig. 6 plots the misfit value (ϵ) between two mapped structures and their difference in enthalpy per atom (ΔHatom). The structures in the test set are comprised of DFT-relaxed entries from the entire AFLOW-ICSD catalog as of 14 August 2020 (60,390)53,54. Compounds are grouped via commensurate atomic elements, stoichiometries, symmetries, and local LFA geometries. Furthermore, only compounds calculated with similar ab initio settings are compared together — such as LDAU parameters, kpoint per reciprocal atom (KPPRA), and pseudopotentials (see Supplementary Note 1 for details) — to prevent extraneous enthalpy differences due to differing parameters. In addition, magnetic systems are excluded since the magnetic moment is not incorporated into the misfit value. For these comparisons, the unit cell volumes are not rescaled, and the best lattice/origin choices are explored (minimizing the misfit value) to show better correlation with the enthalpies. After grouping the structures and identifying one-to-one mappings, misfit values for the remaining 6,795 comparison pairs are calculated. Figure 6a and b show the enthalpy difference ranges 0 − 100 meV/atom and 0 − 10 meV/atom, respectively, highlighting the maximum enthalpy differences at different misfit values.

The misfit value (ϵ) and the difference in enthalpy per atom (\({{\Delta }}{H}_{{\rm{atom}}}={H}_{{\rm{atom}}}^{{\rm{ref}}}-{H}_{{\rm{atom}}}^{{\rm{test}}}\)) for all AFLOW-ICSD entries with similar parameters are shown above. The plots show the misfit values between 0 and 0.2 with two enthalpy difference ranges (for clarity): a 0 − 100 meV/atom and b 0 − 10 meV/atom. The plot with the full misfit domain and enthalpy range is shown in Supplementary Fig. 1. Candidate misfit thresholds are chosen based on the acceptable maximum enthalpy deviation between match structures. For example, misfit values below ϵ = 0.025 and ϵ = 0.1 (black vertical lines) are expected to yield enthalpy differences no larger than ΔHatom ≈ 2 meV/atom and ΔHatom ≈ 5 meV/atom, respectively (black horizonal lines). As the misfit values increase beyond ϵ > 0.1, the spread of the data points also increases. A large jump in the maximum enthalpy difference occurs at approximately ϵ = 0.125, indicating matched structures near this value and beyond are not guaranteed to have similar enthalpies.
In general, the misfit value correlates with the enthalpy difference for ϵ≤0.1: as the misfit value decreases, the enthalpy difference also reduces. For ϵ > 0.1, the enthalpy spread widens with the misfit. Some comparison-pairs exhibit large misfit values, but have similar enthalpies. This follows intuition since it is possible for significantly differing structures to have similar properties. The sparsity of the data points for large values of ϵ is attributed to the lack of one-to-one mappings as structures become increasingly dissimilar. This suggests XtalFinder and the misfit criteria are better suited to quantifying similar structures, rather than relating disparate structures. Figure 6 reveals possible thresholds for ϵmatch based on the maximum enthalpy difference allowed for mapped structures. For ϵ≤0.1, the enthalpy differences per atom are all below 5 meV/atom, with the exception of one comparison-pair. Reducing the misfit cutoff reasonably guarantees the enthalpy differences will also decrease; e.g. enthalpies will be within 1 meV/atom and 2 meV/atom for misfit values below 3.58 × 10−3 and 0.025, respectively. The maximum enthalpy difference jumps significantly (to approximately 50 meV/atom) near ϵ = 0.125. Thus, matching structures with misfits beyond ϵ = 0.1 are not guaranteed to exhibit similar enthalpies. This value is in agreement with Burzlaff and Malinovsky’s proposed threshold. By default, XtalFinder employs a threshold of ϵmatch = 0.1 to ensure similar materials match within approximately 5 meV/atom. The threshold is also used for comparing prototypes; two prototypes that match within ϵmatch = 0.1, are expected to have enthalpies within 5 meV/atom when decorated with the same atomic elements. Users can adjust to stricter (or looser) thresholds for matching; however, ϵmatch ≫ 0.1 is not guaranteed to yield small enthalpy differences between matched structures.
Functionality differences with other codes
In addition to XtalFinder, other structure comparison tools are available to the materials science community: Structure Matcher11, XTALCOMP10, SPAP12, CMPZ13, CRYCOM9, STRUCTURE-TIDY (via Platon)14, and COMPSTRU15. A summary of the offered functionalities related to automatic comparisons and structure prototyping is indicated in Table 2 and described below.
Source code availability
The source codes are available for the following packages: XtalFinder (via AFLOW), Structure Matcher (via Pymatgen), and XTALCOMP (via XTALOPT). Pre-compiled binaries for SPAP on different operating systems are available with the CALYPSO software55,56. Source codes for the other software: CMPZ (implemented in KPLOT57), CRYCOM, STRUCTURE-TIDY, and COMPSTRU (online) are not available. Therefore, the latter packages are not convenient for merging into user-workflows.
Input file formats
The different structure file formats for each comparison code are listed below; XtalFinder: VASP (POSCAR)27, FHI-AIMS28, QUANTUM ESPRESSO29, ABINIT30, ELK31, and CIF; Structure Matcher: POSCAR, CIF, ABINIT, and a Pymatgen object; XTALCOMP: C++ object (and POSCARs via online tool); SPAP: POSCAR (and CIFs via CIF2Cell58); CMPZ: KPLOT structure files; CRYCOM: FDAT files (native to the Cambridge Structural Database (CSD)59); STRUCTURE-TIDY: creates structures based on space group input, unit cell parameters, and positions of atoms; and COMPSTRU: CIF.
Symmetry analysis
XtalFinder is the only package coupled with an internal symmetry calculator (AFLOW-SYM20). CRYCOM, STRUCTURE-TIDY, and COMPSTRU require a symmetry input (space group number) to perform the comparison, but lack methods to calculate the symmetry internally. CMPZ allows a symmetry input; however it is not required to perform the comparison. Structure Matcher, XTALCOMP, and SPAP do not consider symmetry in their structural analyses.
Multiple comparisons
The only packages that offer comparison of multiple materials in a single command are XtalFinder and Structure Matcher. Other software, such as XTALCOMP and SPAP, showcase comparison results performed on multiple structures, but multi-comparison routines are not available to users. To achieve similar functionality, users need to implement external regrouping procedures.
Decoration-type comparisons
XtalFinder is the only code that automatically determines the unique (and equivalent) atom decorations for a given crystal structure. With other packages, users must externally generate, organize, and compare the subsequent decorations. Beyond the lack of routines to generate decorations, the codes find incorrect equivalent decoration groups. In the BiITe (ICSD #10500) example discussed in the "Decoration-type” comparison mode section, Structure Matcher’s group_structures function (with ltol=0.2, stol=0.17, angle_tol=5.0) identifies groupings that violate divisor theory:
-
ABC = BAC (rms=0.1196) = CBA (rms=0.0194),
-
CAB = ACB (rms=0.1196), and
-
BCA (no equivalent decorations).
A similar discrepancy occurs for the remaining codes depending on the structure input order and comparison tolerance(s). XtalFinder checks consistency with Lagrange’s theorem to validate permissible decoration groupings; a burden that falls to users of the other packages.
Furthermore, XtalFinder calculates Wyckoff positions a priori to check if decorations are commensurate based on symmetry, i.e. mapped positions have the same multiplicity and similar site symmetries. Without this validation, positions with differing site symmetries can be mistaken as equivalent. For example, the GaPu compound (ICSD #103930, space group #139) has three Wyckoff positions: Ga 8 h m.2m, Pu 4 d \(\bar{4}\)m2, and Pu 4 e 4mm (before and after relaxation). From symmetry, the Ga and Pu sites cannot be swapped to yield degenerate compounds. Despite the symmetry restrictions, Structure Matcher incorrectly groups the decorations (group_structures) into equivalent bins using their default tolerance values (i.e. ltol=0.2, stol=0.3, angle_tol=5). XtalFinder — with its symmetry analysis coupled with mapping routines — correctly distinguishes these decorations and is the only viable option to establish consistent unique/duplicate decorations for crystalline prototypes.
Database comparisons
XtalFinder is the only module that features a method for comparing input structures to a database of materials, namely AFLOW.org. The API functionality coupled with XtalFinder ensures comparisons are performed with the most current version of the database, incorporating new materials as they are calculated. Furthermore, XtalFinder users can compare structures at various relaxation steps (with the --relaxation_step option). Users of the other packages need to extract relevant structures (e.g. similar compositions, space group, Wyckoff positions, and local atomic geometries at particular relaxation steps) by-hand or code auxiliary scripts to perform similar functionality.
Prototype comparisons
XtalFinder compares to all prototype structures in AFLOW: the Prototype Encyclopedia, the High-Throughput Quantum Computing library, and initial geometries in the AFLOW.org repository. Similar to the database comparisons, XtalFinder automatically includes new prototype structures as they are added to AFLOW. Structure Matcher only compares structures against a static subset of AFLOW prototypes (i.e. the Prototype Encyclopedia via AflowPrototypeMatcher60). Moreover, XtalFinder provides the internal degrees of freedom for any structure (via the prototyping routines); functionality all existing codes currently lack. To compare to these prototype representations, users of other packages need to convert the degrees of freedom — including expansion of the corresponding Wyckoff positions — into a structure file a priori.
Speed and scaling considerations
Comparison speeds were evaluated for packages that could be compiled locally on a Linux machine: XtalFinder (V3.2), Structure Matcher (V2020.4.2), and XTALCOMP (downloaded from GitHub on 14 Apr. 2020). The benchmarks were run with a single processor on a 2.60 GHz Intel(R) Xeon(R) Gold 6142 CPU machine, and the respective default tolerances were used for all codes. Pairwise comparison times are similar between the packages; on the order of milliseconds. For a 1,494 pairwise comparison test set, XtalFinder averaged 282 milliseconds/comparison, Structure Matcher averaged 689 milliseconds/comparison, and XTALCOMP averaged 33 milliseconds/comparison. The test set is comprised of ICSD unaries (original geometries) with 1-105 atoms per unit cell and varying symmetries; along with a mix of equivalent and inequivalent structures. XTALCOMP is the fastest, but at the cost of limited scope and functionality: XTALCOMP does not scale volumes and quits immediately if the volumes/lattice vectors are different sizes. Therefore, XTALCOMP finds fewer matches (18), while XtalFinder and Structure Matcher find more (approximately 450). For large, skewed cells, XtalFinder can be slower since it does not convert to Minkowski, Niggli, and/or primitive cells by default to preserve the input representations (unlike Structure Matcher and XTALCOMP). To increase speed in XtalFinder, lattice transformations are available with the relevant options for Minkowski (--minkowski), Niggli (--niggli), and primitive (--primitive) reductions.
For multiple comparisons, XtalFinder scales more efficiently with the number of compounds when compared to other software. XtalFinder groups structures into near isoconfigurational sets via symmetry and local atomic geometries (both calculated internally), eliminating unnecessary mapping comparisons between dissimilar structures. All other codes do not use symmetry or local geometry analyses to optimize groupings. Structure Matcher — and other straightforward extensions to pairwise comparisons — only groups by composition and executes more mapping procedures. For an ensemble of 600 structures modeling a disordered 5-metal carbide61,62, XtalFinder partitions immediately into 54 groups via the symmetry and local geometry comparisons, while Structure Matcher puts all 600 structures into one large group. Consequently, XtalFinder executes 546 structure mappings, and Structure Matcher performs 17,640 mapping attempts before arriving at the same solution.
While all benchmarks were performed serially, XtalFinder routines are parallelized and users can specify the number of threads for the analyses (--np=x), offering additional speed over other packages for large-scale automatic comparisons requiring little or no user input. Therefore, XtalFinder will be more performant, especially when comparing more structures.
Comparison accuracy
As shown in Fig. 6, the XtalFinder misfit value decreases with the enthalpy difference between matched compounds, validating its accuracy. Comparisons with Structure Matcher are less accurate — and at times qualitatively incorrect — due to conversions of structures to an “average lattice”11, matching significantly differing lattices with no penalty on the rms value. For example, Se (ICSD #104187, space group #229) and Se (ICSD #57181, space group #166) are classified as distinct by XtalFinder because the lattices are considerably dissimilar (ϵlatt = 0.15), consistent with the space groups. Despite having different symmetries, Structure Matcher inaccurately finds rms = 0 between the structures. This distorts their rms value, and it cannot be used to correlate properties of matched compounds, e.g. enthalpy. Conversely, XTALCOMP is qualitatively accurate, but it lacks a quantitative similarity metric (the return type is a Boolean). Furthermore, XTALCOMP comparisons neglect volume scaling between structures, an essential feature for identifying prototypes. XtalFinder is the only comparison software suitable for quantitatively measuring similarity of materials and prototypes.
Overall, XtalFinder is optimized for prototype detection and structural comparison within large datasets. In addition, it is designed to be accessible to the broader materials science community for integration into user workflows.
Unique prototypes in the AFLOW-ICSD catalog
With XtalFinder, unique compounds and prototypes have been identified in the ICSD catalog of the AFLOW.org repository. Table 3 shows the statistics for the original (reported by the ICSD53) and DFT-relaxed geometries (via the AFLOW standard32) for 60,390 entries. Material-type comparisons and suppressing volume scaling reveal the number of unique compounds. Subsequent structure-type comparisons (allows for volume scaling) determine the number of distinct prototypes. The representative compound for each prototype is chosen as the entry with the lowest ICSD number, since it is generally the oldest among the compounds (and less likely to be removed from the ICSD). The unique atom decorations for each prototype are determined via the decoration-type comparison. Moreover, the prototypes are cast into the AFLOW prototype designation form, exposing its degrees of freedom. Finally, the prototypes are compared to the Prototype Encyclopedia17,18 to distinguish between existing and new structures. For the subsequent comparisons, the matching threshold is chosen as ϵmatch = 0.1 to group similar compounds (and prototypes when decorated with alike atoms) that are expected to have enthalpies differing by approximately 5 meV/atom or less (see Fig. 6).
The analysis shows that the original geometry set includes 34,820 unique compounds (57.7% of the total number of entries) and 15,205 prototypes (25.2%). Similarly, the DFT-relaxed set contains 33,544 unique compounds (55.5%) and 14,692 prototypes (24.3%). Based on the symmetry comparisons, there are 8,521 (14.1%) original and 8,493 (14.1%) relaxed distinct isopointal structure-types. In general, the original geometry set has more distinct compounds and prototypes than the DFT-relaxed set. This is attributed to the different volumes (e.g. measured temperatures and pressures) of the original geometries, while the DFT-relaxed geometries represent the ground state configurations, yielding additional degenerate compounds.
Overall, the binaries and ternaries have the highest number of entries, and thus, prototypes. The number of entries/prototypes drops with species n > 3, following statistics regarding the complexity of materials63. Table 4 partitions the prototypes by their symmetry, i.e. Bravais lattices. The number of lower symmetry prototypes (tri, mcl, and mclc) exceed the higher ones (cub, fcc, bcc) because lower symmetry classes have additional degrees of freedom, permitting more geometric diversity. Similar to Table 3, there are generally more original prototypes in each lattice type compared to their relaxed counterparts. However, 347 structures changed lattice symmetry upon relaxation, yielding the following net Bravais lattice type gains/losses: tri (+8), mcl (-17), mclc (+46), orc (-31), orcc (+48), orcf (+1), orci (+5), tet (+17), bct (+8), hex (-87), rhl (-7), cub (+2), fcc (+4), bcc (+3). In particular, the mclc and orcc Bravais lattices had a considerable influx of prototypes, offsetting the expected reduction of prototypes due to DFT geometry optimization.
While AFLOW.org contains a subset of the ICSD catalog, the highest frequency prototypes are consistent with those published for the ICSD64. In particular, XtalFinder and the ICSD both identify the following structures as some of the most common prototypes: Al2MgO4 (spinel, A2BC4_cF56_227_d_a_e-001), CaTiO3 (cubic perovskite, AB3C_cP5_221_a_c_b), GdFeO3 (AB3C_oP20_62_a_cd_c-001), and NaCl (rocksalt, AB_cF8_225_a_b) (see Table 5 and Supplementary Tables 1-14). The criteria for grouping compounds into structure-types described in ref. 64 is more relaxed than XtalFinder (e.g. larger tolerances for c/a and β ranges and user-defined ranges for fractional atomic coordinates). Consequently, XtalFinder finds more distinct prototype structures than the 1,600 (as of January 2007) in ref. 64.
From this analysis, new candidate prototypes have been identified that are missing from the Prototype Encyclopedia (signified by empty rows in the last columns of Table 5 and Supplementary Tables 1-14). The number of new prototypes in the original (relaxed) sets with more than 10 unique compounds exhibiting the structure are: binaries 31 (33), ternaries 168 (177), quaternaries 40 (42), and quinaries 4 (3); while the unaries, senaries, and septenaries have 0 (0). This amounts to 243 distinct crystalline structures that will be incorporated into future installments of the Prototype Encyclopedia. Most entries in the Prototype Encyclopedia stem from experimentally observed structures; therefore, we plan to use the original geometries for prototyping.
Some structures in Table 5 and Supplementary Tables 1-14 are equivalent to the Prototype Encyclopedia prototypes with a different number of atom types. For example, the third most common ternary ABC_hP9_189_f_bc_g (RuSiZr ICSD #16306, original geometry) matches to the binary analog A2B_hP9_189_fg_bc (Fe2P, Strukturbericht: C22)17,18 when the f and g Wyckoff positions are of the same atom type. We classify the prototypes as distinct; similar to distinguishing between the diamond (n = 1) and zincblende (n = 2) structures.
Discussion
Herein, we present XtalFinder: a software for automatically identifying unique prototypes and calculating structural similarity of crystals. The framework performs robust symmetry, local atomic geometry, and geometric structure comparisons. Routines are available to quantify structural similarity for i. compounds (material-type comparisons), ii. prototypes (structure-type), iii. atom decorations (decoration-type), and iv. spin configurations (magnetic-type). The program can analyze multiple structures simultaneously and aggregate them into equivalent groups, with multithreading capabilities available for improving performance. Built-in methods compare input structures to the AFLOW.org repository and the AFLOW prototype libraries for detecting new compounds and structure-types. Crystal prototyping techniques are also introduced to cast structures into a standard designation, facilitating extensions of the Prototype Encyclopedia. A command line and Python interface are provided for easing incorporation into user-workflows. Applying the procedures to the AFLOW-ICSD repository revealed approximately 15,000 prototypes out of over 60,000 ICSD entries, representing over 34,000 unique compounds. Subsequent comparisons with the AFLOW prototype libraries exposed new candidate entries for future iterations of the encyclopedia. Overall, XtalFinder serves as a versatile tool for finding prototypes and comparing crystalline geometries.
Methods
Command-line interface
The XtalFinder command-line calls are detailed below. Function descriptions and options are provided following each command.
Prototype commands.
-
aflow --prototype < file
-
Converts a structure (file) into its standard AFLOW prototype label. The parameter variables (degrees of freedom) and corresponding values are also listed. Information about the label and parameters are described in the refs. 17,18.
Options specific to this command:
--setting=1∣2∣aflow
◇ Specify the space group setting for the conventional cell/Wyckoff positions. The aflow setting follows the choices of the Prototype Encyclopedia: axis-b for monoclinic space groups, rhombohedral setting for rhombohedral space groups, and origin centered on the inversion site for centrosymmetric space groups (default: aflow).
-
-
aflow --proto=<label>.<ABC..>:Ag:C:Cu:... --params=parameter_1,parameter_2,...
-
Generates a geometry file based on the ideal prototype designation (<label>) and parameter values (parameter_1,parameter_2,...). A particular atom decoration can be specified after the label (<ABC...>). By default, the structure is created with fictitious atoms (i.e. A, B, C, D, ...); however, this can be overridden by appending real elements to the label separated by colons (e.g. <label>.<ABC...>:Ag:C:Cu:...). Options specific to this command:
--add_equations
◇ The symbolic version of the geometry file (in terms of the variable degrees of freedom) is printed after the numeric geometry file.
--equations_only
◇ Only print the symbolic version of the geometry file (in terms of the variable degrees of freedom).
-
Comparison commands.
-
aflow --compare_materials
-
Compares compounds comprised of the same atomic species and with commensurate stoichiometric ratios, i.e. material-type comparison, and returns their level of similarity (misfit value). This method identifies unique and duplicate materials. There are three input types:
aflow --compare_materials=<f1>,<f2>,...
◇Append geometry files (<f1>,<f2>,...) to compare.
aflow --compare_materials -D <path>
◇Specify path to directory (<path>) containing geometry files to compare.
aflow --compare_materials -F=<filename>
◇Specify file (<filename>) containing compounds between delimiters [VASP_POSCAR_MODE_EXPLICIT]START and [VASP_POSCAR_MODE_EXPLICIT]STOP. Additional delimiters will be included in later versions.
-
-
aflow --compare_structures
-
Compares compounds with commensurate stoichiometric ratios with no requirement of the atomic species, i.e. structure-type comparison, and returns their level of similarity (misfit value). This method identifies unique and duplicate prototypes. There are three input types:
aflow --compare_structures=<f1>,<f2>,...
◇Append geometry files (<f1>,<f2>,...) to compare.
aflow --compare_structures -D <path>
◇Specify path to directory (<path>) containing geometry files to compare.
aflow --compare_structures -F=<filename>
◇Specify file (<filename>) containing compounds between delimiters [VASP_POSCAR_MODE_EXPLICIT]START and [VASP_POSCAR_MODE_EXPLICIT]STOP. Additional delimiters will be included in later versions.
-
-
aflow --compare2database < file
-
Compares a structure (file) to AFLOW database entries, returning similar compounds and quantifying their levels of similarity (misfit values). Material properties can be extracted from the database (via AFLUX) and printed, highlighting structure-property relationships. Performs material-type comparisons or structure-type comparisons (by adding the --structure_comparison option). Options specific to this command:
--properties=<keyword,keyword,...>
◇ Specify the properties via their API keyword to print the corresponding values with the comparison results.
--catalog=<string>
◇ Restrict the database entries to a specific catalog/library (e.g. ‘lib1’, ‘lib2’, ‘lib3’, ‘icsd’, etc.).
--relaxation_step=<number>
◇ Compare geometries from a particular DFT relaxation step (e.g. 0 : original, 1: relax 1, 2: relax 2, etc.).
-
-
aflow --compare2prototypes < file
-
Compares a structure (file) against the AFLOW prototype libraries, returning similar structures, and quantifying their levels of similarity (misfit values).
--catalog=<string>
◇ Restrict the prototypes to a specific catalog/library (e.g. ‘aflow’ or ‘htqc’).
-
-
aflow --isopointal_prototypes < file
-
Returns prototype labels that are isopointal (i.e. similar space group and Wyckoff positions) to the input structure (file).
--catalog=<string>
◇ Restrict the prototypes to a specific catalog/library (e.g. ‘aflow’ or ‘htqc’).
-
-
aflow --unique_atom_decorations < file
-
Determines the unique and duplicate atom decorations for a given structure.
-
Generic options for all comparison commands (unless indicated otherwise):
-
--misfit_match=
∣ --misfit_match_threshold=<number>
-
Specifies the misfit threshold for matched structures (default: ϵmatch = 0.1).
-
-
--misfit_family=
∣ --misfit_family_threshold=<number>
-
Specifies the misfit threshold for structures in the "same family” (default: ϵfamily = 0.2).
-
-
--np=
∣--num_proc= -
Allocate the number of processors/threads for the task.
-
-
--optimize_match
-
Explore all lattice and origin choices to find the best matching representation, i.e. minimizes misfit value.
-
-
--no_scale_volume
-
Suppresses volume rescaling during structure matching; identifies differences due to volume expansion or compression of a structure.
-
-
--ignore_symmetry
-
Neglects symmetry (both space group and Wyckoff positions) for grouping comparisons.
-
-
--ignore_Wyckoff
-
Neglects Wyckoff symmetry (site symmetry) for filtering comparisons, but considers the space group number.
-
-
--ignore_local_geometry
-
Neglects local LFA geometries for filtering comparisons.
-
-
--minkowski
-
Performs a Minkowski lattice transformation8 on all structures prior to comparison; offering a speed increase.
-
-
--niggli
-
Performs a Niggli lattice transformation7 on all structures prior to comparison; offering a speed increase.
-
-
--primitive∣--primitivize
-
Converts all structures to a primitive form prior to comparison; offering a speed increase.
-
-
--keep_unmatched
-
Retains misfit information of unmatched structures (i.e. ϵ > ϵmatch).
-
-
--match_to_aflow_prototypes
-
Identifies matching AFLOW prototypes to the representative structure. The option does not apply to --unique_atom_decorations or --compare2prototypes (redundant).
-
-
--magmom=
-
Specifies the magnetic moment for each structure (collinear or non-collinear) delimited by colons, signaling a magnetic-type comparison. The option does not apply to --compare_structures since the atom type is neglected. XtalFinder supports three input formats for the magnetic moment: (i) explicit declaration via comma-separated string m1, m2, . . . mn (m1,x, m1,y, m1,z, m2,x, . . . mn,z for non-collinear) (ii) read from a VASP INCAR, or (iii) read from a VASP OUTCAR. Additional magnetic moment readers for other ab initio codes will be available in future versions.
-
-
--add_aflow_prototype_designation
-
Casts representative structure into the AFLOW standard designation. The option does not apply to command --unique_atom_decorations.
-
-
--remove_duplicate_compounds
-
For structure-type comparisons, duplicate compounds are identified first (via a material-type comparison without volume scaling), then remaining unique compounds are compared, removing duplicate bias.
-
-
--print_mapping
-
For comparing two structures, additional comparison information is printed, including atom mappings, distances between matched atoms, and the transformed structures in the closest matching representation.
-
-
--print=text∣json
-
For comparing multiple structures, the results are printed to human-readable text or JSON files, respectively. By default, XtalFinder writes the output to both files.
-
-
--quiet
-
Suppresses the log information for the comparisons.
-
-
--screen_only
-
Prints the comparison results to the screen and does not write to any files.
-
Python environment. In addition to the command-line interface, a Python module is available for inclusion into a variety of workflows. The module mirrors the format used for AFLOW-SYM20 and AFLOW-CHULL65. An XtalFinder function is performed on the input(s) and the results are returned to an XtalFinder class. The module wraps around a local instance of AFLOW, and the path to the AFLOW executable can be specified by: XtalFinder(aflow_executable=‘your_executable’).
By default, the XtalFinder object searches for an AFLOW executable in the PATH. An example Python script is shown below, where an XtalFinder object is initialized and a material-type comparison between two structure files (POSCARs) is performed.
from aflow_xtal_finder import XtalFinder |
from pprint import pprint |
xtal_finder = XtalFinder(aflow_executable=’./aflow’) |
input_files = [‘test1.poscar’,‘test2.poscar’] |
output = xtal_finder.compare_materials(input_files) |
pprint(output) |
The following Python functions are accessible, corresponding to the commands described in the previous section:
-
get_prototype_label(input_file, options)
-
compare_materials(input_files, options)
-
compare_materials_directory(directory, options)
-
compare_materials_file(filename, options)
-
compare_structures(input_files, options)
-
compare_structures_directory(directory, options)
-
compare_structures_file(filename, options)
-
compare2database(input_file, options)
-
compare2prototypes(input_file, options)
-
get_isopointal_prototypes(input_file, options)
-
get_unique_atom_decorations(input_file, options)
The input fields for the Python functions are as follows:
-
input_file
-
A string specifying the path to a structure file, e.g. input_file=‘/home/user/test.poscar’.
-
-
input_files
-
A list of paths (of any size ≥2) to structure files, e.g. input_files=[‘test1.poscar’, ...].
-
-
directory
-
A string specifying the path to directory containing structure files, e.g. directory=‘/home/user/directory’.
-
-
filename
-
A string specifying the path to a file containing structure files separated by a delimiter, e.g. filename=‘/home/user/list_of_structures.txt’.
-
-
options
-
A string specifying non-default functionality (optional), which has the form --<flag> or --<keyword>=<value>, e.g. "--ignore_symmetry --np=8”.
-
Python module.
A Python module for XtalFinder is available in Supplementary Method 1. All output is converted into JavaScript Object Notation (JSON) to ease integration into user workflows.
AFLOW-XtalFinder JSON output details.
The output keywords for the XtalFinder functions are listed below as they appear in the JSON format. The output for multiple comparisons (user-defined sets, comparison to AFLOW prototypes, and comparison to AFLOW.org entries), unique atom decorations, and casting into the AFLOW prototype representation are described.
AFLOW prototype designation.
-
aflow_prototype_label
-
Description: AFLOW label for the structure.
-
Type: string
-
-
aflow_prototype_params_list
-
Description: degrees of freedom (variables) in the lattice and/or Wyckoff positions for the structure.
-
Type: array of strings
-
-
aflow_prototype_params_values
-
Description: values specifying the degrees of freedom for the structure.
-
Type: array of floats
-
Comparison results.
-
structure_representative
-
Description: information for the representative structure for the prototype structure.
-
Type: structure_representative object
-
-
stoichiometry
-
Description: stoichiometry of the prototype structure.
-
Type: array of integers
-
-
ntypes
-
Description: number of atom types (species) in the prototype structure.
-
Type: integer
-
-
natoms
-
Description: number of atoms in the unit cell (from the representative structure).
-
Type: integer
-
-
elements
-
Description: atomic elements found in this structure from both the representative and duplicate compounds/structures.
-
Type: array of strings
-
-
space_group
-
Description: space group number for the prototype structure.
-
Type: integer
-
-
grouped_Wyckoff_positions
-
Description: Wyckoff positions grouped by atomic species (corresponding to the representative structure).
-
Type: array of Wyckoff objects
-
-
geometries_LFA
-
Description: local atomic geometries comprised of LFA types only (corresponding to the representative structure).
-
Type: array of local_geometry objects
-
-
property_names
-
Description: API keywords corresponding to material properties (available for comparisons to the AFLOW.org repository only).
-
Type: array of strings
-
-
property_units
-
Description: units, if applicable, for material properties (available for comparisons to the AFLOW.org repository only).
-
Type: array of strings
-
-
structures_duplicate
-
Description: information for the duplicate structures that match with the representative structure, i.e. misfit is less than ϵmatch.
-
Type: array of structure_matched objects
-
-
structures_family
-
Description: information for the structures that are within the same family as the representative structure, i.e. misfit is between ϵmatch and ϵfamily.
-
Type: array of structure_matched objects
-
-
matching_aflow_prototypes
A structure_representative object contains the following:
-
name
-
Description: name of the representative structure for the prototype structure.
-
Type: string
-
-
number_compounds_matching_structure
-
Description: number of compounds that match with the representative structure via a material-type comparison (only for structure-type comparisons that remove duplicate compounds beforehand).
-
Type: integer
-
-
<property>
-
Description: value of the material property requested for the representative structure, where <property> is the corresponding API keyword, e.g. enthalpy_atom. The property keywords are only available for comparisons to the AFLOW.org repository.
-
Type: string
-
A Wyckoff object contains the following:
-
element
-
Description: atomic species on Wyckoff site.
-
Type: string
-
-
type
-
Description: an index corresponding to atomic species, based on alphabetic ordering of element name.
-
Type: integer
-
-
letters
-
Description: Wyckoff letters for the atomic species.
-
Type: array of strings
-
-
multiplicities
-
Description: Wyckoff multiplicities for the atomic species.
-
Type: array of integers
-
-
site_symmetries
-
Description: Wyckoff site symmetries for the atomic species.
-
Type: array of strings
-
A structure_matched object contains the following:
-
name
-
Description: name of the matched structure.
-
Type: string
-
-
misfit
-
Description: value of the misfit between the representative structure and the matched structure.
-
Type: float
-
-
lattice_deviation
-
Description: value of the lattice deviation between the representative structure and the matched structure.
-
Type: float
-
-
coordinate_displacement
-
Description: value of the coordinate displacement between the representative structure and the matched structure.
-
Type: float
-
-
failure
-
Description: value of the figure of failure between the representative structure and the matched structure.
-
Type: float
-
-
number_compounds_matching_structure
-
Description: number of compounds that match with this structure via a material-type comparison (only for structure-type comparisons that remove duplicate compounds beforehand).
-
Type: integer
-
-
<property>
-
Description: value of the material property requested for the matched structures, where <property> is the corresponding API keyword, e.g. enthalpy_atom. The property keywords are only available for comparisons to the AFLOW.org repository.
-
Type: string
-
A local_geometry object contains the following:
-
center_element
-
Description: atomic species at the center of the geometry cluster.
-
Type: string
-
-
center_type
-
Description: index corresponding to atomic species at the center of the geometry cluster; enumeration is based on alphabetic ordering of element name.
-
Type: integer
-
-
neighbor_elements
-
Description: atomic elements of neighbors.
-
Type: array of strings
-
-
neighbor_distances
-
Description: distances of the neighbors from the center atom
-
Type: array of floats
-
-
neighbor_frequencies
-
Description: coordination of the neighbors at the corresponding neighbor distance (within 10%).
-
Type: array of integers
-
-
neighbor_coordinates
-
Description: coordinates of the neighbors that comprise the local atomic geometry; the origin of the system resides on the center atom.
-
Type: 2D array of floats
-
Permutation results.
-
atom_decorations_equivalent
-
Description: groupings of equivalent atom decorations for the structure.
-
Type: 2D array of strings
-
Ideal prototype API keywords.
-
aflow_prototype_label_orig
-
Description: the standard prototype label of the structure (original geometry).
-
Type: string
-
-
aflow_prototype_params_list_orig
-
Description: degrees of freedom (variables) in the lattice and/or Wyckoff positions of the structure (original geometry).
-
Type: array of strings
-
-
aflow_prototype_params_values_orig
-
Description: values specifying the degrees of freedom of the structure (original geometry).
-
Type: array of floats
-
-
aflow_prototype_label_relax
-
Description: the standard prototype label of the structure (DFT-relaxed geometry).
-
Type: string
-
-
aflow_prototype_params_list_relax
-
Description: degrees of freedom (variables) in the lattice and/or Wyckoff positions of the structure (DFT-relaxed geometry).
-
Type: array of strings
-
-
aflow_prototype_params_values_relax
-
Description: values specifying the degrees of freedom of the structure (DFT-relaxed geometry).
-
Type: array of floats
-
Data availability
All crystallographic structure data is freely available and accessible online through AFLOW.org or programmatically via the REST- and AFLUX Search-APIs. The AFLOW prototype information is provided online at http://aflow.org/prototype-encyclopedia, and the corresponding structures can be generated with the AFLOW source code.
Code availability
The XtalFinder module is integrated into the AFLOW software (version 3.2 and later). The source code for AFLOW is available at http://aflow.org/install-aflow/ and http://materials.duke.edu/AFLOW/, and it is compatible with most Linux, macOS, and Microsoft operating systems. The multithreaded capabilities require GNU g++-4.4 or later. Questions and bug reports should be emailed to aflow@groups.io with a subject line containing "XtalFinder”.
References
Ewald, P. P. & Hermann, C. (eds.) Strukturbericht 1913-1928 (Akademische Verlagsgesellschaft M. B. H., Leipzig, 1931).
Villars, P. & Calvert, L. Pearson’s Handbook of Crystallographic Data for Intermetallic Phases. 2nd (ASM International, Materials Park, Ohio, USA, 1991).
Curtarolo, S. et al. The high-throughput highway to computational materials design. Nat. Mater. 12, 191–201 (2013).
Kołcz, A., Chowdhury, A. & Alspector, J. Data duplication: an imbalance problem? In Workshop on Learning from Imbalanced Datasets II, ICML (2003). http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.72.8356.
Muratov, E. N. et al. QSAR without borders. Chem. Soc. Rev. 49, 3525–3564 (2020).
Muratov, E. N. et al. Correction: QSAR without borders. Chem. Soc. Rev. 49, 3716–3716 (2020).
Niggli, P. Handbuch der Experimentalphysik, vol. 7 (Akademische Verlagsgesellschaft, 1928).
Minkowski, H. Geometrie der Zahlen (Teubner-Verlag, 1896).
Dzyabchenko, A. V. Method of crystal-structure similarity searching. Acta Crystallogr. Sect. B 50, 414–425 (1994).
Lonie, D. C. & Zurek, E. Identifying duplicate crystal structures: XTALCOMP, an open-source solution. Comput. Phys. Commun. 183, 690–697 (2012).
Richards, W. D., Dacek, S. & Ong, S. P. Pymatgen: Structure Matcher. http://pymatgen.org/_modules/pymatgen/analysis/structure_matcher.html (2011). (Accessed 20 Jan 2020).
Su, C. et al. Construction of crystal structure prototype database: methods and applications. J. Phys.: Condens. Matter 29, 165901 (2017).
Hundt, R., Schön, J. C. & Jansen, M. CMPZ - an algorithm for the efficient comparison of periodic structures. J. Appl. Crystallogr. 39, 6–16 (2006).
Gelato, L. M. & Parthé, E. STRUCTURE TIDY - a computer program to standardize crystal structure data. J. Appl. Crystallogr. 20, 139–143 (1987).
de la Flor, G., Orobengoa, D., Tasci, E., Perez-Mato, J. M. & Aroyo, M. I. Comparison of structures applying the tools available at the Bilbao Crystallographic Server. J. Appl. Crystallogr. 49, 653–664 (2016).
Lonie, D. C. & Zurek, E. XTALOPT: An open-source evolutionary algorithm for crystal structure prediction. Comput. Phys. Commun. 182, 372–387 (2011).
Mehl, M. J. et al. The AFLOW library of crystallographic prototypes: Part 1. Comput. Mater. Sci. 136, S1–S828 (2017).
Hicks, D. et al. The AFLOW library of crystallographic prototypes: Part 2. Comput. Mater. Sci. 161, S1–S1011 (2019).
Hahn, T. (ed.) International Tables of Crystallography. Volume A: Space-group symmetry (Kluwer Academic publishers, International Union of Crystallography, Chester, England, 2002).
Hicks, D. et al. AFLOW-SYM: platform for the complete, automatic and self-consistent symmetry analysis of crystals. Acta Crystallogr. Sect. A 74, 184–203 (2018).
Burzlaff, H. & Malinovsky, Y. A procedure for the classification of non-organic crystal structures. I. Theoretical background. Acta Crystallogr. Sect. A 53, 217–224 (1997).
Toher, C. et al. The AFLOW fleet for materials discovery. In Andreoni, W. & Yip, S. (eds.) Handbook of Materials Modeling, 1–28 (Springer International Publishing, Cham, Switzerland, 2018).
Toher, C. et al. Combining the AFLOW GIBBS and elastic libraries to efficiently and robustly screen thermomechanical properties of solids. Phys. Rev. Mater. 1, 015401 (2017).
Toher, C. et al. High-throughput computational screening of thermal conductivity, Debye temperature, and Grüneisen parameter using a quasiharmonic Debye model. Phys. Rev. B 90, 174107 (2014).
Curtarolo, S. et al. AFLOW: An automatic framework for high-throughput materials discovery. Comput. Mater. Sci. 58, 218–226 (2012).
Lenz, M.-O. et al. Parametrically constrained geometry relaxations for high-throughput materials science. npj Comput. Mater. 5, 123 (2019).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Blum, V. et al. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 180, 2175–2196 (2009).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21, 395502 (2009).
Gonze, X. et al. First-principles computation of material properties: the ABINIT software project. Comput. Mater. Sci. 25, 478–492 (2002).
The Elk code (2020). http://elk.sourceforge.net/.
Calderon, C. E. et al. The AFLOW standard for high-throughput materials science calculations. Comput. Mater. Sci. 108 Part A, 233–238 (2015).
Hardy, Y., Tan, K. S. & Steeb, W.-H. Computer Algebra with SymbolicC++. (World Scientific, Singapore, 2008).
Lima-de-Faria, J., Hellner, E., Liebau, F., Makovicky, E. & Parthé, E. Nomenclature of inorganic structure types. Report of the International Union of Crystallography Commission on crystallographic nomenclature subcommittee on the nomenclature of inorganic structure types. Acta Crystallogr. Sect. A 46, 1–11 (1990).
Boyle, L. L. & Lawrenson, J. E. The origin dependence of Wyckoff site description of a crystal structure. Acta Crystallogr. Sect. A 29, 353–357 (1973).
Koch, E. & Fischer, W. Automorphismengruppen von raumgruppen und die zuordnung von punktlagen zu konfigurationslagen. Acta Crystallogr. Sect. A 31, 88–95 (1975).
Perim, E. et al. Spectral descriptors for bulk metallic glasses based on the thermodynamics of competing crystalline phases. Nat. Commun. 7, 12315 (2016).
Zimmermann, N. E. R. & Jain, A. Local structure order parameters and site fingerprints for quantification of coordination environment and crystal structure similarity. RSC Adv. 10, 6063–6081 (2020).
Hloucha, M. & Deiters, U. K. Fast coding of the minimum image convention. Mol. Simul. 20, 239–244 (1998).
Pearson, W. B. The Crystal Chemistry and Physics of Metals and Alloys (Wiley-Interscience, 1972).
Parthé, E. Elements of Inorganic Structural Chemistry: a course on selected topics. (K. Sutter Parthé, Petit-Lancy, Switzerland, 1990).
Avery, P., Toher, C., Curtarolo, S. & Zurek, E. XtalOpt Version r12: An open-source evolutionary algorithm for crystal structure prediction. Comput. Phys. Commun. 237, 274–275 (2019).
Oses, C., Toher, C. & Curtarolo, S. High-entropy ceramics. Nat. Rev. Mater. 5, 295–309 (2020).
Beachy, J. A. & Blair, W. D. Abstract Algebra. (Waveland Press, Inc., Long Grove, Illinois, 2006).
Oses, C., Toher, C. & Curtarolo, S. Data-driven design of inorganic materials with the automatic flow framework for materials discovery. MRS Bull. 43, 670–675 (2018).
Draxl, C. & Scheffler, M. NOMAD: The FAIR concept for big data-driven materials science. MRS Bull. 43, 676–682 (2018).
Jain, A. et al. Commentary: The Materials Project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
The High-Throughput Toolkit (httk). http://httk.openmaterialsdb.se/ (Accessed 20 Jan 2020).
Pizzi, G., Cepellotti, A., Sabatini, R., Marzari, N. & Kozinsky, B. AiiDA: automated interactive infrastructure and database for computational science. Comput. Mater. Sci. 111, 218–230 (2016).
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B. & Wolverton, C. Materials design and discovery with high-throughput density functional theory: The Open Quantum Materials Database (OQMD). JOM 65, 1501–1509 (2013).
Taylor, R. H. et al. A RESTful API for exchanging materials data in the AFLOWLIB.org consortium. Comput. Mater. Sci. 93, 178–192 (2014).
Rose, F. et al. AFLUX: The LUX materials search API for the AFLOW data repositories. Comput. Mater. Sci. 137, 362–370 (2017).
Bergerhoff, G., Hundt, R., Sievers, R. & Brown, I. D. The inorganic crystal structure data base. J. Chem. Inf. Comput. Sci. 23, 66–69 (1983).
Curtarolo, S. et al. AFLOWLIB.ORG: A distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235 (2012).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 82, 094116 (2010).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 183, 2063–2070 (2012).
Hundt, R. KPLOT: A Program for Plotting and Investigation of Crystal Structures, Version 9. (Technicum Scientific Publishing, Stuttgart, Germany, 2016).
Björkman, T. CIF2Cell: Generating geometries for electronic structure programs. Comput. Phys. Commun. 182, 1183–1186 (2011).
Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The Cambridge structural database. Acta Crystallogr. Sect. B 72, 171–179 (2016).
Pymatgen: AflowPrototypeMatcher. http://pymatgen.org/pymatgen.analysis.prototypes.html (Accessed 20 Jan 2020).
Yang, K., Oses, C. & Curtarolo, S. Modeling off-stoichiometry materials with a high-throughput ab-initio approach. Chem. Mater. 28, 6484–6492 (2016).
Sarker, P. & Harrington, T. et al. High-entropy high-hardness metal carbides discovered by entropy descriptors. Nat. Commun. 9, 4980 (2018).
Mackay, A. L. On complexity. Crystallogr. Rep. 46, 524–526 (2001).
Allmann, R. & Hinek, R. The introduction of structure types into the inorganic crystal structure database ICSD. Acta Crystallogr. Sect. A 63, 412–417 (2007).
Oses, C. et al. AFLOW-CHULL: Cloud-oriented platform for autonomous phase stability analysis. J. Chem. Inf. Model. 58, 2477–2490 (2018).
Acknowledgements
We thank Corey Oses, Marco Esters, Eric Gossett, Demet Usanmaz, Andriy Smolyanyuk, Rico Friedrich, Yoav Lederer, Amir Natan, Matthias Scheffler, Christian Carbogno, and Xiomara Campilongo for valuable discussions. D.H. acknowledges support from the Department of Defense through the National Defense Science and Engineering Graduate (NDSEG) Fellowship Program. D.C.F acknowledges support from the Duke University Provost’s Postdoctoral Fellowship Program. S.C. acknowledges support by DOD-ONR (N00014-17-1-2090) and the Alexander von Humboldt-Foundation.
Author information
Authors and Affiliations
Contributions
S.C. envisioned the project. D.H. and C.D.S. developed the XtalFinder routines. All authors—D.H., C.T., D.C.F, F.R., C.D.S., O.L., M.J.M., and S.C.—tested the algorithms, discussed the results, and contributed to the writing of this article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hicks, D., Toher, C., Ford, D.C. et al. AFLOW-XtalFinder: a reliable choice to identify crystalline prototypes. npj Comput Mater 7, 30 (2021). https://doi.org/10.1038/s41524-020-00483-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-020-00483-4
This article is cited by
-
Disordered enthalpy–entropy descriptor for high-entropy ceramics discovery
Nature (2024)
-
Enhancing interpretability in the exploration of high-energy conversion efficiency in CsSnBr3−xIx configurations using crystal graph convolutional neural networks and adversarial example methods
Science China Materials (2024)
-
Dataset of theoretical multinary perovskite oxides
Scientific Data (2023)
-
Accelerating the prediction of stable materials with machine learning
Nature Computational Science (2023)
-
Accelerating materials-space exploration for thermal insulators by mapping materials properties via artificial intelligence
npj Computational Materials (2023)
