
Abstract
Transition metal oxides that exhibit a metal-to-insulator transition (MIT) as a function of oxygen vacancy concentration are promising systems to realize energy-efficient platforms for neuromorphic computing. However, the current lack of understanding of the microscopic mechanism driving the MIT hinders the realization of effective and stable devices. Here we investigate defective cobaltites and we unravel the structural, electronic, and magnetic changes responsible for the MIT when oxygen vacancies are introduced in the material. We show that, contrary to accepted views, cooperative structural distortions instead of local bonding changes are responsible for the MIT, and we describe the subtle interdependence of structural and magnetic transitions. Finally, we present a model, based on first principles, to predict the required electric bias to drive the transition, showing good agreement with available measurements and providing a paradigm to establish design rules for low-energy cost devices.
Similar content being viewed by others
Introduction
The search for computing architectures with low-power consumption is an active field of research, and neuromorphic architectures, which aspire to mimic the human brain1, have attracted much attention lately. The realization of neuromorphic devices imposes specific requirements on the materials to be used: the response to an input signal should be dependent on the signal’s intensity, frequency and the previous status of the material, in a way similar to neurons and synapses in the brain, which combine computation and memory in one unit. In addition, the energy consumption to transmit and process signals should be extremely low to be affordable on a large scale; remarkably there are ~1011 neurons and ~1014 synapses in the brain, with a power consumption of ~10 W for daily cognitive tasks2.
In the last decades, several transition metal oxides (TMO) have been proposed as promising resistive switching materials3,4, i.e. systems showing tunable resistance states, induced by an external electrical bias. These TMO exhibit a metal-to-insulator transition (MIT) as a function of pressure, temperature, or doping, which may be designed to mimic the behavior of neurons and synapses in the presence of stimuli5,6,7.
One of such TMO is La1−xSrxCoO3−δ (LSCO)8,9,10, for which a resistivity change on the order of 104 was shown with increasing pressure for x < 0.5 (ref. 10). This MIT process is tunable by dopants, e.g. Sr9,11,12, with a change in resistivity of ~105 for x between 0 and 0.8 (refs. 13,14). Recently, an experimental study15 suggested to control the MIT by tuning the oxygen vacancy concentration in LSCO, and a topotactic transition was realized from a paramagnetic metallic perovskite (δ\(\approx\)0) to an antiferromagnetic (AFM) semiconducting brownmillerite (BM) structure (δ = 0.5). Interestingly, a similar transition was also observed between SrCoO3 and SrCoO2.5 (SCO2.5)16,17 and oxygen vacancy concentrations have been varied in multiple ways in cobaltites, e.g. by depositing oxygen-scavenging metals15, annealing in reducing environment16, using electric fields17,18,19 and with epitaxial strain20.
In spite of extensive work, the mechanisms underlying the MIT in cobaltites remain elusive, thus hampering the control of the properties of these materials to design efficient neuromorphic devices. Here we report an investigation of the MIT in LSCO (and the semiconductor-to-insulator transition in LCO) using first principles calculations; we unravel the complex interplay between structural, electronic, and magnetic properties of the material as the transition is driven from a perovskite to a BM phase, as a function of oxygen vacancies. Our calculations show that cooperative, global structural distortions leading to a decrease of the elastic energy of the solid, which in turn are accompanied by changes in the magnetic state of the material, are the main factors driving the MIT. Our results allow for the identification of general descriptors to design materials for neuromorphic computing applications, including multiple resistive states. Finally, we present a model based on first principles to predict the electric bias necessary to drive the MIT, which shows good agreement with experiment, and is general and applicable to broad classes of TMO.
Results
We used a combination of electronic structure techniques based on density functional theory (DFT) with a Hubbard correction (U). In order to choose the most appropriate energy functional and U parameter, we computed the structural, electronic, and magnetic properties of the perovskite LCO and BM SCO2.5 phases, using U values from 3 to 7 eV and several functionals (LDA21, PBE22, and SCAN23). We found that the PBE functional with U = 3 eV represents the optimal choice to correctly describe most measured properties of both phases, and we adopted the same protocol to study the topotactic transition as a function of oxygen vacancies. (Supplementary Discussion 1 contains a detailed study leading to our choice of the DFT parameters, see Supplementary Fig. 1 and Supplementary Tables 1 and 2).
Structural transitions
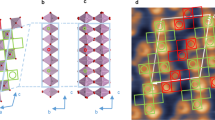
We start by analyzing the structural distortions associated with the topotactic transformation between perovskite and BM phases in LCO and LSCO, as a function of oxygen vacancy concentrations (VO, including 4.2, 8.3, and 12.5%, where VO = δ/3 × 100%). We carried out calculations in a supercell with four layers of octahedral units (Fig. 1), and we considered symmetry-inequivalent oxygen vacancy positions (namely 2 configurations at 4.2% VO, 12 at 8.3% VO, and 1 at 12.5% VO), and different magnetic states at each concentration (ferromagnetic, FM, and several AFM states including G-AFM, A-AFM, and C-AFM) (see Supplementary Figs. 2 and 3). We found that the lowest-energy structure corresponds to ordered vacancies belonging to the same layer (see Supplementary Fig. 4) and we observed a transformation from octahedral CoO6 units to pyramidal CoO5 and then to tetrahedral CoO4 ones within a layer; such transformations then occurred in alternating layers, eventually leading to the BM phase. Our results are consistent with previous in situ TEM observations, which indicated that the perovskite to BM transition occurs as a nucleation process of oxygen vacancies from layer to layer, in both LCO24 and La2/3Sr1/3MnO3 (ref. 25). We also found that the perovskite lattice expands with increasing vacancy concentration, as shown in Fig. 2a; the same trend was observed in various perovskite materials26,27,28. Our calculations show that upon Sr doping, the lattice expansion is less pronounced, consistent with experimental data29 reported for 50% Sr doping (green line in Fig. 2a). Indeed, Sr doping leads to an increase of the Co oxidation state, counterbalancing, at least in part, the effect of the vacancy’s extra electrons. As a result, the Co 3d electrons mainly occupy the t2g manifold (see Supplementary Fig. 5 for energy diagram) and hence the Jahn–Teller (JT) distortion following the introduction of vacancies is smaller compared to LCO, where the eg band is occupied. Another notable change revealed by our calculations occurs in the Co–O–Co angle, which substantially decreases, as a function of VO (Fig. 2b). We expect these results to be only weakly dependent on the distribution of Sr dopants in the crystal. For example, we computed the structural, magnetic, and electronic structure properties of non-defective LSCO samples as a function of Sr and we found that the standard deviation of the calculated lattice parameters for different Sr positions is less than 0.5% (see Supplementary Figs. 6 and 7 and Supplementary Table 3); we also found that all samples are FM metals, with similar magnetic moments of the Co atoms (see Supplementary Fig. 8).

a Side view of the structures at all sampled oxygen vacancy content (shown on the bottom row, corresponding δ value is also shown). The introduction of one oxygen vacancy in the 40 atom perovskite cell (4.2% concentration) considered in our calculations leads to the transformation of an octahedral CoO6 unit into a pyramidal CoO5 unit in a given layer (layer 4); when a second oxygen vacancy is introduced (8.3% concentration) the CoO5 units are transformed into tetrahedral CoO4 ones. Upon introduction of three (12.5%) and four (16.7%) vacancies, an octahedral-to-pyramidal-to-tetrahedral transformation is observed in alternating layers (layer 2) and eventually a topotactic transformation to a brownmillerite (BM) phase is achieved. b Top-down view along the b-axis of the perovskite and BM structures. In the BM structure, octahedra were removed to clearly show the rotation pattern of tetrahedra in alternating layers of the defective perovskite that led to the BM formation. The BM has Pmnb crystal symmetry58 as the tetrahedra in alternating layers (layers 2 and 4) have opposite handed rotation (shown by curved arrows).

a Supercell volume expansion percentage (ΔV) as a function of oxygen stoichiometry value δ; ref_La0.5Sr0.5CoO3 experimental data are from ref. 29. b Average Co–O–Co tilt angle as a function of increasing oxygen vacancy concentration VO (VO = δ/3 × 100%).
Following the analysis of ref. 30 we characterized the distortions of the perovskite phase introduced by oxygen vacancies as a combination of JT distortions, a breathing mode of the octahedral units and a change of the Co–O–Co tilt angle, as shown in Fig. 3. The combined JT/breathing distortion of the octahedra is characterized by an increase of in-plane (ac plane in Fig. 1) and out-of-plane (along the b-axis in Fig. 1) Co–O bond lengths, with the latter being more pronounced (see Supplementary Tables 4 and 5 and Supplementary Fig. 9 for lattice parameters change and bond length and bond angle change).

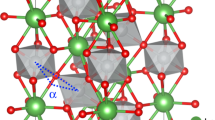
Octahedral units in layers adjacent to oxygen-deficient layers (vacancy position marked by green circle) expand as a combination of Jahn–Teller and breathing mode distortions (expansion indicated by arrows within the octahedra). The octahedra rotate to accommodate the expansion with a rotation angle Φ0. Oxygen ions connecting the octahedral and oxygen-deficient layers move as a result of rotations, shown by the arrows on these oxygen ions. Co ion shifts to the center of the oxygen-deficient polygons, and a slight JT expansion in the in-plane direction occurs in the oxygen-deficient polygons. Red spheres: oxygen ions, blue spheres: Co ions.
The Co–O–Co angle variation, or rotation distortion, is reflected in the decrease of the angle in the out-of-plane direction, more prominent than in other directions. Such a decrease is caused both by the rotation of octahedra and by the Co ion displacement observed in defective polygons.
While our results on JT/breathing distortions are validated, at least qualitatively, by the volume expansion found in experiments15,29, there are no experimental measurements of the Co–O–Co tilt angle. However, there are indirect effects of bond angle changes that can be measured, e.g., the atomic distance change between cations (La/Sr) in the out-of-plane direction. When octahedra rotates, La/Sr moves closer to its neighboring oxygen ion, thus increasing their covalent bonding strength31 and shortening the intra-layer La(Sr)–La separation. We found that such separation decreases (in even number rows in our cell: see Supplementary Fig. 10), in good agreement with the experimental trend reported for LCO24.
In order to further understand the physical picture underlying the distortions found in our calculations, we modeled the atomic deformations of the perovskite using a Hamiltonian written as a sum of three terms:
In Eq. (1) the index r denotes the position of each octahedron in the unit cell; aT, aD, and aR are the stiffness of the Jahn–Teller, breathing, and rotation distortions and Tr, Dr, and Rr are the respective deformation magnitudes. We assumed aT, aD, and aR to be site independent; they were computed as averages over all the octahedra/polygons in the unit cell. The Hamiltonian in Eq. (1) is written in the harmonic approximation.
We first estimated ai from the average vibrational frequencies of the corresponding mode i which we obtained by carrying out ab initio phonon calculations (using the QUANTUM ESPRESSO32,33 code):
Here \(\bar \nu\) is the average wave number of the ith mode and \(\mu = \sqrt {m_1m_2/3}\) is the reduced mass; m1 and m2 denote the mass of Co and O atoms, respectively. We found that aT and aD are on the order of 104 meV Å−2, while aR is two orders of magnitude smaller (see Supplementary Table 6), indicating that the JT/breathing distortion requires much higher energy to occur than rotations. The small energy required to trigger these rotations stems from phonon modes of very low energy, as identified in our phonon calculations as well as detected experimentally34. Following the protocol of ref. 30 (see Supplementary Equations 1 and 2), we rewrote Eq. (1) by applying specific structural constraints, so as to describe the interaction between the JT/breathing mode and rotations. In particular, we rewrote Eq. (1) by applying two deformation constraints: octahedra are corner-sharing and the parallelogram shape is assumed to be maintained. In doing so, we allowed for the coupling between the deformation of neighboring octahedra, and we could describe the distortions of each single octahedron using a combination of different phonon modes. By expressing the coupled distortions in momentum space, we could easily represent phonon–phonon interactions. Figure 4 shows the stiffness of the JT/breathing mode in momentum space, which includes cooperative effects and thus depends on rotation. By increasing the rotation angle Φ0, the stiffness of the JT/breathing mode decreases, showing that rotations can effectively counterbalance the elastic energy increase caused by JT distortions. Coupling between different distortions has been previously observed in other pervoskite systems such as manganites35 and nickelates36. The stiffness shown in Fig. 4 progressively decreases from the perovskite to the BM phase, due to more facile rotations as VO increases. Such cooperative distortion effects occur also in the presence of Sr doping; however, the JT/breathing distortion is less pronounced than in LCO (see Fig. 2a), and so are rotations, as expected (see Fig. 2b).

a With rotation angle Φ0 = 0° and b With rotation angle Φ0 = 10° in perovskite and BM LCO. Φ0 is the octahedral rotation angle defined in Fig. 3, and it is equal to the deviation of the Co–O–Co tilt angle (Fig. 2) from 180° divided by two. The color scale represents the scaled value (E/aT: aT is the Jahn–Teller stiffness parameter aT computed for the perovskite phase; see text) in the perovskite (left panels) and BM phases (right panels). Note the different scale used for Φ0 = 0° and Φ0 = 10°, which shows the notable decrease in energy for Φ0 = 10°. Such decrease is more significant in the brownmillerite than in the perovskite case. L is the octahedral lattice spacing.
Magnetic transitions
The structural deformations described above are accompanied by important changes in the magnetic properties of the system. With increasing VO, we observed a transition from a non-magnetic structure (LCO) or FM (LSCO) to an FM-local AFM (for VO = 4.2% and 8.3% respectively), where some of the Co atoms neighboring oxygen vacancies give rise to an AFM state, while the surrounding ones form an FM state. Eventually the system undergoes a transition to a complete AFM phase. Note that in the spin-polarized case, where one has partially filled d orbitals (full occupation of majority spin channel and partially filled minority spin channel), the super-exchange interaction between Co–O–Co with almost 180°, favors AFM ordering, according to Goodenough–Kanamori–Anderson rules. In turn this ordering implies lower Co–O orbital hybridization and longer Co–O bond length, consistent with a previous study on SrCoO3 (ref. 37). As shown in Fig. 5a, the absolute and total magnetic moment differences increase, consistent with the decrease in total magnetic moment observed in experiments15. Our results are also consistent with those of previous theoretical and experimental studies that reported different magnetic states at different vacancy concentrations37,38. For example, in strained LCO epitaxial film with 11.1% ordered vacancy concentration, first-principle calculations38 predicted a local AFM structure developing next to vacancies embedded in an FM environment.

a Absolute, total magnetic moment difference (ΔM) as a function of oxygen vacancy concentration (VO), averaged over all Co atoms in the cell. Note that the total magnetic moment, i.e. the subtraction of sum of the up-spin from down-spin moments, decreases; instead the absolute magnetic moment, i.e. the sum of the absolute value of spin-up and spin-down moments, increases and the difference between these two quantities increases. Zero corresponds to a non-magnetic (LCO) or an FM (LSCO) configuration; increasing DM leads to a transition to an AFM state. b Supercell volume change of LCO as a function VO for spin-polarized and non-spin-polarized (non-magnetic) configurations. The error bar at 8.3% in the spin-polarized case is the standard deviation of various oxygen vacancy configurations sampled in our calculations. The error bar at 12.5 and 16.7% is the standard deviation of different magnetic structures sampled in our calculations.
Interestingly, if we constrain the system to be non-magnetic (a configuration which is less energetically favorable than that of a magnetic state), we do not observe any volume expansion (see Fig. 5b) or any increase in the Co–O bond length, indicating that the combined JT/breathing distortions are suppressed. This result can be understood noting that the Co 3d–O 2p hybridization is higher (see Supplementary Figs. 11 and 12 of PDOS), thus leading to a shorter Co–O bond length37. As a consequence, the changes of angles in the layer with tetrahedral units and in the out-of-plane direction are greatly suppressed: the out-of-plane Co–O–Co angle is the same as in the perovskite phase, and the in-plane Co–O–Co angle in tetrahedral units approaches 180° (see Table 1), indicating that rotations are inhibited. If the system is instead constrained in a FM state, we observed only a slight modification of the deformation compared to that in the ground state; the volume expansion is within the error bar of our calculations at VO = 16.7% (see Fig. 5b), and the Co–O–Co angle is just slightly larger (<3%) than in the ground state (see Table 1).
Metal-insulator transition
The structural and magnetic transitions identified as a function of increasing V0 are responsible for the MIT (Fig. 6a) found in our calculations and observed in experiments in the case of LSCO15, and for the semiconductor-to-insulator transition in the case of LCO. We emphasize that the MIT is intimately connected to the magnetic state transition described above (see Fig. 5a). Note that doping LCO with Sr decreases the band gap, consistent with Cheng et al.’s14 results obtained by combining experiments and DFT calculations; in LSCO the band gap opening is first observed at a higher VO than in LCO, and in both cases it is clearly associated with the FM-to-AFM transition point. The trends in the density of states (DOS) as a function of VO shown in Fig. 6b reveals in detail how the electronic structure of the system evolves (DOS for LSCO data is shown in Supplementary Fig. 13). Interestingly, a gap opens up first between states associated to oxygen-deficient layers. As oxygen vacancies are added to alternating layers, states originating from oxygen-deficient Co ions (i.e. from Co less than sixfold coordinated) as well as from neighboring octahedral Co are increasingly more localized. This finding is supported by the analysis of Born effective charge (see Supplementary Table 8), which shows that the charge transfer from Co to O decreases with increasing VO, and it is consistent with the observed decrease of the dielectric constant. Furthermore, we observed that the band gap increases as a function of structural deformations leading to pyramidal and then tetrahedral coordination. The main contribution to band edge states (see Supplementary Fig. 11) originates from the octahedral Co’s eg manifold (majority spin channel) for VO ≤ 8.3 %; as VO increases, the main contribution comes from the pyramidal Co’s eg manifold (majority spin channel) for VO = 12.5%, and from the minority spin channel of Co for VO = 16.7%, with contributions from each d orbital of either octahedral Co or tetrahedral Co. These results highlight the importance of configurational as well as valence state changes for a band gap to develop in the system.

a Band gap variation as a function of vacancy concentration VO. b Density of states (DOS) of LCO as a function of VO, showing the band gap opening. The total DOS is further decomposed into projected DOS including the 2p orbitals of oxygen atoms, and the 3d orbitals of octahedral Co, pyramidal Co, and tetrahedral Co atoms. c The corresponding oxygen-deficient structures and Co spin configurations. The color of the dashed border corresponds to the color legend in b and it is used to distinguish Co with different oxygen coordination. High-spin and low-spin octahedral Co is also distinguished (see Supplementary Fig. 15 for magnetic moment of each Co). Pink arrow represents increased valence electrons in the 3d orbital caused by oxygen vacancies (due to both extra valence electrons from the oxygen vacancy as well as the decreases charge transfer from Co to O, see more explanation in the main text).
A detailed Co spin configuration analysis (Fig. 6c and see Supplementary Table 7 and Supplementary Fig. 15 for details) reveals how the electronic structure of the system is related to structural deformations and magnetic state change. We found that the crystal field splitting of oxygen-deficient Co depends not only on the oxygen vacancy configuration but also on the spin configuration of neighboring octahedral sites. For example, the spin-up pyramidal Co site present at VO = 4.2% experiences an octahedral crystal field splitting with six valence electrons, resembling that of its neighboring octahedral Co site; the spin-down pyramidal Co site exhibits instead a pyramidal crystal field splitting and seven electrons. This indicates that the effect of oxygen vacancies on the electronic structure is not a local effect. In a consistent fashion, the variation in the number of electrons belonging to the 3d orbitals of Co caused by oxygen vacancies (see Fig. 6c) occurs not only in oxygen-deficient layers but also in neighboring octahedral layers. To understand this variation in number of electrons, it is important to consider the contribution of charge from oxygen vacancies as well as the decrease of electron transfer from Co to O due to an increase in electron localization of the Co orbitals. The charge variation at oxygen-deficient Co sites is mainly due to the extra electrons, while electron localization influences the charge at octahedral sites the most, and more so for low-spin state Co than spin-polarized one. The relative importance of these effects (extra charges and decrease in electron transfer due to localization) depends on VO, and in the BM phase, both effects contribute equally to determine the electronic structure of tetrahedral and octahedral Co sites. The increase in number of electrons leads to transitions from LS to HS or IS spin states at octahedral Co sites, and triggers JT/breathing distortions in layers with octahedral sites, followed by rotations to decrease the elastic energy. The final, stable spin configurations, magnetic structures, and structural distortions are thus the result of a global redistribution of the electrons in the lowest-energy configurations of the system. This redistribution is influenced both by the oxygen vacancy configuration and by their concentration. Hence the materials electronic structure is intimately connected to the magnetic state transition and to structural deformations.
In addition, we note that in the projected DOS, a fairly large gap is present between states associated with oxygen-deficient layers, as soon as oxygen vacancies are introduced in the system, indicating that electron correlation increases at oxygen-deficient Co sites. The gap between states associated to octahedral layers decreases when a small amount of vacancies is introduced, as it is related to extra electrons occupying the eg orbitals. However, as the concentration of VO increases, the effect of correlation between electrons at octahedral Co sites increases as well, eventually leading to the opening of a gap. The presence of Sr partially compensates the electron doping effect from oxygen vacancies; hence, correlation effects are expected to be weaker and likely weakly influenced by an increase in oxygen vacancy concentration, as reflected by less significant structural distortions and smaller band gap changes in LSCO than in LCO.
To further understand how electronic correlation is affected by the magnetic state and associated structural deformations in the material, we carried out calculations by constraining LCO to be in specific magnetic states, corresponding to structural distortions different from those observed in the ground state (see Supplementary Fig. 12). We found that when the system is constrained to be non-magnetic, the band gap vanishes instead of increasing. The projected DOS shows that the band gap closure is mainly due to the energy levels arising from oxygen-deficient Co moving closer to the Fermi level. Note that in the absence of spin polarization (see Supplementary Fig. 12a), tetrahedral and octahedral Co sites exhibit the same crystal field splitting and hence one observes a decrease in electron correlation relative to the spin-polarized case. When constraining the system in an FM state, we observed instead a finite, albeit smaller band gap, and the difference compared to the AFM ground state (Fig. 6b) is mainly in the localization of the states; in particular, the band stemming from tetrahedrally coordinated Co ions is less localized than in the ground state. Overall our results indicate that due to spin polarization, especially in the presence of AFM ordering, the oxygen-deficient Co sites experience a crystal field splitting different from the one of the octahedral sites, leading to an increase in localization of the d orbitals and thus to an increase in correlation. As a result, defect states introduced by oxygen vacancies are pushed well inside the valence band of the material, thus allowing for a band gap to open.
We further analyzed the effect of structural distortions on the band gap by constraining the angle of the G-AFM BM LCO structure to be close to the value it has in the initial perovskite structure (160° of all Co–O–Co angle). Lattice expansion was allowed in order to stabilize the G-AFM state. We found that the hybridized octahedral Co 3d–O 2p energy levels moved upwards, towards the Fermi level. The crystal field splitting of tetrahedral Co sites becomes similar to the octahedral case, showing the tight link between the system’s crystal field splitting and rotation distortions. These results suggest that lattice expansion alone cannot lead to an MIT, and that rotation is a key factor for the opening of the band gap. Hence in order to have a complete opening of the gap all elements identified here—volume expansion, rotation, and associated magnetic state change— are essential.
From mechanisms to devices
We close by discussing the impact of our results on device applications. Experiments have demonstrated that applying electric fields17, e.g. by the ion-gel gating method29,39, is a promising way to control VO in LSCO and in other perovskite materials such as nickelate where a synaptic behavior was generated by electrical stimuli40; hence it is interesting to estimate the required applied potential to modulate the vacancy concentration. The field applied to a non-metallic material (E0) can be evaluated using the following equation:
where E is the induced field, Ppolar is the permanent polarization vector (which is zero for the non-defective perovskite phase of LCO, but non zero in the presence of defects); \(\varepsilon ^\infty\) is the long-wavelength dielectric constant of the system and Eq. (3) is derived by assuming that the ionic and electronic components of the induced polarization are approximately the same (see Supplementary Discussion 6). Indeed, we know from experiments that the zero-frequency dielectric constant of LCO is about twice as \(\varepsilon ^\infty\)41. We computed \(\varepsilon ^\infty\)using density functional perturbation theory (DFPT) and Ppolar using the Berry phase approach. We obtained the ith component of E as Ei = η/di, where di is a supercell unit length and η is the electrochemical potential necessary to trigger the perovskite to BM phase transition. We computed \(\eta\) by requiring that vacancies can form spontaneously, i.e. that the oxygen vacancy free energy formation (Fvac) equals to zero; this requirement yields the condition: \(\left| \eta \right| = \frac{1}{2} \times \frac{{F_{{\mathrm{vac}}}}}{{n{\mathrm{e}}}}\) where Fvac was obtained by adding to the total energy of formation a vibrational free energy contribution computed from phonon frequencies (see Supplementary Equation 6). Figure 7a reports Fvac/n (free energy per vacancy) as a function of VO for both LCO and LSCO. To estimate the electrochemical potential required for a complete perovskite to BM phase transition, we considered the highest value of Fvac/n, yielding \(\left| \eta \right|\) = 0.6 V for LCO and 0.4 V for LSCO, respectively. We found that Sr doping generally decreases Fvac/n by ~0.4–0.5 eV relative to LCO, thus leading to a lower value of \(\left| \eta \right|\). These results indicate that Sr–VO segregation may occur during the topotactic transformation as the VO formation energy is lowered with Sr doping; however, we do not expect that a lower oxygen vacancy formation energy will affect the MIT mechanism described earlier, since also in the case of 100% segregation (i.e., SCO3−δ), a similar MIT transition has been shown to occur16,37, accompanied by an FM-to-AFM change during the perovskite to BM topotactic transition. We also note that the energy required to create oxygen vacancies is higher than that of the barrier for oxygen diffusions (the migration enthalpy in LCO is estimated to be ~0.7 eV from DFT+U calculations42 and experiments43); thus, the diffusion process is unlikely to be the bottleneck of topotactic phase transitions.

a Computed free energy of formation (Fvac) per oxygen vacancy as a function of oxygen vacancy content VO for LCO and LSCO. b Computed applied electric field and the corresponding voltage required to trigger an electrochemical reaction as a function of VO in LCO (see text). The voltage is averaged over different directions. Dotted and dashed lines in black represent the induced electric field (E) computed from the maximum vacancy free energy shown in b. a, b, and c denote the unit cell axis of LCO.
In Fig. 7b, we show the applied electric field E0 and the corresponding voltage as a function of VO assuming the maximum value of η computed above. Applying an electric field along the out-of-plane axis b leads to the lowest polarization value in the material and thus minimizes the difference between E0 and E. The upper limit of the voltage corresponding to E0 averaged along the three axes is estimated to be ~1.2 V for LCO (at VO = 8.3%) and 0.8 V for LSCO (at VO = 12.5%). The latter value is approximate, as it was obtained by assuming the same polarization for LSCO and LCO at 12.5% VO, and considering the value of η computed for LSCO.
The voltage estimated in our calculations is a promising value, qualitatively similar to that found experimentally (0.5 V) to trigger a lattice expansion in 50% Sr-doped LCO films by the introduction of oxygen vacancies29. It is a value comparable to the operation voltage (0.8 V) in VO2-based artificial neurons, which is considered low44 compared to that of CMOS-based neurons. A typical CMOS component requires a minimum operating voltage around 0.5 V, while a CMOS implementation of a neuron typically requires eight or more active components, thus leading to an increase of the total energy consumption44. Moreover, for an onset of the MIT or a transformation between different resistive states, a complete perovskite to BM phase transition is not necessary; therefore, a small variation of VO near the transition point suffices, e.g., near VO = 8.3% in LCO, which would further decrease the required potential value by ~0.14 V, due to a smaller value of Fvac per vacancy. Varying the Sr concentration may also help modulate the required potential, which we find decreases the upper bound by 0.4 V for Sr = 37.5%. In fact, in the 100% Sr case (SCOx), the relative stability of the perovskite and BM phases is reversed and a voltage of only 30 mV is found to be necessary to trigger the topotactic phase transition17.
Discussion
Using a series of first principles calculations, we characterized, at the microscopic level, the MIT in La1−xSrxCoO3−δ (x = 0 and x = 0.375) as a function of oxygen vacancy concentration (δ from 0 to 0.5). We found that the introduction of oxygen vacancies in the perovskite phase of LCO and LSCO lead to structural distortions, accompanied by a change of the magnetic state of the material and of the Co ion’s oxidation state. In particular, in LCO (LSCO) we observed a non-magnetic (FM) to AFM transition during the topotactic perovskite to BM transformation. Importantly, when constraining the system to be non-magnetic, all structural distortions, including the combined JT/breathing distortions and rotations, are suppressed, thus preventing a transition to an insulating state. We identified several structural transition pathways from the perovskite to the BM, with the lowest-energy pathway well separated (~0.8 eV per vacancy lower) from other metastable pathways. We found that the combined JT/breathing distortions lead to a localization of states near oxygen-deficient Co ions and thus an increased electron correlation effect, enabling the opening of a band gap between states associated to oxygen-deficient layers; the rotations triggered to lower the elastic energy of the system are further responsible for the opening of the gap between states connecting octahedral and tetrahedral layers, eventually leading to an MIT. We also showed that the band gap may be tuned by regulating the oxygen vacancy concentration, which is a necessary prerequisite to form different resistive states. These findings are consistent with the experimentally measured resistivity increase in the material, as a function of oxygen vacancy concentration and can be further validated by future optical measurements. We emphasize that in order to have a complete opening of the gap upon introduction of vacancies, all elements identified in our calculations—volume expansion, lattice distortions, and associated magnetic state change—are essential. The cooperative distortions predicted in our calculations, in particular the rotations of Co–oxygen units may be probed by X-ray and TEM measurements. It is interesting to note that the microscopic mechanism identified here in LSCO differ from that reported for the MIT in defective nickelates45. In nickelates, local electronic structure changes arising from excess electron in different crystal field splitting environments of the Ni ions can explain the band gap opening, in the absence of magnetic state changes and cooperative structural distortions.
Finally, we showed that the topotactic transformation in LCO and LSCO may be induced by an application of an electric field of the order of that found experimentally, and we presented a general model applicable to broad classes of TMOs. We note that a similar non-volatile resistive switching process may also be triggered in VO2 (ref. 46), a broadly studied TMO for neuromorphic applications3,47. However, in this case the Fermi level is moved to the conduction band48 as soon as vacancies are formed, and no intermediate resistive states may be created. In addition, in LSCO the formation of oxygen-deficient layers during the topotactic transformation facilitates the migration of oxygen ions49,50, which in turn increases the switching speed from a metal to an insulator, relative to vanadium oxide compounds.
The results obtained here, especially the interplay between structural, electronic, and magnetic properties identified in our calculations, fully characterize the MIT for the first time, and may be used to engineer specific resistive states in cobaltite films, e.g., by interfacial heterostructures51 and by controlling strain effects. It is well known that strain plays a key role in the presence of interfaces38,52,53,54; the combined structural and magnetic effects identified in our calculations may thus be used to engineer interfaces with desired properties for neuromorphic applications in cobaltites and likely manganites as well55, and in general to understand topotactic transformations in several classes of TMOs.
Methods
DFT+U calculation parameters
We used DFT, the PBE generalized gradient approximation with a Hubbard U correction and the projected augmented wave PP from PSlibrary56, and performed calculations using QUANTUM ESPRESSO (QE) code (V6.2 and V6.4.1)32,33. To obtain the ground state structures for different vacancy concentrations and La/Sr ratios, a \(\sqrt 2 \times 4 \times \sqrt 2\) perovskite supercell (La(Sr)8Co8O24, 40 atoms) was used, which was then optimized for each configurations within the orthorhombic lattice symmetry, using a plane-wave cutoff of 90 Ry and a 6 × 3 × 6 Monkhorst-Pack k-point grid. Sr doping positions were determined by sampling all symmetry-inequivalent positions as A-site cation (see Supplementary Fig. 6).
Data availability
Data that support the findings of this study will be available through the Qresp57 curator.
References
Grollier, J., Guha, S., Ohno, H. & Schuller, I. K. Preface to special topic: new physics and materials for neuromorphic computation. J. Appl. Phys. 124, 151801 (2018).
Jeong, D. S., Kim, K. M., Kim, S., Choi, B. J. & Hwang, C. S. Memristors for energy-efficient new computing paradigms. Adv. Electron. Mater. 2, 1600090 (2016).
Ramanathan, S. Quantum materials for brain sciences and artificial intelligence. MRS Bull. 43, 534–540 (2018).
Wang, Z. et al. Resistive switching materials for information processing. Nat. Rev. Mater. 5, 1–23 (2020).
Li, Y., Wang, Z., Midya, R., Xia, Q. & Yang, J. J. Review of memristor devices in neuromorphic computing: materials sciences and device challenges. J. Phys. D Appl. Phys. 51, 503002 (2018).
del Valle, J., Ramírez, J. G., Rozenberg, M. J. & Schuller, I. K. Challenges in materials and devices for resistive-switching-based neuromorphic computing. J. Appl. Phys. 124, 211101 (2018).
Sawa, A. Resistive switching in transition metal oxides. Mater. Today 11, 28–36 (2008).
Vankó, G., Rueff, J.-P., Mattila, A., Németh, Z. & Shukla, A. Temperature- and pressure-induced spin-state transitions in LaCoO3. Phys. Rev. B 73, 024424 (2006).
Mastin, J., Einarsrud, M.-A. & Grande, T. Structural and thermal properties of La1-xSrxCoO3-δ. Chem. Mater. 18, 6047–6053 (2006).
Lengsdorf, R. et al. Pressure-induced insulating state in (La,Sr)CoO3. Phys. Rev. B 69, 140403 (2004).
Louca, D., Sarrao, J. L., Thompson, J. D., Röder, H. & Kwei, G. H. Correlation of local Jahn-Teller distortions to the magnetic/conductive states of La1-xSrxCoO3. Phys. Rev. B 60, 10378–10382 (1999).
Mineshige, A. et al. Crystal structure and metal–insulator transition of La1−xSrxCoO3. J. Solid State Chem. 121, 423–429 (1996).
Wu, J. & Leighton, C. Glassy ferromagnetism and magnetic phase separation in La1 − xSrxCoO3. Phys. Rev. B 67, 174408 (2003).
Cheng, X. et al. Oxygen evolution reaction on La1–xSrxCoO3 perovskites: a combined experimental and theoretical study of their structural, electronic, and electrochemical properties. Chem. Mater. 27, 7662–7672 (2015).
Gilbert, D. A. et al. Ionic tuning of cobaltites at the nanoscale. Phys. Rev. Mater. 2, 104402 (2018).
Jeen, H. et al. Reversible redox reactions in an epitaxially stabilized SrCoOx oxygen sponge. Nat. Mater. 12, 1057–1063 (2013).
Lu, Q. & Yildiz, B. Voltage-controlled topotactic phase transition in thin-film SrCoOx monitored by in situ X-ray diffraction. Nano Lett. 16, 1186–1193 (2016).
Walter, J., Wang, H., Luo, B., Frisbie, C. D. & Leighton, C. Electrostatic versus electrochemical doping and control of ferromagnetism in ion-gel-gated ultrathin La0.5Sr0.5CoO3−δ. ACS Nano 10, 7799–7810 (2016).
Lu, N. et al. Electric-field control of tri-state phase transformation with a selective dual-ion switch. Nature 546, 124–128 (2017).
Gazquez, J. et al. Lattice mismatch accommodation via oxygen vacancy ordering in epitaxial La0.5Sr0.5CoO3-δ thin films. APL Mater. 1, 012105 (2013).
Perdew, J. P. & Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048–5079 (1981).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Sun, J., Ruzsinszky, A. & Perdew, J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 115, 036402 (2015).
Jang, J. H. et al. In situ observation of oxygen vacancy dynamics and ordering in the epitaxial LaCoO3 system. ACS Nano 11, 6942–6949 (2017).
Yao, L., Inkinen, S. & van Dijken, S. Direct observation of oxygen vacancy-driven structural and resistive phase transitions in La2/3Sr1/3MnO3. Nat. Commun. 8, 1–9 (2017).
Adler, S. B. Chemical expansivity of electrochemical ceramics. J. Am. Chem. Soc. 84, 2117–2119 (2001).
Marrocchelli, D., Perry, N. H. & Bishop, S. R. Understanding chemical expansion in perovskite-structured oxides. Phys. Chem. Chem. Phys. 17, 10028–10039 (2015).
Zuev, A. Y. U., Vylkov, A. I., Petrov, A. N. & Tsvetkov, D. S. Defect structure and defect-induced expansion of undoped oxygen deficient perovskite LaCoO3−δ. Solid State Ion. 179, 1876–1879 (2008).
Walter, J. et al. Ion-gel-gating-induced oxygen vacancy formation in epitaxial La0.5Sr0.5CoO3-δ films from in operando x-ray and neutron scattering. Phys. Rev. Mater. 1, 071403 (2017).
Guzmán-Verri, G. G., Brierley, R. T. & Littlewood, P. B. Cooperative elastic fluctuations provide tuning of the metal–insulator transition. Nature 576, 429–432 (2019).
Pavarini, E., Yamasaki, A., Nuss, J. & Andersen, O. K. How chemistry controls electron localization in 3d1 perovskites: a Wannier-function study. N. J. Phys. 7, 188 (2005).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 29, 465901 (2017).
Ishikawa, A., Nohara, J. & Sugai, S. Raman study of the orbital-phonon coupling in LaCoO3. Phys. Rev. Lett. 93, 136401 (2004).
Lee, J. H., Delaney, K. T., Bousquet, E., Spaldin, N. A. & Rabe, K. M. Strong coupling of Jahn-Teller distortion to oxygen-octahedron rotation and functional properties in epitaxially strained orthorhombic LaMnO3. Phys. Rev. B 88, 174426 (2013).
Mercy, A., Bieder, J., Íñiguez, J. & Ghosez, P. Structurally triggered metal-insulator transition in rare-earth nickelates. Nat. Commun. 8, 1677 (2017).
Lim, J. & Yu, J. Role of oxygen vacancy in the spin-state change and magnetic ordering in SrCoO3−δ. Phys. Rev. B 98, 085106 (2018).
Biškup, N. et al. Insulating ferromagnetic LaCoO3-δ films: a phase induced by ordering of oxygen vacancies. Phys. Rev. Lett. 112, 087202 (2014).
Lu, Q. et al. Bi-directional tuning of thermal transport in SrCoOx with electrochemically induced phase transitions. Nat. Mater. 19, 1–8 (2020).
Shi, J., Ha, S. D., Zhou, Y., Schoofs, F. & Ramanathan, S. A correlated nickelate synaptic transistor. Nat. Commun. 4, 1–9 (2013).
Schmidt, R., Wu, J., Leighton, C. & Terry, I. Dielectric response to the low-temperature magnetic defect structure and spin state transition in polycrystalline LaCoO3. Phys. Rev. B 79, 125105 (2009).
Ritzmann, A. M., Pavone, M., Muñoz-García, A. B., Keith, J. A. & Carter, E. A. Ab initio DFT+U analysis of oxygen transport in LaCoO3: the effect of Co3+ magnetic states. J. Mater. Chem. A 2, 8060–8074 (2014).
Ishigaki, T., Yamauchi, S., Mizusaki, J., Fueki, K. & Tamura, H. Tracer diffusion coefficient of oxide ions in LaCoO3 single crystal. J. Solid State Chem. 54, 100–107 (1984).
Lin, J. et al. Low-voltage artificial neuron using feedback engineered insulator-to-metal-transition devices. In 2016 IEEE International Electron Devices Meeting (IEDM), San Francisco, CA, USA 34.5.1–34.5.4 (2016).
Kotiuga, M. et al. Carrier localization in perovskite nickelates from oxygen vacancies. Proc. Natl Acad. Sci. USA 116, 21992–21997 (2019).
Jeong, J. et al. Suppression of metal-insulator transition in VO2 by electric field–induced oxygen vacancy formation. Science 339, 1402–1405 (2013).
Yang, Z., Ko, C. & Ramanathan, S. Oxide electronics utilizing ultrafast metal-insulator transitions. Annu. Rev. Mater. Res. 41, 337–367 (2011).
Chen, L. et al. First-principles study of the effect of oxygen vacancy and strain on the phase transition temperature of VO2. RSC Adv. 6, 86872–86879 (2016).
Mitra, C., Meyer, T., Lee, H. N. & Reboredo, F. A. Oxygen diffusion pathways in brownmillerite SrCoO2.5: influence of structure and chemical potential. J. Chem. Phys. 141, 084710 (2014).
Zhang, Q. et al. Atomic-resolution imaging of electrically induced oxygen vacancy migration and phase transformation in SrCoO2.5-δ. Nat. Commun. 8, 1–6 (2017).
Lee, S. et al. Strong orbital polarization in a cobaltate-titanate oxide heterostructure. Phys. Rev. Lett. 123, 117201 (2019).
Yang, H. W. et al. Anomalous magnetism in strained La1-xSrxCoO3 epitaxial films (0 ≤ x ≤ 0.5). Sci. Rep. 4, 1–7 (2014).
Rondinelli, J. M. & Spaldin, N. A. Structure and properties of functional oxide thin films: insights from electronic-structure calculations. Adv. Mater. 23, 3363–3381 (2011).
Lee, J. H. & Rabe, K. M. Coupled magnetic-ferroelectric metal-insulator transition in epitaxially strained SrCoO3 from first principles. Phys. Rev. B 107, 067601 (2011).
Zhou, Y., Kouser, S., Borisevich, A. Y., Pantelides, S. T. & May, S. J. Evidence for interfacial octahedral coupling as a route to enhance magnetoresistance in perovskite oxide superlattices. Adv. Mater. Interfaces 7, 1901576 (2020).
Dal Corso, A. Pseudopotentials periodic table: from H to Pu. Comput. Mater. Sci. 95, 337–350 (2014).
Govoni, M. et al. Qresp, a tool for curating, discovering and exploring reproducible scientific papers. Sci. Data 6, 190002 (2019).
Parsons, T. G., D’Hondt, H., Hadermann, J. & Hayward, M. A. Synthesis and structural characterization of La1−x Ax MnO2.5 (A=Ba, Sr, Ca) phases: mapping the variants of the brownmillerite structure. Chem. Mater. 21, 5527–5538 (2009).
Acknowledgements
This research was conducted as part of the Quantum Materials for Energy Efficient Neuromorphic Computing, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Basic Energy Sciences under award DE-SC0019273. We thank Shaobo Cheng, Peter Littlewood, Chris Leighton, Shriram Ramanathan, Yayoi Takamura, Wennie Wang, and Yimei Zhu for many useful discussions and comments on the manuscript. This research used computational resources of the University of Chicago’s Research Computing Center and at the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility supported by the Office of Science of the US Department of Energy.
Author information
Authors and Affiliations
Contributions
Both authors designed the research and wrote the manuscript. S.Z. performed all calculations.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, S., Galli, G. Understanding the metal-to-insulator transition in La1−xSrxCoO3−δ and its applications for neuromorphic computing. npj Comput Mater 6, 170 (2020). https://doi.org/10.1038/s41524-020-00437-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-020-00437-w
This article is cited by
-
Tunable ferroelectricity in oxygen-deficient perovskites with Grenier structure
npj Computational Materials (2023)
