
Abstract
Breast adenomyoepitheliomas (AMEs) are rare epithelial-myoepithelial neoplasms that may occasionally produce myxochondroid matrix, akin to pleomorphic adenomas (PAs). Regardless of their anatomic location, PAs often harbor rearrangements involving HMGA2 or PLAG1. We have recently shown that the repertoire of somatic genetic alterations of AMEs varies according to their estrogen receptor (ER) status; whilst the majority of ER-positive AMEs display mutually exclusive PIK3CA or AKT1 hotspot mutations, up to 60% of ER-negative AMEs harbor concurrent HRAS Q61 hotspot mutations and mutations affecting either PIK3CA or PIK3R1. Here, we hypothesized that a subset of AMEs lacking these somatic genetic alterations could be underpinned by oncogenic fusion genes, in particular those involving HMGA2 or PLAG1. Therefore, we subjected 13 AMEs to RNA-sequencing for fusion discovery (n = 5) and/or fluorescence in situ hybridization (FISH) analysis for HMGA2 and PLAG1 rearrangements (n = 13). RNA-sequencing revealed an HMGA2-WIF1 fusion gene in an ER-positive AME lacking HRAS, PIK3CA and AKT1 somatic mutations. This fusion gene, which has been previously described in salivary gland PAs, results in a chimeric transcript composed of exons 1–5 of HMGA2 and exons 3–10 of WIF1. No additional in-frame fusion genes or HMGA2 or PLAG1 rearrangements were identified in the remaining AMEs analyzed. Our results demonstrate that a subset of AMEs lacking mutations affecting HRAS and PI3K pathway-related genes may harbor HMGA2-WIF1 fusion genes, suggesting that a subset of breast AMEs may be genetically related to PAs or that a subset of AMEs may originate in the context of a PA.
Similar content being viewed by others


Introduction
Breast adenomyoepitheliomas (AMEs) are rare neoplasms with dual epithelial-myoepithelial differentiation,1 composed of gland-like structures containing an inner layer of pink, eosinophilic epithelial cells and an abluminal layer of often clear, myoepithelial cells. AMEs can display a variety of histologic appearances, and be either estrogen receptor (ER)-positive or ER-negative.1,2 Although there is overlap in the histologic features of ER-positive and ER-negative AMEs, we have recently shown that the repertoire of genetic alterations of these tumors vary according to their ER status.3 Whilst ER-negative AMEs harbor HRAS Q61 hotspot mutations co-occurring with mutations affecting PIK3CA or PIK3R1 in up to 60% of cases, the majority of ER-positive AMEs were found to display seemingly mutually exclusive PIK3CA or AKT1 activating hotspot mutations.3
In the spectrum of histologic appearances of AMEs, myxochondroid matrix has been occasionally described.4 This type of matrix bears histologic resemblance to the matrix of pleomorphic adenomas (PAs),1 epithelial-myoepithelial neoplasms that may arise in various anatomic locations, including the breast.5 PAs are underpinned by recurrent gene rearrangements involving HMGA2 or PLAG1 in up to 65% of cases, regardless of their anatomic origin.6,7,8 Due to the overlapping histologic appearances of AMEs and PAs, we sought to determine whether a subset of AMEs, primarily those lacking mutations affecting known drivers (e.g., HRAS or PI3K pathway-related genes), would be genetically related to PAs, and would be underpinned by fusion genes, in particular those involving HMGA2 and PLAG1.
Results
HMGA2-WIF1 fusion gene in an ER-positive AME
Thirteen breast AMEs, whose whole-exome, targeted capture and/or Sanger sequencing and ER status were previously described in Geyer et al,3 were included in this study (Table 1). Six cases were ER-negative and seven were ER-positive. Four ER-negative AMEs harbored concurrent PIK3CA and HRAS mutations (4/6), one harbored an HRAS Q61K mutation and concurrent likely pathogenic PIK3R1 mutations (1/6), and one was HRAS wild-type and harbored a PIK3CA mutation (1/6). Five ER-positive AMEs harbored PIK3CA mutations (5/7), and all were wild-type for HRAS. None of the cases harbored mutations affecting the AKT1 E17 hotspot locus (Fig. 1a and Supplementary Fig. 1). Notably, all HRAS and PIK3CA mutations were classical activating hotspot mutations, except for one PIK3CA mutation (Q546) which targeted a hotspot residue and was predicted to be likely pathogenic (Fig. 1a).

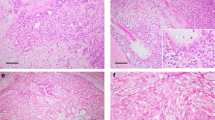
Fusion genes involving HMGA2 or PLAG1 and somatic mutations targeting HRAS, PIK3CA, AKT1 and PIK3R1 in breast adenomyoepitheliomas. a Heatmap depicting fusion gene and somatic mutations targeting HRAS Q61, PIK3CA and AKT E17 hotspot loci and PIK3R1 mutations identified in breast adenomyoepitheliomas (AMEs; n = 13). Cases are shown in columns and genes in rows. Hotspot mutations are annotated as per Chang et al.23 b Representative Sanger sequencing electropherograms of HRAS Q61 and PIK3CA hotspot loci in AM16. c Representative hematoxylin and eosin micrographs of an AME harboring an HMGA2-WIF1 fusion gene (AM16), and micrographs depicting p63 and estrogen receptor expression. Scale bars, 500 μm (upper left), 100 μm (upper right) and 50 μm (middle and lower panels). ER estrogen receptor, FISH fluorescence in situ hybridization, SNV single nucleotide variant, WT wild-type
To determine whether AMEs lacking HRAS Q61 hotspot mutations would harbor fusion genes, we subjected five HRAS wild-type AMEs with available material to RNA-sequencing analysis for an unbiased detection of expressed fusion genes (Supplementary Fig. 1). Using a validated pipeline for the de novo discovery of fusion genes,9 we identified an HMGA2-WIF1 fusion gene in an ER-positive HRAS-/PIK3CA-/AKT1-wild-type AME (AM16) (Figs. 1a-c, Table 1 and Supplementary Table 1). The HMGA2-WIF1 fusion gene identified in AM16 results in a chimeric transcript encompassing all five exons and the initial segment of the 3′ UTR of HMGA2 fused to exons 3–10 of WIF1, and is predicted to be translated to a full length HMGA2 protein and an N-terminal truncated WIF1 protein, with a truncated WIF domain. WIF1 encodes for a tumor suppressor that modulates Wnt signaling, a role that requires an intact WIF domain.10
The AME found to harbor the HMGA2-WIF1 fusion gene (AM16) displayed the typical histologic features of AMEs,3 and constituted a well-circumscribed lesion with pushing borders, surrounded by a thick fibrous capsule. This lesion displayed a nodular architecture and a mixed tubular and papillary growth pattern. No cellular atypia, mitotic activity or necrosis was identified (Fig. 1c). Focal areas with conspicuous stroma with myxoid quality were observed (Fig. 1c). The myoepithelial component was highlighted by p63 on immunohistochemical analysis, and strong ER expression was observed in the epithelial component (Fig. 1c).
Given that in the salivary glands, epithelial-myoepithelial carcinomas, the salivary gland counterpart of breast AME, can occasionally originate in the context of a PA (i.e., the so-called carcinoma ex-PA),11 we sought to define if AM16 would have areas diagnostic of PA. An independent pathology review of all slides available from this AME by five pathologists failed to reveal any areas that would be consistent with a diagnosis of PA.
Breast AMEs lack recurrent HMGA2 or PLAG1 rearrangements
None of the additional AMEs subjected to RNA-sequencing harbored other fusion genes involving gene partners previously described in PAs,12 in myoepitheliomas of other anatomical sites (i.e., EWSR1 and FUS rearrangements),13,14 or in other tumors displaying myoepithelial differentiation (i.e., CRTC1-MAML2 fusion gene in mucoepidermoid carcinomas or MYB and MYBL1 rearrangements in adenoid cystic carcinoma).9,15 No additional likely pathogenic in-frame fusion gene was identified in the cases subjected to RNA-sequencing analysis (Supplementary Table 1).
Given that HMGA2 and PLAG1 rearrangements have been described in other neoplasms with epithelial-myoepithelial differentiation, in particular in PAs, we sought to define whether AMEs may harbor fusion genes known to underpin PAs. We subjected the five AMEs analyzed by RNA-sequencing and all the other AMEs included in this study (n = 8) to FISH using HMGA2 and PLAG1 dual-color break apart probes (Table 1 and Supplementary Fig. 1). This analysis confirmed the presence of an HMGA2 rearrangement in both the epithelial and myoepithelial components of AM16 (Fig. 2b) and did not reveal any additional AMEs harboring HMGA2 or PLAG1 rearrangements (Table 1).

HMGA2-WIF1 fusion gene identified in the epithelial and myoepithelial cells of a breast adenomyoepithelioma. a Schematic representation of the HMGA2-WIF1 fusion transcript identified in AM16, including the exons and domains involved. HMGA2 is on the (+) DNA strand and WIF1 on the (−) DNA strand. The breakpoints of the 5’ and 3’ partner genes are represented as black vertical lines. Eight spanning reads were found to cross the genomic breakpoint of the HMGA2-WIF1 chimeric transcript and are depicted aligned to the predicted junction sequence. b Representative hematoxylin and eosin and FISH micrographs of the epithelial and myoepithelial components of AM16 using HMGA2 dual-color break apart probes (red, 5′ HMGA2; green, 3′ HMGA2). aa aminoacid, AcD acidic domain, DBD DNA binding domain, E epithelium, M myoepithelium, SpD spacer domain. Scale bar, 50 μm
Discussion
We have previously shown that approximately 60% ER-positive AMEs harbor PIK3CA or AKT1 mutations, whereas up to 60% of ER-negative AMEs are characterized by HRAS Q61 mutations concurrent with mutations affecting genes of the PI3K signaling pathway.3 Given the occasional histologic similarities between AMEs and PAs, and the fact that a subset of AMEs lack a known driver genetic alteration in the form of somatic mutations affecting protein coding genes, we posited that a subset of AMEs may harbor oncogenic fusion genes previously described in other myoepithelial lesions including PAs. Our analyses resulted in the identification of an ER-positive HRAS-/PIK3CA-/AKT1-wild type AME harboring an HMGA2-WIF1 fusion gene, which has been described in PAs and carcinomas ex-PA of the salivary gland.10,16
The high-mobility group AT-hook 2 (HMGA2) gene encodes for a transcriptional regulator of genes involved in cell proliferation and cell death.17 HMGA2 overexpression plays a key role in oncogenic transformation through several mechanisms, such as the induction of E2F1 and AP1 activity, promotion of cyclin A expression, inactivation of p53-dependent apoptosis, and activation of the TGF-β signaling pathway.17,18 The Wnt signaling pathway is regulated by secreted antagonists that bind to Wnt proteins, preventing ligand-receptor interactions,19 such as WIF1.20 WIF1 consists of an N-terminal secretion signal, five EGF-like domains, a hydrophilic C-terminus, and a WIF domain, which is required for binding to Wnt proteins and for the tumor suppressor properties of WIF1.19 The HMGA2-WIF1 fusion gene identified in AM16 has been shown to result in increased HMGA2 expression, presumably due to loss of regulatory sites in its 3′ UTR,21 and decreased WIF1 expression.10 The HMGA2-WIF1 chimeric transcript identified in our study is predicted to encode a full length HMGA2 protein and an N-terminally truncated WIF1 protein harboring a truncated WIF domain. Given that the HMGA2 breakpoint maps to its 3’ UTR after the stop codon, it is possible that WIF1 may not even be translated, akin to a rearrangement involving the same HMGA2 and WIF1 exons previously described in a PA arising in the salivary gland.16 In addition, HMGA2 and WIF1 display opposite transcriptional orientations, and this fusion gene may stem from a cryptic paracentric inversion (Fig. 2a).16
Taken together, the HMGA2-WIF1 fusion identified here might result in increased expression of HMGA2, with ensuing activation of TGF-β signaling, along with derepression of Wnt signaling. Taken together, our findings suggest that a subset of AMEs lacking genetic alterations involving genes of the RAS-MAPK pathway may be underpinned by fusion genes resulting in the activation of alternative signaling pathways, such as TGF-β and Wnt.
One could posit that the AME harboring the HMGA2-WIF1 fusion gene described in this study would, in fact, constitute a breast PA. This case was independently reviewed by five breast pathologists who concurred in the diagnosis of AME. It should be noted, however, that despite being a bona fide AME, this case focally displayed increased myxoid stroma, bearing some resemblance to, but not fulfilling the diagnostic criteria for, a breast PA. Another potential explanation for the presence of this fusion gene in AM16 is that it would constitute the breast equivalent of the salivary gland epithelial-myoepithelial carcinoma ex-PA.11 The central pathology review of all slides available from this case failed to reveal any areas diagnostic of PA. Therefore, our findings suggest that a subset of AMEs share not only morphologic features with PAs, but may also resemble PAs at the genetic level. We cannot rule out, however, that AM16 developed in the context of a PA, which was subsequently obliterated by the outgrowth of the AME.
The FISH analysis of AM16 revealed the presence of the HMGA2-WIF1 both in the epithelial and myoepithelial cells of the tumor (Fig. 2b). This observation is consistent with the notion that in AMEs, both the epithelial and myoepithelial components are neoplastic and clonally related, even though in this AME (AM16), the epithelial component was ER-positive, whereas the myoepithelial component was ER-negative.
Our study was several limitations, such as the small size of our cohort and the fact that we included only two HRAS-/PIK3CA-/AKT1-wild type AMEs, given that no additional material from other AMEs was available for transcriptomic or FISH analysis. The limited sample size of our study precludes definitive conclusions regarding the relationship between ER status and the presence of the HMGA2-WIF1 fusion gene in AMEs to be drawn. Despite these limitations, our findings demonstrate that AMEs lacking mutations affecting HRAS, PIK3CA and AKT1 may harbor the HMGA2-WIF1 fusion gene, previously described in salivary gland PAs and carcinomas ex-PA,10,16 suggesting that a subset of AMEs may be genetically related to PAs or that AMEs may originate in the context of breast PAs.
Methods
Cases and DNA sequencing data
This study was approved by the Institutional Review Boards (IRBs) and local research ethics committees of the authors’ institutions. Patient consent was obtained if required by the approved IRB protocols. In this study we included thirteen breast AMEs, retrieved from the authors’ institutions and previously described by Geyer et al.3 Whole-exome sequencing, MSK-IMPACT and Sanger sequencing data, and immunohistochemical data were retrieved from Geyer et al.3 ER status was assessed by immunohistochemistry according to the current ASCO/CAP guidelines.22 Hotspot mutations are annotated as per Chang et al.23 For power calculations, if we posited that an HRAS-wild type AMEs would be underpinned by a recurrent fusion gene and that this fusion gene would be present in ≥ 70% of cases akin to recurrent fusion genes in other tumor types,9,24,25,26 sequencing analysis of five samples would confer 80% power for its detection.
RNA-sequencing and the identification of fusion transcripts
RNA-sequencing was performed on five HRAS-wild type AMEs according to standard protocols employed at the Integrated Genomics Operation of Memorial Sloan Kettering Cancer Center (MSKCC).27 In brief, paired-end massively parallel RNA-sequencing (2 × 50 bp) was performed on a HiSeq2000 (Illumina), as previously described.28 Read pairs supporting chimeric transcripts were identified using deFuse,29 and INTEGRATE,30 followed by exclusion of candidate fusion transcripts found in a set of 287 normal breast tissues from the TCGA dataset,31 as previously described.28 The Bayesian probability of the remaining candidate fusion genes, supported by at least two spanning reads, to constitute drivers was annotated using OncoFuse,32 as previously described.28
Fluorescence in situ hybridization (FISH)
All cases included in this study (n = 13) were subjected to FISH analysis for HMGA2 and PLGA1 using dual-color break-apart probes following validated protocols at the MSKCC Molecular Cytogenetics Core, as previously described.33 The probe mix consisted of bacterial artificial chromosome (BAC) clones mapping to 5′ HMGA2 (RP11-230G5, RP11-662G15; red) and 3′ HMGA2 (RP11-937C6, RP11-167E10; green), and BAC clones mapping to 5′ PLAG1 (RP11-92A9, RP11-111I18; red) and 3′ PLAG1 (RP11-144E19, RP11-246A9; green). A minimum of 50 interphase nuclei were analyzed for HMGA2 or PLAG1 rearrangements. Cases were considered positive for rearrangement if separation of the 5′ (red) and 3′ (green) signals (>2 signal width apart) was identified in >15% tumor cells. FISH analyses were performed with observers blinded to the results of the RNA-sequencing analysis.
Data availability
RNA-sequencing data that support the findings of this study have been deposited in the NCBI Sequence Read Archive (SRA) under the accession code SRP158271. WES and MSK-IMPACT sequencing data retrieved from Geyer et al3 are available in SRA under the accession numbers SRP065277 and SRP065302, respectively. All available data are available from the authors.
References
Yoon, J. Y. & Chitale, D. Adenomyoepithelioma of the breast: a brief diagnostic review. Arch. Pathol. Lab. Med. 137, 725–729 (2013).
Lakhani S. R., Ellis I. O., Schnitt S. J., Tan P. H., van de Vijver M. J. WHO Classification of Breast Tumors. (International Agency for Research Cancer (IARC), Lyon, 2012).
Geyer, F. C. et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat. Commun. 9, 1816 (2018).
McLaren, B. K., Smith, J., Schuyler, P. A., Dupont, W. D. & Page, D. L. Adenomyoepithelioma: clinical, histologic, and immunohistologic evaluation of a series of related lesions. Am. J. Surg. Pathol. 29, 1294–1299 (2005).
Foschini, M. P. et al. The morphological spectrum of salivary gland type tumours of the breast. Pathology 49, 215–227 (2017).
Stenman, G. Fusion oncogenes in salivary gland tumors: molecular and clinical consequences. Head. Neck Pathol. 7(Suppl 1), S12–S19 (2013).
Bahrami, A., Dalton, J. D., Krane, J. F. & Fletcher, C. D. A subset of cutaneous and soft tissue mixed tumors are genetically linked to their salivary gland counterpart. Genes Chromosomes Cancer 51, 140–148 (2012).
Pareja, F. et al. Pleomorphic adenoma and mucoepidermoid carcinoma of the breast are underpinned by fusion genes. Mod. Pathol. 31, 98–98 (2018).
Kim, J. et al. MYBL1 rearrangements and MYB amplification in breast adenoid cystic carcinomas lacking the MYB-NFIB fusion gene. J. Pathol. 244, 143–150 (2018).
Queimado, L., Lopes, C. S. & Reis, A. M. WIF1, an inhibitor of the Wnt pathway, is rearranged in salivary gland tumors. Genes Chromosomes Cancer 46, 215–225 (2007).
Antony, J., Gopalan, V., Smith, R. A. & Lam, A. K. Carcinoma ex pleomorphic adenoma: a comprehensive review of clinical, pathological and molecular data. Head. Neck Pathol. 6, 1–9 (2012).
Antonescu, C. R. et al. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer 52, 675–682 (2013).
Huang, S. C. et al. Novel FUS-KLF17 and EWSR1-KLF17 fusions in myoepithelial tumors. Genes Chromosomes Cancer 54, 267–275 (2015).
Antonescu, C. R. et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer 49, 1114–1124 (2010).
Tonon, G. et al. t(11;19)(q21; p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat. Genet. 33, 208–213 (2003).
Persson, F. et al. High-resolution genomic profiling of adenomas and carcinomas of the salivary glands reveals amplification, rearrangement, and fusion of HMGA2. Genes Chromosomes Cancer 48, 69–82 (2009).
Fusco, A. & Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 7, 899–910 (2007).
Morishita, A. et al. HMGA2 is a driver of tumor metastasis. Cancer Res 73, 4289–4299 (2013).
Malinauskas, T., Aricescu, A. R., Lu, W., Siebold, C. & Jones, E. Y. Modular mechanism of Wnt signaling inhibition by Wnt inhibitory factor 1. Nat. Struct. Mol. Biol. 18, 886–893 (2011).
Hsieh, J. C. et al. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature 398, 431–436 (1999).
Mayr, C., Hemann, M. T. & Bartel, D. P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315, 1576–1579 (2007).
Hammond, M. E. et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 28, 2784–2795 (2010).
Chang, M. T. et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163 (2016).
Martelotto, L. G. et al. Genomic landscape of adenoid cystic carcinoma of the breast. J. Pathol. 237, 179–189 (2015).
Wetterskog, D. et al. Adenoid cystic carcinomas constitute a genomically distinct subgroup of triple-negative and basal-like breast cancers. J. Pathol. 226, 84–96 (2012).
Ashworth, A., Lord, C. J. & Reis-Filho, J. S. Genetic interactions in cancer progression and treatment. Cell 145, 30–38 (2011).
Weinreb, I. et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat. Genet. 46, 1166–1169 (2014).
Piscuoglio, S. et al. Integrative genomic and transcriptomic characterization of papillary carcinomas of the breast. Mol. Oncol. 8, 1588–1602 (2014).
McPherson, A. et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 7, e1001138 (2011).
Zhang, J. et al. INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 26, 108–118 (2015).
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Shugay, M., Ortiz de Mendibil, I., Vizmanos, J. L. & Novo, F. J. Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 29, 2539–2546 (2013).
Piscuoglio, S. et al. Uterine adenosarcomas are mesenchymal neoplasms. J. Pathol. 238, 381–388 (2016).
Acknowledgements
This study was funded by the Breast Cancer Research Foundation. Research reported in this publication was funded in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (grant No P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
B.W. and J.S.R.-F. conceived the study. H.Y.W., Z.V., J.P., B.P.R., I.O.E., E.B. and E.A.R. provided tissue samples. F.P., F.C.G, M.E., A.P.M.S., E.A.R., I.O.E. and J.S.R.-F. performed the pathology review. Data acquisition, interpretation and analysis were performed by F.P., F.C.G., D.N.B., A.L., A.P.M.S., R.G.-M., A.d.C.P., P.S. and A.A.J. The manuscript was initially drafted by F.P. and J.S.R.-F., and all authors edited and approved the final draft of the manuscript. F.P. and F.C.G. contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
J.S.R.-F. reports personal/consultancy fees from VolitionRx, Page.AI, Goldman Sachs, Grail, Ventana Medical Systems and Genentech, outside the submitted work. The remaining authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pareja, F., Geyer, F.C., Brown, D.N. et al. Assessment of HMGA2 and PLAG1 rearrangements in breast adenomyoepitheliomas. npj Breast Cancer 5, 6 (2019). https://doi.org/10.1038/s41523-018-0101-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-018-0101-7
This article is cited by
-
The clinical behavior and genomic features of the so-called adenoid cystic carcinomas of the solid variant with basaloid features
Modern Pathology (2022)
-
Triple-negative breast carcinomas of low malignant potential: review on diagnostic criteria and differential diagnoses
Virchows Archiv (2022)
-
Problematic breast tumors reassessed in light of novel molecular data
Modern Pathology (2021)
-
Pleomorphic adenomas and mucoepidermoid carcinomas of the breast are underpinned by fusion genes
npj Breast Cancer (2020)
-
New Insights into Tumor Heterogeneity: A Case of Solid-Oncocytic Epithelial-Myoepithelial Carcinoma of the Parotid Gland Harboring a HRAS and Heterogeneous Terminating ARID1A Mutation
Head and Neck Pathology (2020)
