
Abstract
Here an improved carrot reference genome and resequencing of 630 carrot accessions were used to investigate carrot domestication and improvement. The study demonstrated that carrot was domesticated during the Early Middle Ages in the region spanning western Asia to central Asia, and orange carrot was selected during the Renaissance period, probably in western Europe. A progressive reduction of genetic diversity accompanied this process. Genes controlling circadian clock/flowering and carotenoid accumulation were under selection during domestication and improvement. Three recessive genes, at the REC, Or and Y2 quantitative trait loci, were essential to select for the high α- and β-carotene orange phenotype. All three genes control high α- and β-carotene accumulation through molecular mechanisms that regulate the interactions between the carotenoid biosynthetic pathway, the photosynthetic system and chloroplast biogenesis. Overall, this study elucidated carrot domestication and breeding history and carotenoid genetics at a molecular level.
Similar content being viewed by others


Main
Carrot (Daucus carota L., 2n = 2x = 18) is known for being among the richest sources of dietary provitamin A carotenoids, α- and β-carotene. Carrot is grown globally, and production has risen steadily during the past 50 years1, with extensive adaptation to Asia, Europe and the Americas, including subtropical climates. The adaptability, nutritional value and diversification of carrot for fresh and processed markets (for example, as a natural colourant) have been the driving forces for this growth1,2. These attributes raise expectations that new cultivars can be developed to meet market demands and sustain expanded production under increasingly challenging environmental growing conditions. Advancing research that can enable the implementation of molecular-assisted breeding strategies is critical to support these efforts.
Carrot germplasm collections include an array of cultivars, landraces and wild carrots, which harbour a wide range of phenotypic diversity useful for breeding3. This crop is propagated via seed, and, as a primarily outcrossing species, hybridization within and between carrot populations is common, which facilitates gene flow within carrot germplasm4. It is currently well accepted that cultivated carrot germplasm can be separated into two major groups: Eastern and Western4. The Eastern group includes the first domesticated carrots, which were purple or yellow and originated in the region spanning Asia Minor and central Asia. According to historical records, Eastern carrots were used as a food crop in the Iranian Plateau and Persia in the tenth century4. The Western group, primarily represented by orange carrots, first appeared in Europe during the seventeenth century and quickly became the predominant carrot type grown and consumed globally5. Recent molecular studies clearly separated Wild, Eastern and Western carrot populations and indicated Eastern carrots as the progenitor of Western carrots4,6. Despite recent advances in understanding the genetic structure of the carrot germplasm and phylogenetic relationships between Eastern, Western and Wild carrot populations, the demographic events that characterized carrot domestication and improvement have not been investigated. Furthermore, previous studies have indicated that after carrot domestication, a genetic bottleneck was either absent or marginal6,7,8. Due to the lack of whole-genome-wide analysis, the impact of domestication and improvement on genetic diversity within carrot germplasm remains unresolved.
The selection of orange carrots in the 1500s resulted in carrots that accumulate high levels of α- and β-carotene, which, as later discovered in the 1800s and 1900s, improved the nutritional value of the crop. Indeed, ‘carotene’, the first carotenoid discovered, was initially isolated from carrot juice extracts in the 1800s and was observed to be medically active9. The most health benefit of carrot was demonstrated with the discovery of vitamin A in 191310 and the observation that dietary carotenoids from plants can prevent vitamin A deficiency11. Numerous studies have demonstrated additional health benefits associated with carotenoids12, which probably contributed to the increased popularity of orange carrots and their consumption. For instance, carrot represents the most abundant plant source of the provitamin A carotenoids, α- and β-carotene, in the US diet today13. Given the importance of these compounds, increasing the α- and β-carotene content in orange carrots and studying the genetic mechanism controlling their accumulation have been primary targets of carrot breeding and genetic studies14. To date, two loci named Or and Y2 have been associated with high α- and β-carotene and thus the appearance of an orange phenotype7,14,15,16,17. An Orange like gene homologue (Or-like) was identified as candidate gene controlling the Or locus, while several candidate genes have been identified in the genomic region associated with the Y2 locus7,16. Findings from these previous studies indicate that none of the proposed candidate genes encode the biosynthetic enzymes in the carotenoid pathway. Instead, they suggest that the accumulation of high α- and β-carotene in carrots is regulated through the light-response feedback mechanism and chloroplast biogenesis7. The rapid increase in the popularity of orange carrot probably led to the fixation of many alleles responsible for carotenoid presence, but the roles of loci controlling carotenoid accumulation and other important domestication and improvement traits in carrot have been only partially evaluated using reduced sequence representation methods (for example, GBS and DarT)7,18 and biparental populations. As a result, within the Or and Y2 loci, candidate genes and causal mutations have not been fully confirmed.
To advance knowledge about carrot domestication and modern breeding, we present an improved carrot genome assembly of the double haploid orange Nantes-type carrot DH1, alongside a large-scale resequencing study that represents a global collection of carrot germplasm. These data enabled us to uncover the demographic events that characterized carrot domestication and improvement and the genes that were selected during these processes. The outcomes of this study and the DH1 v.3 genome will provide improved genomic insights into traits important for carrot domestication and improvement.
Results
An improved carrot genome assembly and annotation
The new DH1 v.3.0 (hereafter DH1 v.3) assembly was developed using long-read (PacBio and Oxford Nanopore) and Illumina Hi-C sequence data (Supplementary Tables 1–3). The assembly spans 440.7 Mb, assembled into nine chromosomes that represent ~93% of the estimated genome size (473 Mb)15 (Table 1, Extended Data Fig. 1 and Supplementary Tables 4 and 5). Quality assessment for assembly contiguity, gene space coverage and sequence contaminations confirmed that the assembly reached high-quality standards (Supplementary Note, Extended Data Figs. 2 and 3, and Supplementary Tables 6–8). The overall N50 was 51 Mb, the contig N50 was over 6.0 Mb and the longest contig was over 28.0 Mb, covering much of the long arm of chromosome 4 (Fig. 1a and Extended Data Fig. 2). Compared with the DH1 v.2 assembly15, developed using Illumina short-read sequencing technology, DH1 v.3 has a >4-fold higher scaffold, a 193-fold higher contig N50 (Table 1) and about 21% newly anchored sequences. Also, a moderate number of sequence corrections were made around centromeric regions (Fig. 1a and Extended Data Fig. 2). As a result of these improvements, the DH1 v.3 assembly includes about 53.1 Mb (11.3%) more repetitive sequences (Supplementary Tables 9 and 10), largely represented by relatively young long terminal repeat (LTR) elements located in centromeric and pericentromeric regions (Fig. 1a, Supplementary Note and Extended Data Figs. 4–6) and a much higher LTR Assembly Index (22.88 versus 5.09) (Extended Data Fig. 7).

a, Circos display of the DH1 v.3 genomic features: (i) the chromosome (chr) coordinates in Mb; (ii) gene frequency (bin size, 100 kb) (red line) and alignments of v.2 contigs versus v.3 chromosomes (blue heat map); (iii) gaps (Ns) in the v.3 genome assembly; (iv) the telomeric repeat frequency histogram (×100) (red) and the centromeric repeat frequency histogram (blue); (v) heat map representing the distribution of gypsy+copia transposable elements (TEs) (bin size, 250 kb); (vi) heat map representing new v.3 genes (bin size, 250 kb); (vii) the distribution of new transcription factors; and (viii) the distribution of new resistance genes. b, Schematic representation of the genomic region spanning DcMYB6 in the DH1 v.2 and v.3 genome assemblies. DcMYB6 was not assembled at the chromosome level in DH1 v.2, probably due to complex repetitive sequences flanking the gene. The region including DcMYB6 was fully assembled in the DH1 v.3 assembly.
In total, 36,211 protein-encoding genes were predicted in the DH1 v.3 genome (Table 1, Supplementary Tables 11 and 12 and Supplementary Note). Over 99.5% of the predicted genes had a match with Single Copy Ortholog, and 99.3% could be annotated (Supplementary Tables 13 and 14). Isoform analysis indicated that 15,723 predicted genes had more than one isoform, which can potentially change protein function by altering the conserved protein domains (Supplementary Note, Supplementary Tables 15 and 16, and Extended Data Figs. 8 and 9). In v.3, 4,103 additional genes were predicted compared with v.2, of which 3,084 were located in newly assembled sequences (Fig. 1a and Supplementary Table 17) and 98.2% were expressed, confirming the reliability of these predictions. Genes located in new regions were particularly enriched for gene families involved in electron transport (for example, CB5-B, PSBO-2 and CICDH)19,20,21 and functioning in highly conserved processes such as photosynthesis or regulation of redox homeostasis (Supplementary Table 18). Comparing the alignments of genes in DH1 v.3 and DH1 v.2, 19,353 gene models had an identical start and end position, and 16,858 genes (48%) either were new in DH1 v.3 or had a different start or end position (Supplementary Table 19). IsoSeq reads confirmed the correctness of the new gene predictions (Extended Data Fig. 10).
To exemplify the improvement of the DH1 v.3 assembly, we reanalysed a region on chromosome 3 encompassing a MYB-TF named DcMYB6 (DCAR_000385) that regulates anthocyanin accumulation in carrot root22 and that, in the v.2 assembly, was assembled into a short contig and not anchored to the chromosome sequences. In DH1 v.3, the region spanning DcMYB6 was fully assembled into chromosome 3, and the regions flanking it were found to be composed of repetitive DNA carrying insertions of full-length DcReina and DcAthila nested into an older copy of DcRetand (Fig. 1b). The presence of nested LTRs from younger lineages that attained high copy numbers in the carrot genome (Fig. 1b) made it intractable to assemble this contig into chromosome 3 using the DH1 v.2 short-read assembly strategy and is now fully resolved using the longer read data in v.3. As a result, DcMYB6 could be associated with a putative anthocyanin quantitative trait locus (QTL)23. In addition, the improved annotation method for the DH1 v.3 genome captured predictions for 1,037 new transcription factors and 917 new resistance genes (Fig. 1a, Table 1 and Supplementary Tables 20–23).
Overall, the characterization of the DH1 v.3 genome highlighted previously unknown features of the carrot genome, as well as the advantages that a higher-quality genome annotation can provide for the identification and characterization of biologically and economically relevant genes.
Carrot population structure and phylogeny
A total of 630 carrot accessions, including wild carrots (n = 95), cultivars and landraces (n = 533), and outgroups (n = 2, D. syrticus and D. sahariensis), were resequenced to investigate carrot population dynamics, clustering, gene flow and demographic history (Supplementary Tables 24–26). These accessions were chosen to represent diverse geographic origins and breeding histories and to capture the extensive variation in traits associated with domestication and improvement, such as root colour, shapes, annual/biennial flowering and presence/absence of lateral branching. Resequencing resulted in the identification of 25,375,112 single nucleotide polymorphisms (SNPs), with 1,599,287 located within coding regions.
Population structure was inferred using a randomly sampled set of 168,410 linkage disequilibrium (LD)-pruned SNPs. Clustering analysis identified the strongest support for K = 5 populations (Fig. 2a and Supplementary Table 26). Population I, which includes wild carrots from Africa, Asia, Europe, and North and South America, is referred to as the Wild population (Fig. 2a,b and Supplementary Fig. 1a,b). Populations II and III, referred to as Landrace-A and Landrace-B, respectively, represent the Eastern carrots and include accessions with somewhat undomesticated phenotypes such as non-uniformity within accessions or non-smooth roots. However, these populations also had clearly domesticated characteristics including reduced lateral root branching and the presence of anthocyanin or carotenoid pigmentation (Supplementary Fig. 1b). Accessions belonging to Landrace-A represent carrots from central and eastern Asia, while Landrace-B accessions represent carrots from western and southern Asia in the geographic area spanning from Turkey to India (Fig. 2b). In addition to carrot accessions with landrace phenotypes, the Landrace-A population included 15 wild accessions (hereafter Landrace-AW) (Supplementary Fig. 1b), perhaps derived from intercrosses with cultivated carrots, all from central Asia, where farmers’ seed production is often very close to wild carrot populations (Supplementary Fig. 1b). Further analysis of gene flow and demographic history (Supplementary Note and Supplementary Figs. 2 and 3) indicated that Landrace-AW probably represents a feral lineage of carrot that escaped from cultivation and re-established in the wild. Two additional populations (IV and V), named the Early cultivar and the Improved cultivar, represent Western carrots, which originated mostly in Europe and North America (Fig. 2a,b). Accessions belonging to these populations exhibit morphological phenotypes similar to modern carrot cultivars, such as uniform root shape and the accumulation of high amounts of orange carotenoid pigments (Supplementary Fig. 1b). Early cultivars represent the ‘Horn’ and ‘Long Orange’ carrot market types that were the founders of Western orange carrot. Improved cultivars represent orange market-type cultivars such as ‘Nantes’, ‘Amsterdam Forcing’, ‘Chantenay’ and ‘Danver’, which were developed between the eighteenth and nineteenth centuries in response to the increasing demand for orange carrots in Europe and globally. Over 261 (41%) accessions harbour >10% alleles derived from more than two populations (Supplementary Table 26), indicating a high level of inter-population admixture that reflects the outcrossing nature of carrot24. To avoid bias due to potential ancestry admixture, downstream analyses were also conducted using low-admixture samples (ancestry coefficient >0.9 for a given reference population).

a, Population structure of 630 carrot accessions. The bar plot represents the percentage of membership (q) for each group identified at K = 5. The colour designations for each population (I–V) illustrated in a are used to represent accessions in all the other panels (b–d). b, Geographic distribution of the accessions according to the greatest proportion of ancestry at K = 5. The inset represents the distribution at the regional level of accessions grouped as Early cultivar, Landrace-A, Landrace-B and Wild populations that were located in more defined geographic regions. The Improved cultivars were spread across the world and are not represented in this inset. c, Neighbour-joining phylogenetic tree of 353 samples with <10% admixture proportions (low-admixture set). The consensus tree resulted from a bootstrap test (1,000 replicates). The branch tip colours represent the root colour phenotypes, and the outer ring corresponds to the population identity of each sample. The tree was rooted using D. syrticus as the outgroup. The numbers next to each carrot represent the following carrot cultivars/market types: (1) ‘Yellow Belgian’, (2) ‘Early Half-Long Horn’ (Yellow), (3) ‘Early Half-Long Horn’ (Orange), (4) ‘Long Orange’, (5) ‘Chantenay’, (6) ‘Altringham’, (7) ‘Amsterdam’, (8) ‘Oxheart’, (9) ‘Nantes’ and (10) ‘Amsterdam Forcing’. d, PCA of accessions (n = 630). PC1 and PC2 account for 4.0% and 2.8% of the total variation, respectively.
Phylogenetic analysis and principal component analysis (PCA) support the separation of five populations (Fig. 2c,d and Supplementary Figs. 4–6). Wild and cultivated accessions formed two distinct clades, except for five wild accessions, Landrace-AW (Fig. 2c). Landrace-A and Landrace-B populations were distinct from accessions belonging to the Early cultivar and Improved cultivar populations (Fig. 2c,d). These results suggest that the Landrace-A and Landrace-B populations share a common origin (Fig. 2c), which was reinforced by the low FST estimate (FST = 0.06) between Landrace-A and Landrace-B, indicating a low amount of differentiation between these two populations (Fig. 3a and Supplementary Table 27). Gene flow was detected between these two populations (Supplementary Table 28) and probably contributed to this low differentiation. The Improved cultivar and Early cultivar populations clustered into a separate sister clade and formed two distinct subclades, with the Early cultivar clade being ancestral to the Improved cultivar clade. Interestingly, a group of yellow carrots from the Netherlands and Poland clustered at the base of all the Early and Improved cultivars, which supports the hypothesis that the ‘Long Orange’ and ‘Horn’ types were selected in Europe from yellow carrots5 and that these populations formed the basis of Western and modern orange carrot varieties. The topology of the phylogenetic tree suggests that Western carrots are not directly descended from Eastern carrots but share a common ancestor with wild carrots, possibly due to hybridization between these populations. Supporting this hypothesis, evidence of gene flow between Early cultivars and Wild populations was detected using f4-statistics and TreeMix25 analysis (Supplementary Table 28 and Supplementary Fig. 2), with a TreeMix migration edge indicating that gene flow occurred from Early cultivars into Wild accessions. This result was also reinforced by FST estimates, which indicated that, among cultivated and landrace accessions, Early cultivars have the least amount of differentiation (FST = 0.12) from the Wild population (Fig. 3a and Supplementary Table 27). Relationships among carrot populations were further clarified using outgroup f3-statistics, represented as f3(reference population, test population; outgroup), using wild samples from D. carota subspecies as the outgroup population (subsp. gummifer, maximus carota and maritimus carota). The results support the relationships inferred from the phylogeny, with the Wild accessions having diverged from a common ancestor first, followed by the Landrace-A and Landrace-B populations, and lastly the Early and Improved cultivar populations (Supplementary Fig. 7).

a, Nucleotide diversity and FST values between carrot populations using the low-admixture dataset (n = 353). The numbers in the circles represent π values; the numbers outside the circles represent FST values. The π values for each population represent the 10−3 decimal scale (for example, 0.00986 for Wild). b, LD decay across the five carrot populations (low-admixture set). c, Effective population size trajectories for the carrot populations estimated using SMC++, with the estimate uncertainty displayed as lighter lines for ten bootstrap replicates. Both the x and y axes are on a log10 scale. d, Inferred divergence times for pairs of carrot populations. Point estimates based on all data are displayed as solid black diamonds, with uncertainty displayed for ten bootstrapped replicates (the lighter points and violin plots, with a cross to denote the median of the bootstrapped estimates). The colours indicate the population that is being compared against. The y axis is on a log10 scale. The grey boxes in c,d indicate the hypothesized window of carrot domestication from 522 to 1,100 years ago.
Carrot genetic diversity
Analysis of genetic diversity within the low-admixture set indicated that nucleotide diversity was substantially higher for wild carrots (π = 9.86 × 10−3) than for landraces (π = 5.85 × 10−3 to 5.86 × 10−3) and cultivars (π = 5.81 × 10−3 to 5.86 × 10−3) (Fig. 3a and Supplementary Table 29). Similar results were obtained using the full set (Supplementary Table 29). Among cultivated accessions, nucleotide diversity was lowest in the Improved cultivars (π = 5.81 × 10−3), which reflects their status as highly selected populations (Fig. 3a). Additionally, a survey of the half-life of LD decay occurred at 57 nucleotides (nt) in the Wild population, while the Early cultivar and Improved cultivar populations exhibited an LD decay half-life of 315 nt and 348 nt, respectively (Fig. 3b). The slower rate of LD decay in cultivated carrot populations suggests a substantial decrease in genetic diversity following domestication and improvement.
Carrot demographic history
To investigate the demographic history of each carrot population, SMC++26 was used to infer population size histories (Fig. 3c). Individuals used in this analysis were restricted to samples with low admixture. Effective population size (Ne) trajectories support a shared bottleneck followed by recent expansion. We observed equivalent or increased Ne in modern populations relative to ancestral Ne in the Landrace-A, Landrace-B, Early cultivar and Improved cultivar populations, with minima occurring at ~1,360, 1,206, 953 and 895 years ago, respectively. This result is consistent with historical documents, which place the period of carrot domestication in central Asia (Landrace-A region) between the ninth and tenth centuries, approximately 1,200 years before present, and the selection and improvement of Western orange carrots (Early and Improved cultivars) in the sixteenth and seventeenth centuries, between 500 and 600 years before present5,27 (Fig. 3c). No corresponding bottleneck was observed in the Wild population, further supporting the idea that the observed reduction in Ne probably coincides with the period of domestication in the landrace and cultivar populations.
To estimate divergence between populations, SMC++ uses a ‘clean split’ model, which assumes there is no gene flow following a split between populations. When post-split gene flow occurs, the model is expected to underestimate divergence times28. When estimating divergence among carrot populations, the deepest splits were observed for the landrace and cultivated populations compared with the Wild population, with median estimates of divergence ranging from ~10,804 to 14,970 years ago (Fig. 3d). Subsequent divergence times between Early cultivars and Landrace-A and Early cultivars and Landrace-B were estimated at 2,803, and 1,998 years ago (median), respectively (Fig. 3d). Bootstrapped estimates support the most recent split occurring between the Early and Improved cultivar populations (median of ~788 years ago).
Selective sweeps for carrot domestication and improvement
To identify selective sweeps, pairwise scans were performed between the five populations. Selective sweeps identified between the Wild population and Landrace-A and Landrace-B were considered as those involved in domestication, while those between Landraces A and B and the Early and Improved cultivars were involved in improvement (Supplementary Table 30). In total, 18 distinct genomic regions were identified as selective sweeps (Fig. 4 and Supplementary Table 30). Analysis for genes underlying the selective sweeps identified several enriched gene families, including those related to photoperiodism and circadian clock regulation, control of flower development, photosynthesis, and regulation of isoprenoid metabolic processes (Supplementary Note and Supplementary Table 31).

a, Selective sweeps and candidate genes contrasting the Wild and Landrace-B populations (domestication sweeps). b, Selective sweeps and candidate genes contrasting the Landrace-B and Early cultivar populations (improvement sweeps). c, Selective sweeps and candidate genes contrasting the Early cultivar and Improved cultivar populations (improvement sweeps). Previously known loci are indicated with square horizontal brackets. The carrot pictures and short descriptions presented on the right side of each panel represent a possible change in plant characteristics that occurred during domestication and cultivar improvement. Each selective sweep panel on the right side (a–c) is a representative comparison among the populations that correspond to that specific step (domestication, selection or improvement).
Delayed flowering is a critical trait for domestication because the taproot becomes fibrous and inedible once flowering occurs. Interestingly, within a selective sweep associated with domestication, genes involved in circadian clock regulation and flowering time (including homologues of CCA1 HIKING EXPEDITION (CHE)29, TCP23 (ref. 30) and TCP7 (ref. 31)) were enriched (Fig. 4a and Supplementary Tables 31 and 32). This region overlaps with the region spanning the vernalization (Vrn1) locus previously mapped in carrot (Supplementary Table 32)32.
Multiple selective sweeps associated with improvement also harboured homologue genes involved in flowering time regulation (KHZ1, FBH4, AREB3, LWD1 and CIB4)33,34,35,36,37. As domesticated carrot spread into multiple geographic regions, selection for genes involved in flowering time regulation continued to play a critical role in adaptation to multiple environments (Fig. 4b,c and Supplementary Table 32).
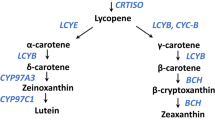
The increasing accumulation of carotenoids in the taproot has been a major focus of modern carrot breeding. The QTL Bt.3.1 (ref. 17) on chromosome 3 co-localized with the primary improvement sweep identified on chromosome 3 that harbours the Or-like gene (Fig. 4b and Supplementary Table 32). Or genes control chromoplast biogenesis and enhance the preferential accumulation of β-carotene38,39,40. Another improvement selective sweep harboured the gene ELIP1, which is known to interact with Or to regulate chloroplast biogenesis41. The Y2 QTL16 on chromosome 7 overlaps with a selective sweep that harbours DCAR_730022, a gene that was identified here (see below) as a new candidate gene controlling this QTL. DCAR_730022 shares homology to EXECUTER1, which mediates the response to singlet oxygen within the chloroplast42,43. Breeding for high-carotenoid phenotypes may have indirectly led to the selection of genes involved in plastid biogenesis and the cross-talk between the photosynthetic system and carotenoids accumulating in the carrot root.
Genome-wide association analysis for carotenoids
Carotenoid accumulation was investigated using genome-wide association (GWA) analyses of the visual taproot phenotypes for 601 accessions and with the relative carotenoid content of 435 accessions (Supplementary Note and Supplementary Table 33). The most significant loci were mapped in chromosomes 2, 3 and 7 and were associated with taproot colour and the ratios of α + β-carotene and lutein to total carotenoids, while four weaker loci were identified in chromosomes 5 and 9 and were associated only with root colour (Fig. 5a,b and Supplementary Table 34).

a, Manhattan plot of GWA analysis for the ratio of α + β-carotene relative to total carotenoids. b, Manhattan plot of GWA analysis for taproot colour. c, Overlap of the significant locus identified on chromosome 7 with the previously mapped Y2 locus16 and a 38.8-kb region containing the top 30% of markers most associated with carotenoid accumulation. d, Comparative analysis of the region spanning the Y2 locus in DH1 and Lunar-White, a white accession with the wild Y2 allele. The Y2 candidate gene (DCAR_730022) is highlighted in orange. The structure of DCAR_730022 in DH1 and Lunar-White and a 17-kb insertion detected in the DH1 Y2 gene are illustrated. e, Overlap of the significant locus identified on chromosome 3 with the previously mapped Or locus7,17. A region of 43 kb was identified here as the most significantly associated with Or that harbours the gene controlling this locus. Predicted genes within the locus identified on chromosome 3 are illustrated, and the Or-like gene (DCAR_310369) is highlighted in orange. f, The effects of the three different alleles (AA, AB and BB) at the REC1, Or and EX1 candidate genes on the ratio of α + β-carotene concentration to total carotenoid content. Statistical analysis was performed using the F statistical test. The box plots represent the 25th, 50th and 75th percentiles, and the upper and lower whiskers represent 1.5× the 75th and 25th percentiles, respectively. For REC1, n(CC) = 14, n(AC) = 32 and n(AA) = 360; for Or, n(GG) = 28, n(AG) = 27 and n(AA) = 351; and for EX1, n(TT) = 59, n(AT) = 27 and n(TT) = 320. g, REC1, Or and EX1 allelic interaction analysis. The data are presented as the mean plus or minus the s.e.m. For B_A_B_, n = 11; for B_B_A_, n = 11; for B_B_B_, n = 12; for A_B_B_, n = 16; for A_A_B_, n = 19; for A_B_A_, n = 20; for B_A_A_, n = 23; and for A_A_A_, n = 294. h, Neighbour-joining phylogenetic tree of the SNPs identified at the REC1, Or and EX1 genes across the low-admixture set. The consensus trees were constructed from 1,000 bootstrap replicates. The outer ring corresponds to the allele detected for each sample (AA, AB or BB). The inner ring represents the population identity of each sample. The asterisks in f,g indicate allelic groups that were significantly different (P < 0.01); NS indicates allelic groups that were not significantly different.
The most significant locus detected on chromosome 7 overlapped with the fine-mapped Y2 QTL region that controls the orange phenotype in carrot (Fig. 5c)16. The region spanning the top 30% of the most significant SNPs included two candidate genes, DCAR_730021 and DCAR_730022 (Fig. 5d and Supplementary Tables 35 and 36). DCAR_730022 was downregulated in orange samples harbouring the recessive Y2 allele and harboured SNPs with stronger associations (Supplementary Note and Supplementary Tables 37 and 38). Also, an insertion of a Helitron disrupting the DCAR_730022 coding sequence (CDS) was identified in DH1 and 97% of the orange accessions (Fig. 5d, Supplementary Note and Supplementary Table 39). Transcriptional interactome network analysis identified DCAR_730022 as a key link in the interaction between genes involved in ‘Photosystem PSII associated light-harvesting complex’, including ‘singlet oxygen response’ (1O2) along with isoprenoid biosynthetic pathways (Supplementary Tables 40 and 41 and Supplementary Fig. 8). In line with these results, DCAR_730022 shares partial homology to EXECUTER1 (EX1), which is known to be involved in activating the enzymatic 1O2 stress response program in plants to repair photosystem II43,44. Interestingly, the non-enzymatic breakdown of β-carotene, a 1O2 scavenger, represents the alternative mechanism of reactive oxygen quenching in photosystem II45,46. Considering these results, it is plausible that a non-functional EX1-like gene in genotypes carrying the insertion, such as DH1, could cause the plant to maintain high levels of β-carotene biosynthesis to quench 1O2. This possible mechanism, its expression and the disruption of the CDS in orange samples (which is compatible with a recessive mutation like Y2) provide compelling evidence for pursuing functional validation of DCAR_730022 as the Y2 candidate gene.
The significant associations mapped on chromosome 3 overlap with the previously identified Or locus (Fig. 5b and Supplementary Table 34)7,17. A survey of the region within 30% of the top-scoring SNPs yielded six genes (Fig. 5e and Supplementary Table 35). The gene DCAR_310369, orthologous to the Arabidopsis Or-like gene, was the only gene located within this region that has been associated with carotenoid accumulation in carrot and other species7,39 (Supplementary Table 36). Recent work in carrot demonstrated that knocking down the expression of DCAR_310369 in an orange carrot genotype resulted in yellow carrot47. Notably, this gene was not differentially expressed between yellow carrots carrying the dominant allele and orange carrots carrying the recessive allele (Supplementary Note and Supplementary Tables 42 and 43), suggesting that its function may be controlled at the protein level as reported in other plant systems38,40.
The locus mapped in chromosome 2 represents a new locus related to carotenoid accumulation in carrot (Fig. 5a,b and Supplementary Table 34). The region harbours 26 positional candidate genes and includes one gene, DCAR_206039, homologous to Arabidopsis reduced chloroplast coverage 1 (REC1)48. A REC1 orthologous gene in Mimulus (RCP2) directly affects carotenoid content49,50 (Supplementary Note and Supplementary Table 35).
Carotenoid gene effects and interactions
Next, SNPs detected within EX1-like (DCAR_730022), Or-like (DCAR_310369) and REC1-like (DCAR_206039) were used to evaluate their effects and interactions in relation to the ratios of α + β-carotene content to total carotenoid content and visual orange phenotypes. Single marker effect analysis indicated that Or-like, EX1-like and REC1-like contribute to a significant (P < 0.001) increase of the α-carotene and β-carotene concentration. The results also indicated that the recessive alleles for all three genes (hereafter cultivated, A, REC1_A, Or_A and EX1_A; Fig. 5f) as opposed to the dominant wild alleles (hereafter wild, B, REC1_B, Or_B and EX1_B; Fig. 5f) condition carotenoid accumulation. The recessive genetic model for EX1-like fully agrees with previous studies performed in multiple mapping populations14,16,17. Also, for Or-like, the allele associated with the recessive model in this analysis (homozygous TT at position 551) corresponds to the allele coding for leucine (named DcOR3Leu), which has been proved to control the orange phenotype in carrot47. These observations confirm the robustness of the results presented here at the population level.
Two-way epistatic interactions exist between the three loci (P < 0.001) except REC1 and Or. Also, a three-way interaction among all the alleles was significant (P < 0.05). On the basis of analysis of variance for allele interaction and the ratios of α + β-carotene to total carotenoids, the genotypes could be separated into three groups (P < 0.05) (Fig. 5g). Group 1 included genotypes that either harbour only one of the recessive alleles or are missing all of them (for example, REC1_B/Or_B/EX1_B). Among these genotypes, only one was orange, and the fraction of α + β-carotene was very low (<0.1%) or not detected (Fig. 5g). Group 2 included genotypes that harboured the REC1 recessive allele and either the Or or EX1 wild allele (for example, A_A_B_). Among these genotypes, 25% were orange (mostly pale orange), with a fraction of α + β-carotene that was significantly higher (average 0.24%) than group 1 (Supplementary Figs. 9 and 10). Group 3 included genotypes that harboured recessive alleles for all three genes or harboured Or and EX1 recessive alleles (for example, A_A_A_). Among these genotypes, 96% were orange (nearly all dark orange), and the fraction of α + β-carotene in these genotypes was the highest (average 0.78%) (Supplementary Figs. 9 and 10). Overall, these results demonstrate that the recessive alleles at both Y2 and Or are strictly needed to select orange carrot with high concentrations of α + β-carotene, and a recessive allele at the REC1 locus contributes to reaching the highest concentrations of α + β-carotene.
To gain some preliminary insight into the selection process of these three genes, we carried out phylogenetic analysis with SNPs spanning the REC1, Or and EX1 genes (CDS) from the low-admixture set. The results indicated a clear separation of genotypes that harbour the recessive alleles, found in cultivated accessions, from those that harbour the dominant wild alleles (Fig. 5h). The clades including the cultivated alleles included nearly all orange genotypes as well as a limited number of non-orange genotypes (for example, purple). Relative to the five populations, for all three genes, the phylogenies clustered the same populations of domesticated carrot (Landrace-A, Landrace-B, and the Early and Improved cultivars) together. These results indicate that the origin of the orange cultivated alleles for all three genes is monophyletic; each gene was probably selected once and rapidly fixed as soon as the orange phenotype was selected. This assertion is also supported by the shared genetic bottleneck identified in the demographic analysis.
Discussion
Historical documents and previous studies indicate that carrot germplasm can be separated into three major groups (Eastern, Western and Wild carrots) and suggest that Eastern carrots were domesticated in central Asia6,15 and formed the basis of Western carrots5,6,27,51. However, the demographic events that characterized carrot domestication and improvement have not been assessed to support this hypothesis. In this study, an improved carrot genome assembly and resequencing of 630 diverse carrot accessions that represent the global distribution of carrot germplasm were used to reconstruct a detailed picture of carrot domestication and improvement, as well as the consequences of these selection processes for the genetic makeup of this important crop.
The separation between Wild, Eastern and Western populations was confirmed. Eastern and Western carrots were further separated into subpopulations, named here Landrace-A and Landrace-B for Eastern carrots, and Early cultivars and Improved cultivars for Western carrots. Phylogenetic analysis indicated that the progenitor of Western carrots shared its ancestry with Eastern and Wild carrots, in contrast to the standing hypothesis of Eastern carrots as the progenitor of Western carrots. However, gene flow analysis indicated that the signature of wild ancestry detected in the Early cultivars was confounded by hybridization between Early cultivars and the Wild population, particularly due to the movement of alleles from cultivated to wild populations. Considering that carrot is an outcrossing species and that wild carrot is often found in areas of cultivated carrot seed production, gene flow between wild and cultivated carrots can easily occur52. On the basis of these results and observations, this study still lends support to the hypothesis that Eastern carrots are the progenitor of Western carrots. However, we cannot exclude the possibility that Western carrots originated from an unsampled or extinct population. Furthermore, given that Landrace-A and Landrace-B represent sister populations and have evidence of gene flow between them, the origin of Western carrots cannot be specifically traced to one of the two populations.
Population divergence estimates strongly support the documented chronological history of carrot domestication and improvement. Demographic analysis indicates that recent population expansion in Eastern carrots began ~1,300 years ago, with the more recent expansion of orange Western carrot cultivars estimated to have begun about 800 years ago. These estimates closely match existing timelines from historical records, which indicate that Eastern carrots were documented in central Asia between 1,100 and 1,500 years ago5,27,51,53. On the basis of historical records and our demographic analysis, carrot domestication can be placed between the sixth and tenth centuries, during the Early Middle Ages. The distribution of the Landrace-A and Landrace-B populations coincides with the separation between western-southern Asia (Turkey, Iran and India) and central-eastern Asia (Afghanistan, Tajikistan, Uzbekistan, Pakistan, China and Japan), and overlaps with Asia Minor and central Asia, respectively. Divergence time estimates support the separation of the Landrace-A population from wild carrots earlier than Landrace-B, suggesting that the domestication of central Asian carrots pre-dated the spread of carrot in Asia Minor.
The more recent population expansion detected for the Early and Improved cultivar samples began about 800–900 years ago. This estimate matches the selection and documented spread of Western orange carrot between the sixteenth and eighteenth centuries53. Historical records also indicate that between the twelfth and fifteenth centuries, yellow and purple carrot were used in Spain, Italy, France, Germany, England and the Netherlands5. However, yellow carrots became more popular in Europe and probably established the basis of Western carrot5,53. This chronological reconstruction based on molecular and historical data was corroborated by the phylogenetic analysis, which placed a number of Western yellow carrots as the founders of the Early cultivars at the base of the market types ‘Horn’ and ‘Long Orange’, which are known to be the founders of the orange carrot types5. Clustering of ‘Yellow Belgian’ and other yellow carrots from the Netherlands as the progenitor of all Western orange carrots provides strong support for one of the most debated hypotheses proposed in 19635, which suggests a Dutch (or perhaps Belgian) origin of Western orange carrots that were selected from yellow domesticated carrots.
As demonstrated by our phylogenetic analysis, Early cultivars were the founders of the Improved cultivars. These results coincide with historical records indicating that, after the selection of the orange phenotype occurred in Europe, orange carrots became very popular, and new cultivars with reduced high intra-cultivar uniformity and with specific root shapes or market types (for example, ‘Nantes’, ‘Amsterdam Forcing’ and ‘Chantenay’) were developed during the seventeenth and eighteenth centuries to meet the growing global demand5,27,54.
Previous studies have indicated that after domestication cultivated carrot experienced limited or no reduction of genetic diversity6,7,15. In contrast, our estimates of nucleotide diversity and effective population size suggest that a progressive reduction of genetic diversity accompanied carrot domestication and improvement. The higher SNP density and sequences captured (especially intergenic regions) in the DH1 v.3 genome assembly probably contributed to resolving controversial results from previous studies. As demonstrated in this study, strong selection pressure was detected for domestication traits such as vernalization and improvement traits such as orange roots. Given that these phenotypes are under the control of recessive alleles15,16,32, they were probably used as a visual tool for carrot breeders to keep cultivated carrot relatively free from outcross contamination by wild species. This process probably contributed to the reduction of genetic diversity in cultivated carrot.
Selective sweep analysis identified selection and/or fixation for genes related to flowering and high carotenoid pigmentation. These results are consistent with our knowledge about the traits selected during carrot domestication and improvement and support the role of conscious and/or unconscious selection by farmers and breeders on traits of economic value. For instance, delayed flowering in carrot is strictly needed to produce a nutrient-rich edible root55. The finding that genes controlling flowering time were enriched within the selective sweep regions demonstrates that this trait played an important role during the initial domestication and improvement of carrot and probably enabled their adaptation to and cultivation in different regions of the world. The overlap of a major domestication selective sweep with Vrn32, a vernalization locus previously mapped in chromosome 2, provides strong support for these results.
The GWA and selective sweep results suggest that the high-carotenoid phenotype in modern carrot cultivars is the result of a complex interaction between the response to light perception, plastid biogenesis and development, and carotenoid biosynthesis. The importance of previously mapped loci (Y2 and Or) in regulating orange carotenoid accumulation in carrot roots was confirmed, and a new candidate locus (named here REC1) was mapped on chromosome 2. The previously characterized Or-like gene was confirmed to be the gene controlling the Or locus7,17, and two new candidate genes, EX1-like and REC1-like, were identified for the Y2 and REC loci, respectively. Although the role of EX1-like and REC1-like will need to be verified through functional analysis, the rapid LD decay detected in carrot populations provides high resolution for gene mapping and support for their candidacy. For instance, the recessive genetic model established for EX1 at the Y2 locus matches the results from previous studies7,16,17. Other evidence supporting the role of these genes in controlling carotenoid accumulation includes gene expression analysis (EX1), causal mutation analysis (Or and EX1) and functional annotation indicating that all three genes belong to gene families that regulate or mediate the interaction between the carotenoid biosynthetic pathway, the photosynthetic systems and chloroplast biogenesis.
The large-scale population genomic analysis performed here provides an example investigation of the selection process underlying the orange phenotype at the gene level. The results indicate that the recessive cultivated alleles at all three genes—REC1, Or and EX1—were essential to select the orange phenotype, and each cultivated allele was selected once and rapidly fixed. Or and EX1 were essential to reach the highest fraction of α + β-carotene, while EX1 or Or in combination with REC1 led to the accumulation of a low-medium fraction of α + β-carotene that is mostly associated with a pale-orange root phenotype. As these genes are located on different chromosomes, carrots with different REC1, Or and EX1 cultivated allele combinations may have been developed independently. As a result, multiple orange phenotypes may have been developed in parallel. Carrots with a lower fraction of α + β-carotene and a pale-orange phenotype probably pre-dated or paralleled the selection of the dark-orange phenotype. Interestingly, this hypothesis is supported by historical documents indicating that in the seventeenth century, both types of orange carrots (pale and dark orange) were clearly identified27. Due to their reciprocal epistatic effect on the orange colour, once this trait was selected, the orange alleles were fixed.
This study elucidated the demographic history of carrot domestication and breeding and demonstrated that selection for the REC, Y2 and Or QTLs established the basis for modern-day orange carrot. The new DH1 v.3 genome provides a valuable resource to advance genetic mapping, comparative genomics and gene cloning studies. Building on these findings, future work based on long-read sequencing technology and phased genomes can further trace the ancestry of the REC1, Or and EX1 genes. This foundational work will enable further studies on the genetic mechanisms regulating carotene accumulation in carrot, with potential applications to other crops.
Methods
Sequencing and de novo assembly
For de novo assembly of the DH1 genome (doubled haploid orange Nantes type carrot, NCBI Biosample SAMN03216637), sequencing was performed with Pacific Biosciences (PacBio), Oxford Nanopore and Hi-C sequencing technologies (see the Supplementary Note and Supplementary Tables 1–3 for more details). A detailed description of the genome assembly method is described in the Supplementary Note and Supplementary Table 45 and illustrated in Extended Data Fig. 1. A list of the software and parameters used has also been made available through GitHub (https://github.com/dsenalik/Carrot_Genome_DH1_v3).
Assembly quality verification
A comprehensive analysis was carried out to evaluate the quality of the final carrot DH1 v.3 genome assembly. Fastq-Screen (v.0.4.14)56 and GC content distribution estimates were used to assess the presence of sequence contaminations (see the Supplementary Note for more details).
The correctness of the assembled sequences was evaluated by estimating the mapping distance between a set of 4,717 Bacterial Artificial Chromosome End Sequencing (BES) that unambiguously aligned with both ends to the DH1 v.3 genome assembly and that were not used during the assembly process. The fraction of Paired-end (PE) data that aligned within the expected library insert size should reflect the fraction of assembled sequences that are consistently contiguous and correctly assembled. Also, a linkage map that included 3,242 markers57 not used for genome assembly was used to independently verify the order of the sequences. Marker sequences were mapped using BWA mem58 (see the Supplementary Note for the parameter and filtering settings).
Gene space coverage was assessed using carrot expressed sequence tags59, DH1 IsoSeq full-length transcripts generated in this study and 20 sets of publicly available DH1 Illumina transcriptome data. Expressed sequence tags were mapped using BWA mem, StringTie (v.1.3.5)60 was used to map the Illumina transcriptome data and GMAP (v.2021-08-25) was used to map the IsoSeq sequences (see the Supplementary Note for the parameter and filtering settings).
Repetitive sequences annotation
De novo identification of carrot repetitive DNA was carried out with RepeatModeler (v.2.0.1) (http://www.repeatmasker.org/RepeatModeler/). The annotation of the consensus sequences was performed using a curated database of carrot LTR retrotransposons, Helitrons and MITE61, carrot satellite repeats15 and dicot plant repeats from RepBase (v.23.05)62 and DANTE (v.1.1.0)63,64,65. Masking was performed using RepeatMasker (v.4.1.0; http://www.repeatmasker.org) (see the Supplementary Note for the parameter and filtering settings). Identification, annotation and age analysis of LTR retrotransposons was performed as described by Kwolek et al.66 (see the Supplementary Note for the parameter and filtering settings). The quality of the assembled repetitive sequences was evaluated using the LTR Assembly Index, as recommended for comparison between assemblies of the same species67. For comparative analysis, all the repetitive sequence analyses were also performed using the DH1 v.2 genome assembly using the same methods outlined above. Carrot centromeric and telomeric repeats15,68 were mapped to the DH1 v.3 assembly using Blastn with the default parameters and dust set to ‘no’.
Gene prediction and genome annotation
A multi-step approach was used to predict the most comprehensive gene model catalogue for the carrot genome v.3. MAKER (v.3.01.03)69 and GeMoMa (v.1.6)70 were used to perform gene prediction based on the integration of de novo gene prediction and evidence-based predictions. For MAKER, carrot expressed sequence tags59, DH1 Illumina and IsoSeq transcriptome sequences, gene models obtained from five closely related or model species (Supplementary Table 12), and proteins from Uniprot-sprot were used as transcript evidence. AUGUSTUS (v.2.5.5)71 and SNAP (commit of 3 June 2019)72 were used for de novo prediction (see the Supplementary Note for the details). Through this analysis, MAKER predicted 28,721 gene models. Next, GeMoMa was used to improve the quality of the splice junction sites predicted by MAKER and to predict the gene models that were not predicted by MAKER. The datasets included as input in GeMoMa were the predicted genes from the five related species or model species used for the MAKER prediction, the final gene models produced from the MAKER pipeline and splice sites mined from the mapping of the DH1 Illumina transcriptome data (see the Supplementary Note for the details) on DH1 v.3. This analysis produced an intermediate set of 32,625 gene models. A final step was performed to refine all gene models and predict any missing models. In this step, gene models predicted on the DH1 v.2 assembly15, named DCARv2 (32, 112) and RefSeq (44, 484), were transferred/re-predicted to the DH1 v.3 genome assembly using GMAP73 and GenomeThreader (v.2021-08-25)74. DCARv2 or RefSeq gene models that were not predicted by MAKER + GeMoMa, that had experimental evidence and that were not masked were considered as new gene models. In those cases where the structure of the RefSeq and DCARv2 gene models were not in agreement, the correct structure was manually inspected using the experimental evidence. Finally, high-quality IsoSeq transcripts were mapped to the DH1 v.3 assembly using GMAP and GenomeThreader. Those transcripts mapping with appropriate gene structure and not predicted in the previous steps were added to the gene model catalogue. In total, 3,586 gene models were added by manual curation and polishing, which resulted in a total of 36,211 gene models in the DH1 v.3 gene model catalogue (DCAR v.3.0 Gene Prediction) (Supplementary Tables 11 and 12).
Blast2Go75 was used to annotate the predicted gene models obtained from the last step using the NCBI, KEGG, InterPro and GO databases. PlantTFcat (downloaded in December 2020)76 and PRGdb (v.3.0)77 were used to predict the transcription factors and resistance genes in v.3 gene models, respectively, as well as the DCARv2 genes for comparison purposes. To assess the completeness of annotation, the predicted gene models were searched against the BUSCO (v.3)78 plant dataset (embryophyta_odb9) (Supplementary Table 13). An in silico search for the prediction of candidate microRNAs and small nuclear RNAs in the assembled genome was conducted by INFERNAL (v.1.1.2)79.
Resequencing and phenotyping
For resequencing, a set of 542 cultivated carrots from the National Plant Germplasm System were grown from seed at the Hancock Agricultural Research Station (Hancock, WI, USA) during the summer of 2018 (Supplementary Table 22). An additional set of 88 wild carrots, chosen from the National Plant Germplasm System to represent multiple geographic origins, were grown from seed at the University of Wisconsin–Madison Walnut Street Greenhouse during the winter of 2018 (Supplementary Table 22). Roots were harvested with the tops attached, and mature leaf tissue was collected from each sample. Genomic DNA of each sample was extracted from lyophilized leaf tissue using the Machery-Nagel NucleoSpin Plant II Core kit. Paired-end libraries were sequenced on a NovaSeq6000 sequencer (Illumina) at the University of California, Davis, Genome Center in Davis, California.
Phenotyping for the resequencing material was performed on the basis of visual appearance and high-performance liquid chromatography (HPLC). At harvest, the presence of extensive lateral roots, root pigmentation and evidence of bolting were recorded and used as indicators to confirm the classification of accessions as wild. Visual colour scoring was completed for 630 carrot accessions by taking a cross-section of the taproot and assigning categorical scores of white, yellow, orange, red and purple (Supplementary Table 24). The concentrations of α-carotene, β-carotene, lutein and lycopene were quantitatively measured via HPLC in 528 accessions within three weeks of harvest. Within two weeks of harvest, slices were taken at mid-root, lyophilized and processed as in refs. 80,81 (see the Supplementary Note for the details). The HPLC data were filtered to remove samples with inconsistencies between technical replicates. Other samples were removed from downstream analyses if the HPLC data were not representative of the visual score. Carotenoid concentrations were reported in μg per g dry weight of tissue. This resulted in a set of 435 accessions with HPLC scores that were used for GWA analyses. Considering that the focus of this study was orange carotenoids and that α-carotene and β-carotene represent the major carotenoids in orange carrot, the ratio of α-carotene and β-carotene concentration was calculated relative to the total carotenoid concentration on a per-sample basis (Supplementary Fig. 8, Supplementary Table 27 and Supplementary Note). This method ensured that data across HPLC runs were normalized. The classification of Early and Improved cultivars in the different carrot root types was based on the description of the typical carrot shapes in ref. 82.
Variant calls
Illumina reads from the 630 resequenced carrot accessions were mapped to the assembled genome with BWA (v.0.7.17–r1188) using the BWA-MEM algorithm. These alignments were used for variant calling following the Genome Analysis Toolkit (GATK, v.4.0.7.0) best practices83. Low-quality variants were removed using the following filters: minDP > 5, MQ < 40, FS > 60, QD < 2, MQRankSum < −12.5 and ReadPosRankSum < −8.0. Indels and non-biallelic sites were removed, and sample genotypes were filtered for a minimum GQ > 20. Finally, BCFtools (v.1.9)84 was used to remove singletons and sites with more than 20% missing data, leaving 23,375,112 SNPs across 630 samples. Removing variants with a minor allele frequency (MAF) <0.05 retained 5,393,228 SNPs across 630 samples, indicating that the majority of variants occur at a low frequency. For accurate estimates of nucleotide diversity, an allsites VCF that included invariant sites was also generated, with the same filtering criteria applied to SNPs and by removing low-quality invariant sites on the basis of the following filters: minDP > 5, QUAL < 30, MQ < 40, MQRankSum < −12.5 and ReadPosRankSum < −8.0.
Population structure, phylogenetic analysis and PCA
To infer population ancestry, 300,981 SNPs were randomly sampled and LD pruned with a window size of 50 kb, a step size of five variants and a variance inflation factor of 2 using the command indep 50 5 2 in PLINK (v.1.90b3.44)85, resulting in 168,410 LD-pruned SNPs. Population structure was characterized using ADMIXTURE (v.1.3.0)86,87 on this LD-pruned SNP set. ADMIXTURE was run for K = 1 through K = 10 with a random number seed generated from the current time using the command admixture -s time. The coefficient of variation values for K = 1 through K = 10 were compared, and the K with the lowest coefficient of variation was chosen as the most optimal fit. Using this approach, the strongest support was identified for K = 5, but results at K = 6 were also explored (Supplementary Fig. 1).
Population genetic analyses were performed on a core set of 353 low-admixture samples, defined here as an ancestry coefficient >0.9 for a given reference population: wild (n = 52), Landrace-A (n = 30), Landrace-B (n = 73), Early cultivar (n = 42) and Improved cultivar (n = 156) (Supplementary Table 24). The phylogenetic analysis was performed on both the full set of all 630 samples and on the low-admixture set for comparison. For the low-admixture set, a neighbour-joining phylogeny was constructed with 110,780 LD-pruned SNPs using PHYLIP (v.3.696)88. A consensus of 1,000 bootstrap replicates was used to construct the resulting phylogeny. D. syrticus was used as an outgroup89. The resulting consensus tree was fitted over the original tree using a Perl script90. The phylogeny was visualized using the R package ggtree91. The same methodology was used for the full set of 630 samples, except for 10,000 LD-pruned SNPs and 100 replicates being used to construct the phylogeny.
PCA was performed using the function snpgdsPCA implemented in the R package SNPRelate (v.1.20.1)92 on the LD-pruned set of 168,410 SNPs with all 630 samples and for the set of 353 low-admixture samples.
Gene flow, f 3-statistic and f 4-statistic analysis
Gene flow between populations was inferred by running TreeMix (v.1.12)93 on 26,670 LD-pruned SNPs with no missing data for the 353 low-admixture samples. The model was run with 100 replicates, each with 1,000 bootstraps for one to five migration edges. The most optimal number of migration edges was identified using OptM (v.0.1.6)25. Additionally, gene flow was assessed using f4-statistics by running the qpDstat program in AdmixTools v.7.0.2 (ref. 94). Population comparisons were set up as f4(outgroup, population X; population Y, population Z), where the outgroup included samples of D. sahariensis and D. syrticus and is not expected to have admixture with the test populations. Gene flow between test populations was considered significant if Z-scores had absolute values >3, with high negative values suggesting gene flow between test populations X and Y and high positive values suggesting gene flow between test populations X and Z.
To further clarify the relationships and relative divergence times among carrot subpopulations, outgroup f3-statistics were used to estimate the amount of shared genetic drift between pairs of populations relative to a distant outgroup comprising wild samples from related D. carota subspecies, which are genetically equidistant to the pair of populations being compared. The qp3Pop program in AdmixTools v.7.0.2 (ref. 94) was used to compute outgroup f3-statistics using the structure f3(reference population, test population; outgroup) with the option inbreed set to ‘YES’. Higher f3 values indicated a higher degree of genetic similarity and a longer shared branch length between the reference and test populations relative to the outgroup.
Genetic diversity, F ST and LD analysis
Pairwise FST and π were calculated within 100-kb windows using Pixy (v.1.2.7.beta1)95 and an allsites (variant and invariant sites) VCF as the input file (see https://github.com/dsenalik/Carrot_Genome_DH1_v3 for the details and parameters). Pairwise values were calculated for comparison of domesticated, improved and wild populations using the low-admixture set.
LD decay was calculated using 5,393,228 SNPs filtered for MAF < 0.05 among samples identified to have low-admixture proportions from each of the five populations. LD decay was calculated for all SNPs within 1-Mb windows using the command OutStat implemented in PopLDdecay (v.3.31)96.
Demographic analysis
Estimates of effective population size history and divergence times were obtained using SMC++ software (v.1.15.2)26 (https://github.com/popgenmethods/smcpp), which uses a coalescent hidden Markov model to leverage information on LD and the site frequency spectrum from unphased genomic data. To reduce confounding due to gene flow, samples used in this analysis were restricted to the individuals with low admixture. The full set of 23,375,112 quality-filtered SNPs was included for demographic analysis to avoid excluding low-frequency sites and was filtered to exclude sites with ≥10% missing genotype calls using the command “view -e ‘F_MISSING > = 0.1’ -Oz” in bcftools (v.1.10.2)97. The resulting VCF file was converted to SMC format using the vcf2smc command in SMC++ and by treating repetitive sites identified by RepeatMasker (v.3.2.9) as missing data. To estimate a composite likelihood for population size histories and divergence times, distinct datasets were generated for each population by conditioning allele order across five randomly selected distinguished individuals. Population size history was estimated using the estimate command with the default parameters and a per-base-pair-per-generation mutation rate of µ = 4 × 10−8 as reported for Lactuca sativa98, which was the closest related species with a reported estimate for mutation rate. Divergence times were estimated by first generating a joint site frequency spectrum for each population pair using the vcf2smc command, followed by the split command. Estimate uncertainty for population size trajectories and divergence times was determined using a bootstrap approach in which ten replicates of the input genomic data for each distinguished individual were resampled in 5-Mb blocks. The code for the estimation of effective population size and divergence times using SMC++ (v.1.15.2) is available at https://github.com/mishaploid/carrot-demography.
Genome-wide scans for signatures of selection
To identify regions of the genome that have undergone selection during domestication and improvement, we compared FST, the ratio of nucleotide diversity and selective sweeps among pairwise comparisons of wild, domesticated and improved populations. Pixy software (v.1.2.7beta)95 was used to calculate FST and π across 100-kb windows for the low-admixture samples. An allsites VCF was used as the input to adequately distinguish between uncallable and invariant sites. XP-CLR (v.1.0)99 was then applied to identify variants that increased in frequency at a rate that is higher than by chance alone. XP-CLR scores were calculated using a set of one million variants filtered for MAF < 0.05, among all samples within the low-admixture dataset (Supplementary Table 24). XP-CLR scores were computed in a 0.05 cM window with a maximum of 100 SNPs per window and a 1-kb sliding window. If two SNPs were found to be highly correlated (>0.9), then their contribution to XP-CLR was downweighted. The top 2% of nucleotide diversity ratios between each of the five populations, the top 2% of FST values identified between each population and the genomic windows harbouring the top 1% of XP-CLR SNPs were merged, and regions that overlapped between all three analyses were identified as selective sweeps.
GWA analysis
The phenotypic data for GWA analyses included HPLC data for the fraction of α + β-carotene and lutein to total carotenoids in addition to visual colour scores. The genotypic data were prepared and GWA analyses were completed on the US Department of Agriculture SCINet High Performance server. The genotypic data were filtered with vcftools (v.0.1.16)100 for sequencing depth of >5, MAF > 0.05, missing data <0.3, removal of indels, allele >2, and heterozygosity >0.3 and <0.7. Missing data from the genotypic file were imputed with Beagle (v.5.0) with the default settings101. The genotypic file was formatted in hapmap format with Tassel (v.5)102.
GWA analysis was completed using the R package GAPIT (2020.10.24 Gapit) v.3.0 (ref. 103) with multiple models tested, including a mixed linear model, multiple mixed linear models, and Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway104. The mixed linear model provided the best fit. Due to the number of SNPs used in the analyses, a random subset of 125,000 markers was used to complete a PCA and kinship analysis to account for population structure and relatedness, respectively. GAPIT code was also updated for computational speed by only writing results for the 100,000 markers most associated with each trait. The significance threshold was calculated using a modified Bonferroni correction in the R package simpleM105 for P < 0.05. Manhattan plots for the GWA results were generated using the R package qqman (v.0.1.8)106.
RNA-seq analysis for Or and Y2
RNA-seq analysis was used to investigate the transcriptome profile of candidate genes underlying the Or and Y2 loci mapped by GWA analysis. For the Or locus, RNA was extracted from three biological replicates of eight genotypes that were selected from a mapping population segregating for Or17(see the Supplementary Note for the details). Four genotypes represented plants that were homozygous for the orange cultivated allele (Or_A), and four represented plants that were homozygous for the wild allele (Or_B). Sequencing libraries were prepared using a TruSeq Stranded mRNA kit (Illumina), and the libraries were sequenced on a NovaSeq 6000 sequencer at the University of Wisconsin Biotechnology Center in Madison, Wisconsin. Transcriptome sequencing generated 1,091,729,253 reads across 24 samples (Supplementary Table 30).
For the Y2 locus, existing RNA-seq data available in NCBI (BioProject PRJNA350691) were used in this study16. Transcriptome data represent six genotypes selected from a mapping population segregating for Y2 (ref. 16). Three yellow genotypes were homozygous for the Y2 wild allele, and three orange genotypes were homozygous for the Y2 cultivated allele.
RNA-seq reads were first cleaned for adapter sequences using TRIMMOMATIC (v.0.36)107 and were then aligned to the reference genome using the package Rsubread (v.2.14.1)108. FeatureCounts (v.2.14.1)109 was then used to compute count matrices for each sample, and the results were then analysed using Limma (v.3.56.1)110. The analyses were performed in R v.3.5.0 (R Core Team, 2013). A log-fold-change testing threshold of 1.1 was used to identify genes with a substantial difference in observed log2-fold-change. The transcriptional interactome network analysis was performed as described in the Supplementary Note.
Genetic effect and interaction analysis
Alternative genetic effects including additive effects, dominance, recessiveness and over-dominance for the ratio of α-carotene and β-carotene to total carotenoids were evaluated at a biallelic SNP locus (with reference and alternative alleles—for example, G and T) using SNPs with the maximum effect at the candidate genes REC1, Or and EX1, identified at QTLs mapped on chromosomes 2, 3 and 7, respectively. The SNPs and their locations in the DH1 v.3 genome used for this analysis were the following: A/C at position ch2_28364045, T/C at position chr3_5070341 and A/T at position chr7_39186121. To test for the additive and non-additive (dominance, recessiveness and over-dominance) effects, the SNPs were coded as 0 for homozygous reference allele (for example, AA), 1 for heterozygous (for example, AC) and 2 for the homozygous alternative allele (for example, CC). The allelic models were described by ref. 111.
All possible allele combinations were constructed for testing their interaction effect on the ratio of α-carotene and β-carotene to total carotenoids. To perform the analysis, the minimum number of alleles for each possible combination was set to five. All these analysis were performed in R using the lm function112.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The DH1 v.3 genome is available at CarrotOmics.org (ref. 113). All sequence data generated for this study were deposited in NCBI, under the umbrella BioProject PRJNA285926. The component BioProjects consist of PRJNA798760 for the reads used in the genome assembly, PRJNA865166 for the RNA-seq BioSamples and reads, and PRJNA865653 for the resequenced BioSamples and reads. The assembled genome sequences are available as accession numbers CP093343 through CP093353. The previously published reads used in this study are also available from the umbrella BioProject. Specific BioProject, BioSample and SRA accessions are also listed in the Supplementary Tables, where additional details for each dataset are provided. The Lunar White nucleotide sequences were deposited in NCBI under the name BankIt2620219 lunar_white_DCAR_730022_region, accession no. OP407851.
Code availability
The list of the software and parameters used in this study are available through GitHub (https://github.com/dsenalik/Carrot_Genome_DH1_v3).
References
Simon, P. W. in The Carrot Genome (eds Simon, P. et al.) 1–8 (Springer International, 2019).
Iorizzo, M. et al. Carrot anthocyanins genetics and genomics: status and perspectives to improve its application for the food colorant industry. Genes (Basel) 11, 906 (2020).
Allender, C. in The Carrot Genome (eds Simon, P. et al.) 93–100 (Springer International, 2019).
Ellison, S. in The Carrot Genome (eds Simon, P. et al.) 77–91 (Springer International, 2019).
Banga, O. Origin and distribution of the western cultivated carrot. Genet. Agrar. 17, 357–370 (1963).
Iorizzo, M. et al. Genetic structure and domestication of carrot (Daucus carota subsp. sativus) (Apiaceae). Am. J. Bot. 100, 930–938 (2013).
Ellison, S. L. et al. Carotenoid presence is associated with the Or gene in domesticated carrot. Genetics 210, 1497–1508 (2018).
Rong, J. et al. New insights into domestication of carrot from root transcriptome analyses. BMC Genomics 15, 895 (2014).
Sourkes, T. L. The discovery and early history of carotene. Bull. Hist. Chem. 34, 32–38 (2009).
Mccollum, E. V. & Davis, M. The necessity of certain lipins in the diet during growth. Nutr. Rev. 31, 280–281 (1973).
Steenbock, H. White corn vs. yellow corn and a probable relation between the fat-soluble vitamine and yellow plant pigments. Science 50, 352–353 (1919).
Ahmad, T. et al. Phytochemicals in Daucus carota and their health benefits—review article. Foods 8, 424 (2019).
Simon, P. W., Pollak, L. M., Clevidence, B. A., Holden, J. M. & Haytowitz, D. B. Plant breeding for human nutritional quality. In Plant Breed. Rev. (ed Janick J.) 31, 325–392 (2009).
Simon, P. W., Geoffriau, E., Ellison, S. & Iorizzo, M. in The Carrot Genome (eds Simon, P. et al.) 247–260 (Springer International, 2019).
Iorizzo, M. et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 48, 657–666 (2016).
Ellison, S., Senalik, D., Bostan, H., Iorizzo, M. & Simon, P. Fine mapping, transcriptome analysis, and marker development for Y(2), the gene that conditions β-carotene accumulation in carrot (Daucus carota L.). G3 (Bethesda) 7, 2665–2675 (2017).
Coe, K. M., Ellison, S., Senalik, D., Dawson, J. & Simon, P. The influence of the Or and Carotene Hydroxylase genes on carotenoid accumulation in orange carrots [Daucus carota (L.)]. Theor. Appl. Genet. 134, 3351–3362 (2021).
Grzebelus, D. et al. Diversity, genetic mapping, and signatures of domestication in the carrot (Daucus carota L.) genome, as revealed by Diversity Arrays Technology (DArT) markers. Mol. Breed. 33, 625–637 (2014).
Dwyer, S. A. et al. Antisense reductions in the PsbO protein of photosystem II leads to decreased quantum yield but similar maximal photosynthetic rates. J. Exp. Bot. 63, 4781–4795 (2012).
Maggio, C., Barbante, A., Ferro, F., Frigerio, L. & Pedrazzini, E. Intracellular sorting of the tail-anchored protein cytochrome b5 in plants: a comparative study using different isoforms from rabbit and Arabidopsis. J. Exp. Bot. 58, 1365–1379 (2007).
Zhang, D., Zhao, Y., Wang, J., Zhao, P. & Xu, S. BRS1 mediates plant redox regulation and cold responses. BMC Plant Biol. 21, 268 (2021).
Xu, Z.-S., Feng, K., Que, F., Wang, F. & Xiong, A.-S. A MYB transcription factor, DcMYB6, is involved in regulating anthocyanin biosynthesis in purple carrot taproots. Sci. Rep. 7, 45324 (2017).
Iorizzo, M. et al. A cluster of MYB transcription factors regulates anthocyanin biosynthesis in carrot (Daucus carota L.) root and petiole. Front. Plant Sci. https://doi.org/10.3389/fpls.2018.01927 (2019).
Simon, P. W. in The Carrot Genome (eds Simon, P. et al.) 137–147 (Springer International, 2019).
Fitak, R. R. OptM: estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protoc. 6, bpab017 (2021).
Terhorst, J., Kamm, J. A. & Song, Y. S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 49, 303–309 (2017).
Banga, O. The development of the original European carrot material. Euphytica 6, 64–76 (1957).
Marcus, J. H. et al. Genetic history from the Middle Neolithic to present on the Mediterranean island of Sardinia. Nat. Commun. 11, 939 (2020).
Pruneda-Paz, J. L., Breton, G., Para, A. & Kay, S. A. A functional genomics approach reveals CHE as a component of the Arabidopsis circadian clock. Science 323, 1481–1485 (2009).
Balsemão-Pires, E., Andrade, L. R. & Sachetto-Martins, G. Functional study of TCP23 in Arabidopsis thaliana during plant development. Plant Physiol. Biochem. 67, 120–125 (2013).
Li, X. et al. TCP7 interacts with Nuclear Factor-Ys to promote flowering by directly regulating SOC1 in Arabidopsis. Plant J. 108, 1493–1506 (2021).
Alessandro, M. S., Galmarini, C. R., Iorizzo, M. & Simon, P. W. Molecular mapping of vernalization requirement and fertility restoration genes in carrot. Theor. Appl. Genet. 126, 415–423 (2013).
Yan, Z., Jia, J., Yan, X., Shi, H. & Han, Y. Arabidopsis KHZ1 and KHZ2, two novel non-tandem CCCH zinc-finger and K-homolog domain proteins, have redundant roles in the regulation of flowering and senescence. Plant Mol. Biol. 95, 549–565 (2017).
Ito, S. et al. FLOWERING BHLH transcriptional activators control expression of the photoperiodic flowering regulator CONSTANS in Arabidopsis. Proc. Natl Acad. Sci. USA 109, 3582–3587 (2012).
Wu, J.-F. et al. LWD–TCP complex activates the morning gene CCA1 in Arabidopsis. Nat. Commun. 7, 13181 (2016).
Martignago, D. et al. The bZIP transcription factor AREB3 mediates FT signalling and floral transition at the Arabidopsis shoot apical meristem. PLoS Genet. 19, e1010766 (2023).
Liu, Y., Li, X., Li, K., Liu, H. & Lin, C. Multiple bHLH proteins form heterodimers to mediate CRY2-dependent regulation of flowering-time in Arabidopsis. PLoS Genet. 9, e1003861 (2013).
Lu, S. et al. The cauliflower Or gene encodes a DnaJ cysteine-rich domain-containing protein that mediates high levels of β-carotene accumulation. Plant Cell 18, 3594–3605 (2006).
Tzuri, G. et al. A ‘golden’ SNP in CmOr governs the fruit flesh color of melon (Cucumis melo). Plant J. 82, 267–279 (2015).
Zhou, X. et al. Arabidopsis OR proteins are the major posttranscriptional regulators of phytoene synthase in controlling carotenoid biosynthesis. Proc. Natl Acad. Sci. USA 112, 3558–3563 (2015).
Sun, T. et al. ORANGE represses chloroplast biogenesis in etiolated Arabidopsis cotyledons via interaction with TCP14. Plant Cell 31, 2996–3014 (2019).
Kim, C. et al. Chloroplasts of Arabidopsis are the source and a primary target of a plant-specific programmed cell death signaling pathway. Plant Cell 24, 3026–3039 (2012).
Lee, K. P., Kim, C., Landgraf, F. & Apel, K. EXECUTER1- and EXECUTER2-dependent transfer of stress-related signals from the plastid to the nucleus of Arabidopsis thaliana. Proc. Natl Acad. Sci. USA 104, 10270–10275 (2007).
Wagner, D. et al. The genetic basis of singlet oxygen-induced stress responses of Arabidopsis thaliana. Science 306, 1183–1185 (2004).
Ramel, F. et al. Carotenoid oxidation products are stress signals that mediate gene responses to singlet oxygen in plants. Proc. Natl Acad. Sci. USA 109, 5535–5540 (2012).
Wang, L. et al. Singlet oxygen- and EXECUTER1-mediated signaling is initiated in grana margins and depends on the protease FtsH2. Proc. Natl Acad. Sci. USA 113, E3792–E3800 (2016).
Zhang, Y. M. et al. Plastid diversity and chromoplast biogenesis in differently coloured carrots: role of the DcOR3(Leu) gene. Planta 256, 104 (2022).
Larkin, R. M. et al. Reduced chloroplast coverage genes from Arabidopsis thaliana help to establish the size of the chloroplast compartment. Proc. Natl Acad. Sci. USA 113, E1116–E1125 (2016).
Stanley, L. E. et al. A tetratricopeptide repeat protein regulates carotenoid biosynthesis and chromoplast development in monkeyflowers (Mimulus). Plant Cell 32, 1536–1555 (2020).
Liang, M. et al. Taxon-specific, phased siRNAs underlie a speciation locus in monkeyflowers. Science 379, 576–582 (2023).
Banga, O. Origin of the European cultivated carrot. Euphytica 6, 54–63 (1957).
Simon, P. Domestication, historical development, and modern breeding of carrot. Plant Breed. Rev. 19, 157–190 (2000).
Stolarczyk, J. & Janick, J. Carrot: history and iconography. Chron. Hortic. 51, 13–18 (2011).
Banga, O. Main Types of the Western Carotene Carrot and Their Origin. (W. E. J. Tjeenk Willink, 1963).
Linke, B., Alessandro, M. S., Galmarini, C. R. & Nothnagel, T. in The Carrot Genome (eds Simon, P. et al.) 27–57 (Springer International, 2019).
Wingett, S. & Andrews, S. FastQ Screen: a tool for multi-genome mapping and quality control [version 2; peer review: 4 approved]. F1000Res. https://doi.org/10.12688/f1000research.15931.2 (2018).
Bannoud, F. et al. Genetic and transcription profile analysis of tissue-specific anthocyanin pigmentation in carrot root phloem. Genes (Basel) https://doi.org/10.3390/genes12101464 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Iorizzo, M. et al. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics 12, 389 (2011).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Macko-Podgórni, A., Machaj, G. & Grzebelus, D. A global landscape of miniature inverted-repeat transposable elements in the carrot genome. Genes (Basel) 12, 859 (2021).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Novák, P., Neumann, P. & Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 11, 378 (2010).
Novák, P., Neumann, P., Pech, J., Steinhaisl, J. & Macas, J. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29, 792–793 (2013).
Neumann, P., Novák, P., Hoštáková, N. & Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 10, 1 (2019).
Kwolek, K. et al. Diverse and mobile: eccDNA-based identification of carrot low-copy-number LTR retrotransposons active in callus cultures. Plant J. 110, 1811–1828 (2022).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Iovene, M. et al. Comparative FISH mapping of Daucus species (Apiaceae family). Chromosome Res. 19, 493–506 (2011).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 491 (2011).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol. Biol. 1962, 161–177 (2019).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467 (2005).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59 (2004).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 47, 965–978 (2005).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Dai, X., Sinharoy, S., Udvardi, M. & Zhao, P. X. PlantTFcat: an online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinform. 14, 321 (2013).
Osuna-Cruz, C. M. et al. PRGdb 3.0: a comprehensive platform for prediction and analysis of plant disease resistance genes. Nucleic Acids Res. 46, D1197–D1201 (2018).
Waterhouse, R. M. et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Simon, P. W. & Wolff, X. Y. Carotenes in typical and dark orange carrots. J. Agric. Food Chem. 35, 1017–1022 (1987).
Simon, P. et al. High carotene mass carrot population. Hort. Sci. 24, 174–175 (1989).
Rubatzky, V. E., Quiros, C. F. & Simon, P. W. Carrots and Related Vegetable Umbelliferae (CABI, 1999).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Narasimhan, V. et al. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 32, 1749–1751 (2016).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Felsenstein, J. PHYLIP (Phylogeny Inference Package) v.3.5 c (Univ. of Washington, 1993).
Arbizu, C. I., Ellison, S. L., Senalik, D., Simon, P. W. & Spooner, D. M. Genotyping-by-sequencing provides the discriminating power to investigate the subspecies of Daucus carota (Apiaceae). BMC Evol. Biol. 16, 234 (2016).
Shimada, M. & Nishida, T. A modification of the PHYLIP program: a solution for the redundant cluster problem, and an implementation of an automatic bootstrapping on trees inferred from original data. Mol. Phylogenet. Evol. https://doi.org/10.1016/j.ympev.2017.02.012 (2017).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T.-Y. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Korunes, K. L. & Samuk, K. pixy: unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Mol. Ecol. Resour. 21, 1359–1368 (2021).
Zhang, C., Dong, S. S., Xu, J. Y., He, W. M. & Yang, T. L. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788 (2019).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Zhang, L. et al. RNA sequencing provides insights into the evolution of lettuce and the regulation of flavonoid biosynthesis. Nat. Commun. 8, 2264 (2017).
Chen, H., Patterson, N. & Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 20, 393–402 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Bradbury, P. J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Lipka, A. E. et al. GAPIT: genome association and prediction integrated tool. Bioinformatics 28, 2397–2399 (2012).
Huang, M., Liu, X., Zhou, Y., Summers, R. M. & Zhang, Z. BLINK: a package for the next level of genome-wide association studies with both individuals and markers in the millions. GigaScience https://doi.org/10.1093/gigascience/giy154 (2019).
Gao, X., Starmer, J. & Martin, E. R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 32, 361–369 (2008).
Turner, S. qqman: An R package for visualizing GWAS results using Q-Q and Manhattan plots. R package version 0.1.8 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Tsepilov, Y. A. et al. Nonadditive effects of genes in human metabolomics. Genetics 200, 707–718 (2015).
R Core Team (2022) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org
Rolling, W. R. et al. CarrotOmics: a genetics and comparative genomics database for carrot (Daucus carota). Database https://doi.org/10.1093/database/baac079 (2022).
Acknowledgements
This project was supported by the National Institute of Food and Agriculture, US Department of Agriculture, under award nos 2016-51181-25400 and 2022-51181-38321. M.I., H.B., M.F.M. and J.C. were also supported by the US Department of Agriculture National Institute of Food and Agriculture, Hatch project no. 1008691, and S.T.-H. was supported by the NSF Postdoctoral Research Fellowship in Biology, grant no. 1711347. A.M.-P. and D.G. were funded by the Polish National Science Center, grant no. 2019/33/B/NZ9/ 00757 (OPUS17). W.R. was partially supported by the US Department of Agriculture National Institute of Food and Agriculture project no. 5090-21000-069-061-I. We thank J. Ross-Ibarra for helpful advice on the analyses of demographic history and nucleotide diversity.
Author information
Authors and Affiliations
Contributions
P.S., M.I., S.E. and A.V.D. conceptualized the project. K.C., H.B., S.E., W.R., S.T.-H., A.M.-P., D.S., J.C., P.S. and M.I. devised the methodology. K.C., H.B., W.R., S.T.-H., R.S., A.M.-P., D.S., S.L., J.C., M.F.M. and M.I. conducted the investigation. K.C., H.B., W.R., S.T.-H., A.M.-P., D.G., D.S., J.C., M.F.M. and M.I. visualized the data. P.S. and M.I. supervised the project. M.I., K.C., H.B., W.R., S.T.-H., A.M.-P. and D.S. wrote the original draft of the manuscript. M.I., K.C., H.B., W.R., S.T.-H., A.M.-P., D.S., S.L., J.C., M.F.M., D.G., A.V.D., J.D., S.E. and P.S. reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Plants thanks Xiaowu Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Scheme of the carrot genome assembly.
In Phase I newly sequenced long reads (PacBio and Nanopore) and Hi-C reads from DH1 were used for de-novo assembly, nucleotide error correction (polishing), scaffolding and correcting chimeric sequences. These steps generated contigs and scaffolds. In Phase II, unambiguously aligned sequences from mapped molecular markers, BAC end sequences, Hi-C sequences and 10, 20 and 40 kb Illumina MPE were used to correct chimeric regions and anchor the genome assembly. These steps generated anchored and un-anchored contigs and scaffolds. In Phase III, the assembly obtained from Phase II at scaffold and contig or scaftig level was used to perform a guided genome assembly and to fill additional gaps. These steps produced the carrot DH1 assembly v3.0, that includes nine pseudomolecules or chromosomes.
Extended Data Fig. 2 Comparison of genome assemblies with genetic map, and Hi-C data.
a) Alignment of the integrated linkage map (LG), and the DH1-v2 assembled chromosomes with the DH1-v3 assembled chromosomes. Black dots in the DH1-v3 chromosome scheme represent the approximate location of the centromere repeats. Gray lines between the DH1-v3 and DH1-v2 indicate collinear sequences. Red lines between the DH1-v3 and DH1-v2 indicate non-collinear sequences. b) Heat map of Hi-C contact information along the DH1-v3 chromosomes. Pixel colors represent different normalized counts of Hi-C links between 30-kb non-overlapping windows for all 9 chromosomes (Chr) on a logarithmic scale. Green lines represent the boundaries of individual contigs. Numbers in green represent the extra sequence in Mb that was new assembled compared to DH1 v2 assembly, into each chromosome.
Extended Data Fig. 3 DH1 v3 assembly quality verification using a carrot linkage map.
Comparison of the genetic map of population 32421 to the physical map of the DH1 v3 genome assembly.
Extended Data Fig. 4 Comparison of mobile elements annotated in the DH1 v2 and v3 genome assemblies.
Comparison of the number (a, b) and size (c, d) of full-length LTR retrotransposons (a, c) and TIR DNA transposons (b, d) between v2 (blue) and v3 (red) assemblies.
Extended Data Fig. 5 GC content estimated in the DH1 v2 and v3 genome assemblies.
a) GC content in the v2, v3 genomes and in the newly assembled sequences in the v3 genome. b) GC content in the genes predicted in the v3 genome and in newly assembled sequences. Note, for each fraction of genome and genes evaluated in this analysis (for example v2 genome, v3 genes) the frequency of bins for each GC level (1% GC windows) was rescaled independently setting the minimum number of bins to 0 and maximum number of bins to 100, and plotted on the y axis. The calculation was carried out using mapminmax function implemented in Matlab.
Extended Data Fig. 6 Transposable element age estimates.
Age (Myr)distribution of full-length LTR retrotransposon lineages in DH1 v2 (blue) and v3 (red) carrot genome assemblies.UCn(v3)=1894, n(v2)= 2137; DcTork n(v3)= 52, n(v2)= 48; DcTakay n(v3)= 547, n(v2)= 383; DcTAR n(v3)= 139, n(v2)= 114; DcSIRE n(v3)= 2225, n(v2)= 1106; DcRetand n(v3)= 1184, n(v2)= 731; DcReina n(v3)= 157, n(v2)= 158; DcIvana n(v3)= 228, n(v2)= 213; DcIkeros n(v3)= 10, n(v2)= 8; DcCRM n(v3)= 22, n(v2)= 17; DcBianca n(v3)= 113, n(v2)= 103; DcAthila n(v3)= 918, n(v2)= 477; DcAngela n(v3)= 292, n(v2)= 222; DcAlesia n(v3)=8, n(v2)=7; DcAle n(v3)= 274, n(v2)= 252. The boxplot represents the 25th, 50th and 75th percentiles with the upper and lower whisker 1.5x the 75th and 25th percentiles, respectively.
Extended Data Fig. 7 DH1 v2 and v3 assemblies’ quality verification using LTR Assembly Index (LAI).
Normalized (a) and RAW (b) LAI lineages in DH1 v2 (blue) and v3 (red) carrot genome assemblies.
Extended Data Fig. 8 Gene isoforms.
Schematic representation of the type of isoforms detected in carrot DH1 v3 genome.
Extended Data Fig. 9 Validation of alternative splicing events in DH1 by qRT-PCR.
Eight loci were analyzed, each expressing two isoforms generated from alternative splicing event. Glutathione S-transferase (GST), Terpene synthase (TPS), FAD-dependent hydroxylase (Hpxo) ubiquitin-like-specific protease 2B (ULP2B), MYB1R, Universal phosphorylated stress protein (PHOS34), TBC1 and Fbox. For each locus, the top scheme of the panel show the distribution of exons (dark grey rectangles), skipped exons (light grey rectangles) and retained introns (light grey lines) for both isoform, as well as the percentage of unique circular consensus sequence (CCS) detected in the IsoSeq libraries. The corresponding functional domain(s) predicted using Pfam (pfam.xfam.org) is displayed below each isoform. qRT-PCR reactions were designed to selectively amplify only one isoform. The position of the amplicon is indicated for each isoform by an orange or yellow line. Their level of expression was normalized to the ACTIN housekeeping gene using the ∆Ct method.
Extended Data Fig. 10 Comparison between the structure of the genes predicted in the v2 and v3 genome assemblies.
a) Example of a gene predicted in v3 and not predicted in v2; b) example of a merged gene, predicted as one gene in v2 and two genes in v3; c) example of a split gene, predicted as one gene in v3 and two genes in v2. The quality of the predicted genes was supported by experimental evidence, including IsoSeq and Illumina transcriptome sequences.
Supplementary information
Supplementary Information
Supplementary Figs 1–10 and Note.
Supplementary Data 1
Supplementary Tables 1–46.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Coe, K., Bostan, H., Rolling, W. et al. Population genomics identifies genetic signatures of carrot domestication and improvement and uncovers the origin of high-carotenoid orange carrots. Nat. Plants 9, 1643–1658 (2023). https://doi.org/10.1038/s41477-023-01526-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41477-023-01526-6
This article is cited by
-
The changing colour of carrot
Nature Plants (2023)
