
Abstract
The pre-training molecular representation learning (MRL) has shown considerable potential in computer-aided drug discovery. Recently, many multimodal pre-training MRL methods have been presented, incorporating multimodal molecular data for pre-training and achieving high-accuracy predictions in downstream tasks. However, most current methods require completeness of modality for molecular data in the pre-training phase and often overlook their adaptation to real-world scenarios where, for example, molecular modalities except 2D topological graphs (2D modality) are often unavailable. In this study, we propose a multimodal pre-training MRL framework called M2UMol, which separately matches 2D modality to multiple modalities and undergoes pre-training jointly with a modality classifier. In this way, M2UMol elegantly transfers multimodal knowledge into the 2D modal encoder and allows for inputting incomplete modalities in the pre-training stage. Moreover, in downstream tasks with only the 2D modality given, M2UMol enables the precise simulation of molecular multimodal information based on the pre-trained 2D modal encoder. Comprehensive experimental results show the superior performance of M2UMol in a wide range of molecular tasks with higher efficiency in pre-training than pioneer models and demonstrate the validity of the multimodal knowledge transfer. Furthermore, we developed a user-friendly package based on M2UMol, integrating molecular representation learning, key functional group analysis, molecular multimodal retrieval, etc. It may be conveniently used in diverse fields related to drug discovery and promises to facilitate the process of developing drugs. Our code, pre-trained weights of M2UMol, and the package are available at https://github.com/Zhankun-Xiong/M2UMol.
Similar content being viewed by others

Introduction
In the last decades, artificial intelligence (AI) technologies, particularly machine learning, have got a wide range of applications in drug discovery, such as molecular property prediction1,2, drug-drug interaction (DDI) prediction3,4,5 and drug-target interaction (DTI) prediction6,7, etc. Among these applications, a fundamental challenge is the featurization of molecules into numerical vectors, i.e., molecular representations. Traditional molecular representations, such as descriptor-based features8, are handcrafted features heavily limited to human-summarized physicochemical domain knowledge. By contrast, Molecular Representation Learning (MRL) has been viewed as a promising approach, which leverages deep learning to automatically mine latent knowledge from molecules and produce superior molecular representations that significantly outperform traditional handcrafted features in important tasks of drug discovery9,10,11.
The MRL methods typically consider molecular input data in various formats (modalities), including one-dimensional (1D) molecular sequences12,13, often represented by the Simplified Molecular-Input Line-Entry System (SMILES)14; two-dimensional (2D) topological graphs15,16 with atoms as vertices and covalent bonds as edges; and three-dimensional (3D) conformer graphs17,18 derived from molecule conformations, which can be encoded into molecular representations by various encoders. Commonly, many MRL methods train the encoders on specific-task labeled molecules in a supervised manner. Nevertheless, the scarcity of labeled molecular data significantly impacts the quality of molecular representations and especially limits their out-of-distribution (OOD) generalization. In recent years, there has been a surge in the development of pre-training models within the field of MRL. These MRL methods begin their training process on unlabeled molecular data through self-supervised pretext tasks. Subsequently, they undergo fine-tuning using a small set of labeled molecular data tailored to specific downstream tasks, resulting in notably enhanced performance.
The self-supervised pre-training MRL methods are broadly classified into unimodal-based methods and multimodal-based methods. Unimodal pre-training MRL methods focus on a single molecular modality, typically SMILES or graphs, and learn effective molecular representations by a range of self-supervised tasks, such as predicting specific parts of molecules19,20,21,22,23 and contrasting molecules with their augmented counterparts24,25,26. In contrast, multimodal pre-training MRL methods enhance the quality of molecular representations by capturing complementary information from different molecular modalities. These methods can be divided into two categories. The first category can be summarized as a “one-one” paradigm, which aims to model the relationship between two modalities in pre-training to grasp multimodal information. Typical pre-training tasks in this category include aligning the representations of two molecular modalities using contrastive learning27,28,29,30 or generating a molecular modality from another30,31,32. In such frameworks, downstream fine-tuning usually relies on a single pre-trained encoder, typically a 2D molecular encoder. As a result, the single pre-trained encoder cannot fully exploit the multimodal knowledge learned in pre-training. Moreover, this category is inherently limited to two molecular modalities and provides an insufficient understanding of molecules due to the lack of knowledge from more modalities. It is challenging to design pre-training strategies involving three or more molecular modalities. The second category includes recent efforts33,34 which attempt to address these challenges through a “one-others” paradigm. These methods select one modality as the anchor and integrate representations of all other modalities through concatenation or summation. They then adopt contrastive learning to align the representations of the anchor modality with the integrated multimodal representations. In their fine-tuning phase, all the encoders for distinct modalities serve as pre-trained modules, and their learned representations are integrated for downstream tasks. However, these methods often necessitate that the molecules have complete modalities during both the pre-training and fine-tuning phases, resulting in two flaws. On the one hand, they fail to exploit larger-scale molecules with missing modalities for more sufficient pre-training. On the other hand, they are not competent in practical downstream applications, where all modalities are often unavailable except molecular 2D topological graphs (usually converted from SMILES). For more clearly distinguishing the existing multimodal pre-training MRL methods, we summarize their characteristics in Supplementary Table S2, including some details such as modality usage and pre-training strategy.
In this study, we propose a multi-to-uni modal knowledge transfer pre-training MRL method, termed M2UMol. M2UMol first learns 2D molecular representations and multimodal representations through corresponding encoders, and the modal-specific adapters are designed to generate pseudo multimodal representations based on 2D molecular representations. Then M2UMol undergoes pre-training using the two pretext tasks: generated-actual multimodal contrastive learning and modality classification. The former aims at alignment of the generated pseudo multimodal representations with the corresponding actual multimodal representations in three independent vector spaces, guiding the transfer of multimodal knowledge to the 2D modal encoder and enabling the modal-specific adapters to generate more reliable multimodal representations; and the latter attempts to classify the generated representations into three types of actual modalities, guiding the adapters capturing modal-specific knowledge and generating higher-distinguishability representations. In the subsequent fine-tuning phase, the pre-trained 2D encoder and modal-specific adapters are utilized to learn 2D representations and generate 3D/Text/Bio representations, respectively, and these representations are adaptively integrated by a multi-head attention mechanism to obtain the final molecular representations for various downstream tasks.
Comprehensive experimental results validate the favorable performances of M2UMol on both molecular property prediction tasks and molecular interaction prediction tasks, especially under a harsh evaluation scenario of the scaffold split. In addition, compared to models pre-trained on millions of molecules, M2UMol is only pre-trained on 11k molecules with significantly less computational cost and shorter pre-training time (See Supplementary Table S3). It demonstrates the high scalability and efficiency of our designed pre-training strategies. Moreover, we conducted extensive experiments to evaluate the quality of generated multimodal representations and the benefits of multimodal pre-training, and verified that M2UMol can learn discriminative and uniform molecular representations for downstream tasks and provide accurate and interpretable predictions. We also developed a user-friendly package based on M2UMol, which integrates multiple functions including molecular representation learning, multimodal data retrieval, and key structure identification. The application of the M2UMol package can be found in Section 4 of the Supplementary Information.
The contributions of this paper include the following:
-
We propose M2UMol, a multi-to-uni modal knowledge transfer pre-training MRL method, which is tailored to two practical application challenges: incomplete modalities in pre-training and unimodal downstream tasks.
-
The modal-specific adapters are designed to receive the multimodal knowledge in the pre-training stage and generate pseudo multimodal representations solely from 2D molecular representations for supplementing multimodal information.
-
Through the two well-designed multimodal self-supervised tasks, we achieve the guidance of multi-to-uni modal knowledge transfer and empower the generated multimodal representations to be more reliable and keep modal-specific information.
-
Comprehensive experimental results show the superior performances of M2UMol in a wide range of molecular tasks with high efficiency in pre-training, and verify that M2UMol can learn high-quality molecular representations and provide interpretable predictions. Our codes, pre-trained weights, and package are open-source and can be conveniently used in diverse related fields in AI-assisted drug discovery.
Results
Overview of M2UMol
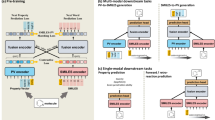
The proposed M2UMol is shown in Fig. 1. In this paper, we constructed a multimodal molecular dataset, which includes 11,571 drug-like molecules with four modalities: 2D topological graphs (2D), 3D conformer graphs (3D), textual descriptions (Text), and biochemical features (Bio) (Fig. 1a), among which biochemical features of molecules that are believed to contain rich domain knowledge, can be considered as an important modality of data, but usually ignored in previous works. M2UMol encompasses four modal encoders and the modal-specific adapters, and undergoes pre-training on the constructed dataset using two innovative self-supervised pretext tasks: generated-actual multimodal contrastive learning and modality classification (Fig. 1b). Then, the pre-trained 2D encoder and the modal-specific adapters, as well as a learnable multi-head attention module, compose our plug-in fine-tuning architecture, which outputs molecular representations for various downstream tasks with only 2D modality (Fig. 1c).

a The four types of molecular multimodal data, and the Venn diagram generated by Evenn tool90 represents the distribution of multimodal data in our constructed pre-training dataset. In the Venn diagram, the area bordered by the red line denotes molecules with incomplete modalities, and the area bordered by the black dashed line denotes molecules with complete modalities. b The 2D/3D/Text/Bio modal data are fed into corresponding encoders to obtain their representations. Then the 2D representations are fed into the designed modal-specific adapters to generate pseudo 3D/Text/Bio representations. The pre-training objective is to minimize the loss of two pretext tasks: the generated-actual multimodal contrastive learning and the modality classification. c The 2D topological graphs are fed into the pre-trained 2D graph encoder and the modal-specific adapters to obtain 2D representations and generate 3D/Text/Bio representations. Then these representations are adaptively integrated by a multi-head attention mechanism to obtain the final molecular representations for various downstream tasks.
It is worth noting that the generated-actual multimodal contrastive learning contains three contrastive learning objectives that aim to separately align the pseudo representations for each modality from {3D, Text and Bio} with the corresponding actual representations. Hence, the molecules with both 2D graph structures and at least one of the three modalities can be used for our pre-training, without requiring complete modalities for molecules. Taking our pre-training dataset as an example, this capability enables M2UMol to scale pre-training from 4325 molecules (the black-bordered area in the Venn diagram of Fig. 1a) with complete modalities to 11,571 molecules (the area bordered by the red line), by incorporating molecules with incomplete modalities. This leads to approximately a threefold increase in pre-training data, which significantly enhances the performance of M2UMol (see Supplementary Fig. S3).
M2UMol outperforms the baseline methods on molecular property prediction and molecular interaction prediction tasks
In this section, we assess the effectiveness of our proposed method M2UMol by comparing it with baseline methods across two types of molecular downstream tasks, including molecular property prediction (MPP) and molecular interaction prediction (MIP). The details about fine-tuning on these downstream tasks can be found in Section 2.3 of the Supplementary Information.
Molecular property prediction (MPP)
Molecular property prediction is a crucial task in drug discovery, beneficial to scientists for screening out molecules with desired properties. This field has been rapidly growing in AI for Science, with a significant number of methods developed, including pre-training MRL methods. We follow most existing methods to evaluate our method M2UMol on scaffold-based splits. The scaffold split is a challenging and realistic evaluation setting, commonly used to assess the OOD generalization capabilities of MRL methods. Here, we compare M2UMol with recent strong pre-training MRL methods for property prediction. The results are shown in Table 1.
A general observation from Table 1 is that multimodal pre-training MRL methods tend to produce better performance than unimodal methods. The performance enhancement is particularly noteworthy when more modalities are incorporated in the pre-training phase, as exemplified by our method M2UMol. Such a trend suggests that the use of more modalities for pre-training may have more potential benefits for downstream molecular property prediction tasks. Among the baselines, M2UMol produces the best performances on 5 out of 8 datasets with average improvements of 5.21%, 10.00%, 10.69%, 5.57%, and 8.79% on AUC, respectively. We attribute the favorable results of M2UMol on unimodal molecular property prediction tasks to our design of transferring multimodal knowledge into 2D molecular representation learning in the pre-training phase and simulating the multimodal information fusion solely based on the 2D modality in the fine-tuning phase. Additionally, as summarized in Supplementary Table S3, M2UMol is pre-trained on only 11k molecules using a single GPU (RTX 3090) within 11 hours, yet still achieves superior predictive performance. This demonstrates the high scalability and efficiency of our model. More results about performance comparison can be found in Section 3.1 of the Supplementary Information.
To sum up, our proposed method, M2UMol, shows promise as an effective tool for predicting molecular properties for those molecules with only 2D modal data available.
Molecular interaction prediction (MIP)
Molecular interactions can provide significant information for sophisticated biological system analysis, which is a powerful resource for drug discovery. Herein, we choose two representative molecular interaction prediction tasks: the DDI prediction task and the DTI prediction task, to further evaluate the performance of M2UMol.
DDIs can arise when multiple drugs are taken simultaneously, leading to unexpected adverse effects, known as DDI events (DDIEs). Predicting DDIEs, typically considered as a multi-classification task within DDI prediction, is crucial for public health security and medicine safety surveillance, garnering much attention in the deep learning and bioinformatics communities. For this task, we focus predominantly on quite challenging scenarios: the cold start split and the scaffold split. For the former scenario, the drugs used for testing are not included in the DDI data on which our method is fine-tuned, while for the latter scenario, not only the testing drugs but also their scaffolds are unseen in the fine-tuning phase. Note that the second scenario is more intractable than the first one, but has been little concerned in previous works. We choose recent advanced DDIE prediction methods, which can be applied to both scenarios, as baselines. The results are shown in Fig. 2a, b. Overall, M2UMol significantly outperforms all baselines under both scenarios. Specifically, compared with the baselines, M2UMol achieves average performance gains of 9.66% and 6.84% in ACC, 11.92% and 7.74% in Macro-F1, 12.03% and 7.47% in Macro-R, as well as 12.27% and 10.43% in Macro-P, respectively, in the cold start split and scaffold split scenarios.

Error bars denote the standard deviation of independent runs, and dots represent individual experimental results. For drug-drug interaction (DDI) prediction, n = 3 independent runs with different random seeds were performed; for drug-target interaction (DTI) prediction, n = 5 independent runs were performed. ACC, AUC, and AUPR refer to accuracy, area under the Receiver Operating Characteristic (ROC) curve, and area under the precision-recall curve, respectively. The values are reported as percentages. a, b The performances of M2UMol and representative DDI prediction methods, including STNN-DDI74, SSI-DDI75, GMPNN76, SA-DDI77, MSAN78, DSN-DDI79 and 3DGT-DDI80, on the DDI dataset66 under the cold start split setting and the scaffold split setting. c, d The performances of M2UMol and representative DTI prediction methods, including Support Vector Machines (SVM)81, Random Forests (RF)82, DeepConv-DTI83, GraphDTA84 and MolTrans85, on BindingDB67 and BioSNAP68 datasets under random split setting and scaffold split setting. Source data are provided as a Source Data file.
DTI prediction is an integral step in drug discovery, which helps to narrow down the search scope of compound candidates. We replace the drug representation learning module in DrugBAN35, a milestone method for predicting DTIs, with our fine-tuning architecture to adapt M2UMol to DTI prediction. Then, we follow two evaluation scenarios in ref. 35, i.e., the random split and the scaffold split, to exhibit the abilities of M2UMol in improving DTI prediction. Figure 2c, d shows the results of different methods in DTI prediction. We observe that compared with DrugBAN, the result of M2UMolunpre is not significantly improved, while the improvement of M2UMol is extra pronounced. To be more specific, M2UMol performs better than DrugBAN with average improvements of 2.60% in AUC and 2.48% in AUPR on two datasets across two evaluation scenarios.
In summary, our proposed method, M2UMol, realizes superior performance on both molecular interaction prediction tasks, showcasing its reliability, flexibility, and robustness, which are attributed to its ability to offer high-quality representations for drug molecules. Its superiority in predicting molecular interactions underscores its extraordinary potential to promote AI-aided drug discovery.
M2UMol acquires diverse knowledge from multimodal data in pre-training
This section shows how M2UMol learns knowledge from unlabeled multimodal data during pre-training. We evaluate the quality of multimodal representations generated by M2UMol and discuss the capability of M2UMol to identify the key structures of molecules without labeled molecular data. In Section 3.2 of the Supplementary Information, we further discuss the importance of the three modalities used, explore multimodal synergy in facilitating cross-modal understanding, and analyze the impact of the five designed components (modality-specific adapters, modality classifier, and other components) on the performance of M2UMol. The results shown in Supplementary Fig. S3 demonstrate that M2UMol can effectively learn multimodal knowledge, addressing the practical application challenges of incomplete modalities in pre-training and unimodal downstream tasks, and thus achieving superior performance.
M2UMol can generate reliable multimodal representations from 2D molecule graphs
For the actual representation of each molecule output from the encoder for each modality (3D/Text/Bio), the modal-specific adapters in M2UMol are to generate the matched pseudo representation solely from the 2D representation of the corresponding molecule. We visualized the actual and generated multimodal representations by t-Distributed Stochastic Neighbor Embedding (t-SNE)36. As shown in Fig. 3a, we observe that both actual modality representations and generated modality representations are distributed separately into three clusters according to the modality types. This observation demonstrates that M2UMol can capture differentiated knowledge of different modalities, which is transferred to the adapters, such that it has the ability to generate distinguishable representations only based on 2D representations. Further, we conducted a multimodal retrieval task37 to assess the quality of the generated multimodal representations. Recall that all actual and generated representations have identical dimensions in our setting, so we consider all the matched generated-actual representation pairs in a common Euclidean space, and we check whether the actual representation in each pair is within the top-k closest representations to the matched generated representation in the common space. We utilized the widely used metric Recall@K29,31,33,37 to evaluate the quality of generated multimodal representations in the multimodal retrieval task. We adopted Recall@K with optional k in {1, 3, 5, 10}. As shown in the bar chart in Fig. 3a, we can observe that: (1) the generated 3D representations are higher-quality than the generated representations of other modalities, likely due to the narrow modality gap between 3D and 2D which both depict molecular structural information; (2) despite the wide modality gap between Text and 2D as well as smaller-scale data of Text than both 3D and Bio, the generated Text representations still approximate to their matched actual representations well (Recall@1 = 0.72, Recall@10 = 0.98), indicating our models’ ability to bridge modality gaps effectively; (3) the quality of the generated Bio representations are relatively poor while acceptable (Recall@1 = 0.35, Recall@10 = 0.76), potentially due to the sparsity of the binary biochemical feature vectors. Overall, the generated and actual representations are matched well in the pre-training phase, which demonstrates the robust capability of M2UMol in generating reliable multimodal representations only using 2D molecule graphs.

a The t-Distributed Stochastic Neighbor Embedding (t-SNE) visualization of the actual and generated multimodal representations, and the Recall@K (abbreviated as R@k) of the multimodal retrieval task on different modalities. Source data are provided as a Source Data file. b The four modalities of the query molecule, and the top 4 modalities closest to the query molecule in different modal representation spaces. c Visualized attention weights of the molecules learned by M2UMol and the three variants (2D+3D, 2D+Text and 2D+Bio modalities for pre-training). The attention weights are extracted from the 2D encoder and normalized. Darker colors indicate higher attention weights. Then, we exhibit the interaction potential maps, where dense probe-point distributions highlight regions with higher interaction potential, thereby identifying molecular key functional groups. They can serve as references to check the model’s ability to capture the functional groups of molecules.
In addition, inspired by previous studies24,38,39, we conducted case studies to show that M2UMol effectively captures semantic information, resulting in similar generated representations for molecules sharing similar characteristics. We selected Diflorasone (ID: DB00223) as the query molecule, and retrieved its top-4 closest molecules in the 2D representation space or in the generated 3D/Text/Bio representation space. The modality data of the retrieved molecules are visualized in Fig. 3b. The top-4 closest molecules in each modality representation space exhibit characteristics akin to Diflorasone. Specifically, in the 2D modality, Diflorasone and the nearest Flumethasone are a pair of conformational isomers, which differ from the other three molecules only in a few groups, such as the substitution of F with CL and CH3 with OH. In 3D modality, Diflorasone and its neighboring molecules have highly similar atomic compositions and structures. In the Text modality, all the listed molecules are corticosteroids with anti-inflammatory effects. In Bio modality, the illustrated molecules target the Glucocorticoid Receptor and can also be metabolized by the enzymes in the CYP3 family, meanwhile sharing similar drug categories. This illustrates M2UMol’s ability to capture the intrinsic relationships between molecules. Moreover, the closest molecules vary across different modalities, which highlights M2UMol’s capability to grasp modality-specific information and generate distinct representations for each modality. This capability ensures the provision of complementary and comprehensive information for each molecule. More results about the multimodal retrieval case studies can be found in Section 3.3 of the Supplementary Information.
M2UMol enables precise focus on the key structures of molecules without labeled data
Since M2UMol uses more modal molecular data than most existing pre-trained MRL models that often include two modalities, a problem naturally arises whether using more modalities in our multi-to-uni modal knowledge transfer pre-training has more benefits for M2UMol to understand key molecular knowledge from a more comprehensive perspective. To answer this problem, we choose several molecules from the pre-training dataset and visualize the attention weights on the atoms and bonds of the molecules learned by our pre-trained M2UMol (2D+3D+Text+Bio) and its three variants pre-trained with partial modalities (2D+3D, 2D+Text, or 2D+Bio) in Fig. 3c. In addition, we exhibited the interaction potential maps computationally generated from Molecular Operating Environment (MOE) (http://www.chemcomp.com/), which provide graphical representations of the regions where chemical probes (Na+, N1+, and O in this case) have favorable interactions with molecular surfaces. Based on the interaction potential maps, we can also pinpoint key active groups of the exemplified molecules serving as the reference for comparative analysis with the attention visualization.
As shown in Fig. 3c, with the progressive inclusion of modalities during the pre-training phase (left to right), M2UMol gradually allocates its attention to specific molecular fragments that are highly consistent with the key active groups identified from the interaction potential maps. We here take Oxymetazoline, an adrenergic agonist, as an example: M2UMol pays much attention to the hydroxy and the imidazoline ring, which are involved in hydrogen bonding interactions and ionic interactions during the binding of Oxymetazoline to adrenergic receptors40, and the other variants disperse their attention. The results demonstrate that M2UMol has the potential to understand molecular structure-activity relationships and identify the key structure of the molecule only based on the multimodal molecular data itself, and the more modalities it incorporates, the more precise the understanding becomes. More details about molecular key structures identification can be found in Section 3.4 of the Supplementary Information.
M2UMol improves performances on downstream tasks with high-quality representations
In this section, we discuss how molecular representations delivered by M2UMol benefit the various downstream tasks. Specifically, we consider the testing samples (i.e., molecules for MPP task; drug-drug or -target pairs for MIP tasks) in the scaffold split scenario, and visualize their representations from M2UMol and M2UMolunpre via t-SNE36. Notice that herein, ’M2UMol’ denotes the pre-trained M2UMol with fine-tuning, and ’M2UMolunpre’ represents our fine-tuning architecture directly trained on downstream tasks with random initialization. As shown in Fig. 4a, we can see that the representations learned by M2UMol for positive and negative samples in two MPP datasets, BBBP and BACE, are more clearly separated into two parts than those from M2UMolunpre. For two DTI datasets, BindingDB and BioSNAP, M2UMol achieves a lower Davies-Bouldin (DB) index (6.4997 and 4.2999) than M2UMolunpre. For the DDI dataset, we choose the drug-drug pairs of the sparsest 10 DDI events within the testing set for the t-SNE visualization, where it is observed that the representations learned by M2UMol are more tightly clustered compared to M2UMolunpre. In summary, the superior performance of our M2UMol on various downstream tasks can be owed to its success in enhancing the representations of molecules with only unimodal inputs in downstream tasks by learning transferable knowledge from pre-training on multimodal molecule data.

a The t-Distributed Stochastic Neighbor Embedding (t-SNE) visualization of the representations of samples in various downstream tasks to see whether samples belonging to the same class would have similar representations. Different colors represent different classes, with a lower Davies-Bouldin (DB) index indicating better clustering separation. b Molecular representations distributions of various downstream tasks are plotted with Gaussian Kernel Density Estimation (KDE) in \({{\mathbb{R}}}^{2}\)(darker colors indicate more points fall in the region) and the corresponding Average Pairwise Gaussian Potential (APGP) score, which measures the average distance between molecular representations. The angle density estimation curves, computed using arctan2(y, x) for each point (x, y), are also plotted. Source data are provided as a Source Data file.
Inspired by previous studies41,42, high-quality representations of molecules should be roughly uniformly distributed on the unit hypersphere, preserving as much data information as possible. Herein, we projected the representations of molecules in testing sets from the above-mentioned M2UMol and M2UMolunpre onto a unit circle by undergoing dimensionality reduction through t-SNE, followed by l2 normalization36. Then, we visualize the density distributions of the representations by using non-parametric Gaussian kernel density estimation (KDE)43 on the circle. Besides, we calculate the average pairwise Gaussian potential41 scores, abbreviated as APGP scores, for quantifying the uniformity of representations. Additionally, we show the density estimations of angles for each point to present the distribution more clearly. As shown in Fig. 4b, for M2UMolunpre, the distributions of the representations consistently exhibit a relatively high degree of clustering, and the angle density estimation curves are sharp with distinct peaks. This may be due to the fact that without knowledge from pre-training, the representations learned by M2UMolunpre only contain limited unimodal information, which may make it difficult to learn unique characteristics when the molecules are similar in structure. As for our pre-trained M2UMol, the distributions of molecular representations become more uniform with lower APGP scores, and the angle density estimation curves are also markedly smoother. This is because after undergoing our designed multi-to-uni knowledge transfer pre-training, the model is able to generate representations of different modalities based solely on the 2D topological graphs of the molecules, which provides more diverse information that enhances learning the unique characteristics of molecules and makes the representations more evenly distributed in the space.
M2UMol uncovers critical structures of molecules related to downstream tasks
In this section, we further conduct the interpretability analysis to investigate M2UMol’s capacity to capture task-induced key structures or functional groups after fine-tuning it on the downstream tasks. To this end, we visualize the attention weights of molecules learned by M2UMol on MMP and DDI prediction tasks.
For MMP, we select testing molecules in the scaffold split scenario from different datasets for visualization analysis, and the results are shown in Fig. 5a. For example, Fluorouracil (PubChem ID: 3385) is a molecule designed for treating tumors, but without the approval of the FDA, and it is labeled as toxic in the Clintox dataset. M2UMol notices the halogen group and two amide groups of Fluorouracil. The former is the key group that causes toxicity of Fluorouracil, and for the latter, a study44 has shown that using Fluorouracil may cause hyperammonemia, which may be related to the two amide groups. Phenylhydrazine (PubChem ID: 7516) is a molecule labeled as toxic in the Toxcast dataset. We can see that the hydrazino is assigned high attention weights by M2UMol, which is proven as the key functional group that can be oxidized by oxyhemoglobin, leading to hemolysis45. The Aromatic heterocycles is labeled as the inhibitors of human b-secretase 1 in BACE datasets, and M2UMol focuses on the imidazole group, which belongs to the aromatic heterocyclic family that shows an inhibitory effect on human b-secretase 1 in a study46. Aspirin (PubChem ID: 2244), a molecule from the BBBP datasets, has the ability to cross the blood-brain barrier. M2UMol focuses on the carboxyl and ester groups, which are acidic and lipophilic functional groups, respectively, and has been proven to help molecules cross the blood-brain barrier47.

We visualize the molecules by RDKit and attach the attention weights learned by M2UMol, which reflect the functional groups' significance to the molecule. Darker colors indicate higher attention weights. a The visualization results of molecules with different properties. b The visualization results of a commonly used analgesic and anti-inflammatory drug, Loxoprofen, and four drugs that interact with it.
For DDI prediction, we choose a commonly used drug for Analgesic and anti-inflammatory in the clinic, namely Loxoprofen, a nonsteroidal anti-inflammatory drug. As shown in Fig. 5b, M2UMol focuses precisely on the carboxyl in the propionic group of Loxoprofen, which is the key active group for the high effectiveness48 and helps inhibit the synthesis of prostaglandins involved in promoting the diuretic effect and antihypertensive effect. Therefore, taking Loxoprofen together with diuretics or antihypertensive medications may potentially decrease their activity49. Here, we randomly choose two diuretic drugs (Furosemide and Etacrynic acid) and two hypotensive drugs (Hydralazine and Practolol) that are predicted by M2UMol to have “decrease the diuretic activities” and “decrease the antihypertensive activities” DDI events with Loxoprofen, respectively, for visualization. As shown in Fig. 5b, for Furosemide, M2UMol focuses on the sulfonyl and amino, which are the key groups resulting in potent diuretic activity50. Besides, M2UMol also focuses on the carboxyl group of Furosemide, which has been proved to be an important active group that has considerable potential for intermolecular interaction51,52. For Etacrynic acid, M2UMol focuses on the halogen group and carboxyl group. The former plays a key role in the inhibition of the Phase II detoxification enzyme53, and the latter is important to the pH suitability and solubility improvement54, which can affect the absorption and effect of drugs. For Hydralazine, the focused hydrazine group is the most reactive portion of the molecule and the part most likely to spontaneously bind to proteins55. For Practolol, acylamino is focused, which was shown to control both the degree of agonism and the cardioselectivity56 which is directly related to the antihypertensive effect, and the focused hydroxyl group can affect the solubility and absorption57, which can affect the effectiveness of the drug. More results about the interpretability analysis can be found in Section 3.5 of the Supplementary Information.
Overall, M2UMol can understand molecular structures from multimodal perspectives, and thus can comprehensively capture key structures or functional groups of molecules, and the visualized results are consistent with human understanding of the structural properties of drug molecules. It confirms that M2UMol has good interpretability and can provide diversified and abundant molecular-related knowledge to assist the drug discovery process.
Discussion
In this study, we presented M2UMol, a multi-to-uni modal knowledge transfer pre-training MRL method, which can effectively learn multimodal knowledge from incomplete multimodal data and is able to generate multimodal representations based on 2D topological graphs, such that it can adapt to various downstream tasks with only 2D modality available. Extensive experiments show that M2UMol can learn high-quality molecular representations and achieve superior performances on various downstream tasks, especially in OOD scenarios. In addition, the results validate that M2UMol can generate reliable multimodal representations from 2D topological graphs and bring about molecular representations containing comprehensive knowledge, which makes it an effective tool aiding in drug discovery.
Benefiting from our well-designed multi-to-uni modal knowledge transfer pre-training framework, M2UMol can learn high-quality molecular representations and achieve superior performance on important molecular tasks, such as molecular property prediction and molecular interaction prediction. In recent years, molecular generation has garnered significant attention due to its potential to accelerate de novo drug design and explore novel chemical space, and has become a critical frontier in computational chemistry and drug discovery. Considering that learning high-quality molecular representations is also the fundamental basis for molecular generation, our future work will focus on extending the superior capabilities of our model in molecular representation learning to molecule generation. We aim to investigate the advantages of multimodal representation learning in molecule generation, and design a multimodal molecule generation model capable of generating novel molecules guided by diverse multimodal inputs, such as the 3D structures of targets’ pockets and textual descriptions about demanded molecular properties.
Methods
Multimodal molecule data construction
For the convenience of the follow-up description and discussion about our pre-training model, we here symbolize the construction of our multimodal molecular data.
2D topological graph (2D)
Let \({{\mathcal{M}}}_{a}\) denote a set of molecules, each of which can be represented as a 2D topological graph with its atoms as nodes and bonds as edges. For a molecule \({m}_{i}\in {{\mathcal{M}}}_{a}\), its corresponding 2D topological graph is denoted as \({{\mathcal{G}}}_{i}\).
3D conformer graph (3D)
Formally, the 3D conformer graph for a molecule contains its original 2D topological graph as well as Cartesian coordinates for all nodes. Let \({{\mathcal{M}}}_{c}\) denote a set of molecules with their 3D conformers available. For a molecule \({m}_{i}\in {{\mathcal{M}}}_{c}\), its corresponding 3D conformer graph is denoted as \({{\mathcal{C}}}_{i}\).
Textual description (text)
For the textual description modality, we extract two categories of drug information from DrugBank: (1) concise summaries containing key properties, mechanisms of action, and usage guidelines; and (2) comprehensive background information covering historical development, discovery processes, and clinical trial outcomes. These texts are concatenated to create a unified textual description \({{\mathcal{T}}}_{i}\) for each molecule \({m}_{i}\in {{\mathcal{M}}}_{t}\), where \({{\mathcal{M}}}_{t}\) is the set of molecules with textual descriptions.
Biochemical feature (Bio)
For the biochemical feature modality, we integrate three types of biochemical features from biological functions for each molecule, i.e., molecules’ drug categories as well as associations with targets and enzymes from DrugBank. Specifically, given a molecule \({m}_{i}\in {{\mathcal{M}}}_{o}\), its biochemical features can be represented as a set of binary vectors \({{\mathcal{O}}}_{i}=\{{{\bf{o}}}_{{i}_{1}},{{\bf{o}}}_{{i}_{2}},{{\bf{o}}}_{{i}_{3}}\}\), where \({{\mathcal{M}}}_{o}\) is the set of molecules with biochemical features and each element value 0 or 1 in \({{\bf{o}}}_{{i}_{1}}\in {\{0,1\}}^{{n}_{1}}\), \({{\bf{o}}}_{{i}_{2}}\in {\{0,1\}}^{{n}_{2}}\) or \({{\bf{o}}}_{{i}_{3}}\in {\{0,1\}}^{{n}_{3}}\) indicates the presence or absence of the drug category/target/enzyme of the molecule. There are 3607 types of drug categories, 4463 types of targets, and 419 types of enzymes, so n1 = 3607, n2 = 4463, n3 = 419.
Note that for each molecule in \({{\mathcal{M}}}_{a}\), its other modalities (3D, Text, and Bio) are not all available (incomplete modalities). Hence, we have \({{\mathcal{M}}}_{c},{{\mathcal{M}}}_{t},{{\mathcal{M}}}_{o}\subset {{\mathcal{M}}}_{a}\).
Multi-to-uni modal knowledge transfer pre-training
In this section, we introduce our multi-to-uni modal knowledge transfer pre-training framework.
Modal encoders
M2UMol employs four independent encoders to process inputs from different modalities. For the 2D encoder fg, GraphGPS58, a transformer-based graph neural network, is utilized to learn molecular 2D embeddings from 2D topological graphs of molecules in \({{\mathcal{M}}}_{a}\). For the 3D encoder fc, we employ ComENet59 for learning molecular 3D embeddings from 3D conformer graphs of molecules in \({{\mathcal{M}}}_{c}\). For the Text encoder ft, we first tokenized the textual descriptions of molecules in \({{\mathcal{M}}}_{t}\) using the pre-trained tokenizer of PubMedBERT60, a large language model pre-trained from scratch using abstracts from PubMed and full-text articles from PubMedCentral. Then the tokens are fed into PubMedBERT, resulting in 768-dimensional vectors from the pooler layer as the Text representations. For the Bio encoder fo, the three types of biochemical features of each molecule in \({{\mathcal{M}}}_{o}\) are first projected into the d-dimensional vectors, and then we take the mean of the vectors as the Bio embedding. Formally, for a molecule \({m}_{i}\in {{\mathcal{M}}}_{a}\), we obtain its 2D embedding by \({{\bf{h}}}_{i}^{g}={f}_{\!g}({{\mathcal{G}}}_{i})\). If the molecule mi also belongs to \({{\mathcal{M}}}_{c}\), \({{\mathcal{M}}}_{t}\) or \({{\mathcal{M}}}_{o}\), we can obtain its 3D, Text or Bio embedding \({{\bf{h}}}^{c}_{i}={f}_{\!c}({{\mathcal{C}}}_{i})\), \({{\bf{h}}}^{t}_{i}={f}_{\!t}({{\mathcal{T}}}_{i})\), or \({{\bf{h}}}^{o}_{i}={f}_{\!o}({{\mathcal{O}}}_{i})\). All these embeddings are d-dimensional vectors. More details about the selection of the encoders and the encoders’ architecture can be found in Section 2.1 of the Supplementary Information.
Modal-specific adapters
The modal-specific adapters are designed to serve as generators capable of generating pseudo representations of other modalities from 2D modality.
Specifically for the molecule mi and its 2D representations \({{\bf{h}}}_{i}^{g}\), modal-specific adapters projects \({{\bf{h}}}_{i}^{g}\) into 3D, Text and Bio modality representation spaces respectively, with projection heads f2D→3D( ⋅ ), f2D→Text( ⋅ ) and f2D→Bio( ⋅ ):
where W1, W2, \({{\bf{W}}}_{3}\,\in {{\mathbb{R}}}^{d\times d}\) are trainable parameter matrices and b1, b2, \({{\bf{b}}}_{3}\,\in {{\mathbb{R}}}^{d}\) denotes trainable bias. \({\widetilde{{\bf{h}}}}_{i}^{c}\in {{\mathbb{R}}}^{d}\), \({\widetilde{{\bf{h}}}}_{i}^{t}\in {{\mathbb{R}}}^{d}\), \({\widetilde{{\bf{h}}}}_{i}^{o}\in {{\mathbb{R}}}^{d}\) denote generated representation of 3D, Text and Bio modalities. More details about the rationale behind the design of the modal-specific adapter can be found in Section 2.2 of the Supplementary Information.
Self-supervised learning tasks
In M2UMol, we designed two self-supervised learning tasks, including the generated-actual multimodal contrastive learning and modality classification.
Generated-actual multimodal contrastive learning
In order to empower the model to generate representations of 3D, Text and Bio modalities which are as similar as possible to actual representations, we align the generated pseudo multimodal representations \({\widetilde{{\bf{h}}}}_{i}^{c},{\widetilde{{\bf{h}}}}_{i}^{t},{\widetilde{{\bf{h}}}}_{i}^{o}\) with actual multimodal representations \({{\bf{h}}}_{i}^{c},{{\bf{h}}}_{i}^{t},{{\bf{h}}}_{i}^{o}\) by contrastive learning (CL).
Let Δ = {c, t, o} denote a set of indices on behalf of the three modalities {3D, Text, Bio}. For every δ ∈ Δ and each molecule \({m}_{i}^{\delta }\in {{\mathcal{M}}}_{\delta }\), we consider its generated/actual representation as the anchor sample, and its actual/generated representation as the positive sample. The generated and actual representations of other molecules in \({{\mathcal{M}}}_{\delta }\) are considered as the negative samples. Then we maximize the mutual information of positive pairs (each positive pair contains an anchor sample and a corresponding positive sample) while minimizing the mutual information of negative pairs (each negative pair contains an anchor sample and a negative sample). We adopt independent CL objectives for each modality, and the total loss is formulated by ref. 61:
where \({\widetilde{{\bf{h}}}}_{i}^{\delta }\) and \({{\bf{h}}}_{i}^{\delta }\) denote the generated representation and actual representation of the molecule \({m}_{i}^{\delta }\) in \({{\mathcal{M}}}_{\delta }\), respectively; \({\rm{E}}({\bf{u}},{\bf{v}})=\exp ({\rm{sim}}({\bf{u}},{\bf{v}})/\tau )\), and here \({\rm{sim}}({\bf{u}},{\bf{v}})={{\bf{u}}}^{\top }{\bf{v}}/\parallel {\bf{u}}\parallel \parallel {\bf{v}}\parallel\) denote the cosine similarity; and τ is the temperature parameter. Note that we contrast the generated and actual representations of each modality independently. Hence, we need each molecule to have at least one modality (3D/Text/Bio) besides 2D, but without the requirement for the presence of all modalities, endowing the model with the ability to train on incomplete-modal molecular data.
Modality classification
Inspired by refs. 62,63, we designed a modality classifier fmcls( ⋅ ) to further enhance the modality specificity and distinguishability contained in the generated multimodal representations.
The modality classifier fmcls( ⋅ ) takes the generated representations \({\widetilde{{\bf{h}}}}_{i}^{c}\), \({\widetilde{{\bf{h}}}}_{i}^{t}\) and \({\widetilde{{\bf{h}}}}_{i}^{o}\) as inputs, and identifies which modality the generated representation belongs to. To optimize the modality classifier fmcls( ⋅ ), we minimize the cross-entropy (CE) loss that is formulated as:
where yi,δ ∈ {0, 1}3 is the one-hot modality label with 1 at the index of modality δ ∈ Δ and 0 elsewhere, and \({\widehat{{\bf{y}}}}_{i,\delta }\in {{\mathbb{R}}}^{3}\) is the predictive values of the modality classifier fmcls( ⋅ ) for \({\widetilde{{\bf{h}}}}_{i}^{\delta }\), i.e., \({\widehat{{\bf{y}}}}_{i,\delta }={f}_{{\rm{mcls}}}({\widetilde{{\bf{h}}}}_{i}^{\delta })\). In the concrete implementation, fmcls( ⋅ ) is a one-layer linear projection followed by the softmax function.
pre-training objective
We optimize the total pre-training loss \({{\mathcal{L}}}_{{\rm{pre-train}}}\) that combines Eq.(2) and Eq.(3) for pre-training our model M2UMol:
where α is a hyperparameter that balances the contribution of the modality classifier.
Fine-tuning with attention-based knowledge fusion
After pre-training, the 2D Modal encoder fg as well as the modal-specific adapter including projection heads f2D→3D, f2D→Text and f2D→Bio in M2UMol are further fine-tuned with the attention-based knowledge fusion strategy on downstream tasks, simulating multimodal fusion with only molecular 2D topological graph inputs.
Attention-based knowledge fusion
For a molecule mi, we first obtain the topological graph and utilize the pre-trained 2D graph encoder fg to learn the 2D representations \({{\bf{h}}}_{i}^{g}\). Then the modal-specific projection heads f2D→3D, f2D→Text and f2D→Bio respectively generate 3D, Text and Bio representations \({\widehat{{\bf{h}}}}_{i}^{c}\), \({\widehat{{\bf{h}}}}_{i}^{t}\) and \({\widehat{{\bf{h}}}}_{i}^{o}\), which are subsequently fused into the final representations hi by a multi-head attention mechanism:
where \({{\bf{h}}}_{i}^{g}\in {{\mathbb{R}}}^{d}\) and \({\bf{P}}=\{{\widetilde{{\bf{h}}}}_{i}^{c},{\widetilde{{\bf{h}}}}_{i}^{t},{\widetilde{{\bf{h}}}}_{i}^{o}\}\in {{\mathbb{R}}}^{3\times d}\); \({{\bf{W}}}^{Q},{{\bf{W}}}^{K},{{\bf{W}}}^{V}\in {{\mathbb{R}}}^{d\times d}\) are trainable parameters and d is the hidden dimension; the projections are parameter matrices \({{\bf{W}}}_{j}^{Q}\in {{\mathbb{R}}}^{d\times {d}_{k}}\), \({{\bf{W}}}_{j}^{K}\in {{\mathbb{R}}}^{d\times {d}_{k}}\), \({{\bf{W}}}_{j}^{V}\in {{\mathbb{R}}}^{d\times {d}_{v}}\) and \({{\bf{W}}}^{O}\in {{\mathbb{R}}}^{d\times d}\), h denotes the number of attention heads and dk = dv = d/h. Finally, hi denotes the final representations of molecular mi, which can be fed into different predictors for various downstream tasks. More details for the fine-tuning on downstream tasks can be found in Section 2.3 of the Supplementary Information.
Experimental setting
The dataset for pre-training
DrugBank64 is a widely utilized and web-enabled database that contains comprehensive molecular information about drugs, providing detailed drug data, including drug targets and drug action information. This database is particularly suitable for constructing multimodal pre-training data. Initially, we downloaded the complete DrugBank database in XML format and extracted 12,227 small molecules with their SMILES, including approved drugs, experimental drugs, withdrawn drugs, and others. Then we obtain their corresponding 2D topological graphs, 3D conformers, textual descriptions, the associations with targets and enzymes, and the drug categories they belong to from the DrugBank database as 2D/3D/Text/Bio modal data. Finally, our dataset contains 11,571 molecules. Out of these molecules, all molecules have their 2D modal data, 9,468 molecules have 3D modal data, 5899 molecules have Text modal data, and 10,581 molecules have Bio modal data. More details about pre-training dataset construction can be found in Section 1.1 of the Supplementary Information.
The datasets for downstream tasks
For molecular property prediction, we adopted 8 types of benchmark datasets from MoleculeNet65 for molecular properties prediction, comprising 678 binary classification tasks. The datasets cover molecular data from a wide range of domains, such as drugs, biology, physics, and chemistry. For DDI prediction, we utilized the DDI data from66, which consists of 191,570 DDIs between 1700 drugs with 86 types of DDI events, and every DDI is associated with a DDI event. For drug-target interaction prediction, we utilize the BindingDB67 and BioSNAP68 dataset, following35. The BindingDB dataset consists of 49,199 DTIs between 14,643 drugs and 2623 target proteins, and the BioSNAP dataset consists of 27,464 DTIs between 4510 drugs and 2181 target proteins. To comprehensively evaluate the performance of M2UMol, we used random split, cold split and scaffold split settings. More details about the downstream datasets and split settings can be found in Section 1.2 of the Supplementary Information.
Baselines
For molecular property prediction, as recent advances in this area are predominantly driven by the pre-trained MRL methods, we utilized two types of pre-training MRL methods as baselines: the first is the multimodal pre-training MRL methods that utilize multimodal data for pre-training (3D-Infomax27, GraphMVP30, MOLEBLEND69, MoleculeSDE32, KV-PLM31, MoleculeSTM128, MoleculeSTM2, MoMu29, MolFM33, and MEMO34); the second is the unimodal pre-training MRL methods that utilize one modality for pre-training (ChemBERTa22, MegaMolBART, Molformer-XL23, EdgePred70, AttrMask20, GPT-GNN19, InfoGraph71, ContextPred20, GraphLoG72, G-Contextal73, G-Motif73, GraphCL25, JOAO26, MolCLR24). For DDI prediction, we mainly choose representative and strong models that are widely adopted in the field as baselines. We summarized four different types of DDI prediction methods which can be applied to cold start and scaffold split settings: 1D fingerprint-based method (STNN-DDI74), 2D topological graph-based methods (SSI-DDI75, GMPNN76, SA-DDI77, MSAN78, DSN-DDI79), and 3D conformer graph-based methods (3DGT-DDI80). For DTI prediction, we compared the performance of M2UMol with five models (SVM81, RF82, DeepConv-DTI83, GraphDTA84, MolTrans85) on DTI prediction following35. More details about baselines can be found in Section 1.3 of the Supplementary Information.
Evaluation metrics
For the molecular property prediction task, we utilize AUROC as the evaluation metric. For the drug-drug interaction prediction task, we use mean accuracy (ACC), macro precision (Macro-P), macro recall (Macro-R), and macro F1 (Macro-F1) as the evaluation metrics. For the drug-target interaction prediction task, we utilize AUROC and AUPRC as the evaluation metrics. For the scaffold split setting, we report the mean and the standard deviation of the metrics of three independent runs with different random seeds. For random and cold start split scenarios, we report the mean and the standard deviation of the metrics of threefold cross-validation. For the multimodal retrieval task, we use Recall@K as the evaluation metric. More details about the evaluation metrics and implementation can be found in Sections 1.4 and 1.5 of the Supplementary Information.
Data availability
The raw data of the pre-training dataset were sourced from the public dataset DrugBank64, available at https://go.drugbank.com/releases/latest. The processed molecular property prediction datasets are available at: http://snap.stanford.edu/gnn-pretrain/data/chem_dataset.zip. The BindingDB67 dataset is available at: https://www.bindingdb.org/bind/index.jspand the BioSNAP68 source is available at: https://github.com/kexinhuang12345/MolTrans/tree/master/dataset/BIOSNAP/full_data. The drug-drug interaction dataset is available at: https://github.com/Zhankun-Xiong/MRCGNN/tree/main/Ryu’s%20dataset. The data generated in this study have been publicly deposited to Hugging Face under https://doi.org/10.57967/hf/7153, and the data version used for this publication is available86. Source data are provided with this paper87. Source data are provided with this paper.
Code availability
The codes, pre-trained model, and the developed package are freely available at https://doi.org/10.5281/zenodo.17798744. The version used for this publication is available88.
References
Walters, W. P. & Barzilay, R. Applications of deep learning in molecule generation and molecular property prediction. Acc. Chem. Res. 54, 263–270 (2020).
Shen, J. & Nicolaou, C. A. Molecular property prediction: Recent trends in the era of artificial intelligence. Drug Discov. Today. Technol. 32, 29–36 (2019).
Ryu, J. Y., Kim, H. U. & Lee, S. Y. Deep learning improves prediction of drug-drug and drug-food interactions. Proc. Natl. Acad. Sci. USA 115, E4304–E4311 (2018).
Lin, X., Quan, Z., Wang, Z.-J., Ma, T. & Zeng, X. Kgnn: Knowledge graph neural network for drug-drug interaction prediction. In IJCAI, 380, 2739–2745 (2020).
Qiu, Y., Zhang, Y., Deng, Y., Liu, S. & Zhang, W. A comprehensive review of computational methods for drug-drug interaction detection. IEEE/ACM Trans. Comput. Biol. Bioinforma. 19, 1968–1985 (2021).
Bagherian, M. et al. Machine learning approaches and databases for prediction of drug–target interaction: a survey paper. Brief. Bioinforma. 22, 247–269 (2021).
Chen, X. et al. Drug–target interaction prediction: databases, web servers and computational models. Brief. Bioinforma. 17, 696–712 (2016).
Todeschini, R. & Consonni, V. Handbook of molecular descriptors (John Wiley & Sons, 2008).
Skoraczyński, G. et al. Predicting the outcomes of organic reactions via machine learning: are current descriptors sufficient? Sci. Rep. 7, 3582 (2017).
Lu, C. et al. Molecular property prediction: a multilevel quantum interactions modeling perspective. In Proceedings of the Thirty-Third AAAI Conference on Artificial Intelligence and Thirty-First Innovative Applications of Artificial Intelligence Conference and Ninth AAAI Symposium on Educational Advances in Artificial Intelligence, 1052–1060 (2019).
Wang, Z. et al. Advanced graph and sequence neural networks for molecular property prediction and drug discovery. Bioinformatics 38, 2579–2586 (2022).
Bjerrum, E. J. Smiles enumeration as data augmentation for neural network modeling of molecules. arXiv preprint arXiv:1703.07076 (2017).
Quan, Z. et al. A system for learning atoms based on long short-term memory recurrent neural networks. In 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 728–733 (IEEE, 2018).
Weininger, D. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In International conference on machine learning, 1263–1272 (PMLR, 2017).
Xiong, Z. et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 63, 8749–8760 (2019).
Schütt, K. et al. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions. Advances in neural information processing systems, 30, (2017).
Gasteiger, J., Groß, J. & Günnemann, S. Directional message passing for molecular graphs. InInternational Conference on Learning Representations (2020).
Hu, Z., Dong, Y., Wang, K., Chang, K.-W. & Sun, Y. Gpt-gnn: Generative pre-training of graph neural networks. In Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 1857–1867 (2020).
Hu, W. et al. Strategies for pre-training graph neural networks. In International Conference on Learning Representations (2020).
Zaidi, S. et al. Pre-training via denoising for molecular property prediction. In International Conference on Learning Representations (2023).
Chithrananda, S., Grand, G. & Ramsundar, B. Chemberta: large-scale self-supervised pretraining for molecular property prediction. arXiv preprint arXiv:2010.09885 (2020).
Ross, J. et al. Large-scale chemical language representations capture molecular structure and properties. Nat. Mach. Intell. 4, 1256–1264 (2022).
Wang, Y., Wang, J., Cao, Z. & Barati Farimani, A. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 4, 279–287 (2022).
You, Y. et al. Graph contrastive learning with augmentations. Adv. neural Inf. Process. Syst. 33, 5812–5823 (2020).
You, Y., Chen, T., Shen, Y. & Wang, Z. Graph contrastive learning automated. In International Conference on Machine Learning, 12121–12132 (PMLR, 2021).
Stärk, H. et al. 3d infomax improves gnns for molecular property prediction. In International Conference on Machine Learning, 20479–20502 (PMLR, 2022).
Liu, S. et al. Multi-modal molecule structure–text model for text-based retrieval and editing. Nat. Mach. Intell. 5, 1447–1457 (2023).
Su, B. et al. A molecular multimodal foundation model associating molecule graphs with natural language. arXiv preprint arXiv:2209.05481 (2022).
Liu, S. et al. Pre-training molecular graph representation with 3d geometry. In International Conference on Learning Representations (2021).
Zeng, Z., Yao, Y., Liu, Z. & Sun, M. A deep-learning system bridging molecule structure and biomedical text with comprehension comparable to human professionals. Nat. Commun. 13, 862 (2022).
Liu, S., Du, W., Ma, Z.-M., Guo, H. & Tang, J. A group symmetric stochastic differential equation model for molecule multi-modal pretraining. In International Conference on Machine Learning, 21497–21526 (PMLR, 2023).
Luo, Y., Yang, K., Hong, M., Liu, X. & Nie, Z. Molfm: A multimodal molecular foundation model. arXiv preprint arXiv:2307.09484 (2023).
Zhu, Y. et al. Featurizations matter: a multiview contrastive learning approach to molecular pretraining. In ICML 2022 2nd AI for Science Workshop (2022).
Bai, P., Miljković, F., John, B. & Lu, H. Interpretable bilinear attention network with domain adaptation improves drug–target prediction. Nat. Mach. Intell. 5, 126–136 (2023).
Van der Maaten, L. & Hinton, G. Visualizing data using t-sne. J. Mach. Learning Res.9, 2579–2605 (2008).
Long, Q., Wang, M. & Li, L. Generative imagination elevates machine translation. In Proceedings of the 2021 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, 5738–5748 (2021).
Xia, J. et al. Mole-bert: Rethinking pre-training graph neural networks for molecules. In The Eleventh International Conference on Learning Representations (2022).
Zang, X., Zhao, X. & Tang, B. Hierarchical molecular graph self-supervised learning for property prediction. Commun. Chem. 6, 34 (2023).
McCune, D. F., Gaivin, R. J., Rorabaugh, B. R. & Perez, D. M. Bulk is a determinant of oxymetazoline affinity for the α1a-adrenergic receptor. Receptors Channels 10, 109–116 (2004).
Wang, T. & Isola, P. Understanding contrastive representation learning through alignment and uniformity on the hypersphere. In International Conference on Machine Learning, 9929–9939 (PMLR, 2020).
Fang, Y. et al. Knowledge graph-enhanced molecular contrastive learning with functional prompt. Nat. Mach. Intel. 5, 542–553 (2023).
BOTEV, Z., GROTOWSKI, J. & KROESE, D. Kernel density estimation via diffusion. Ann. Stat. 38, 2916–2957 (2010).
Chen, M. et al. Dilirank: the largest reference drug list ranked by the risk for developing drug-induced liver injury in humans. Drug Discov. Today 21, 648–653 (2016).
Goldberg, B. & Stern, A. The mechanism of oxidative hemolysis produced by phenylhydrazine. Mol. Pharmacol. 13, 832–839 (1977).
García Marín, I. D. et al. New compounds from heterocyclic amines scaffold with multitarget inhibitory activity on aβ aggregation, ache, and bace1 in the alzheimer disease. Plos one 17, e0269129 (2022).
Di, L. & Kerns, E. H. Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization (Academic Press, 2015).
Black, W. et al. From indomethacin to a selective cox-2 inhibitor: development of indolalkanoic acids as potent and selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett. 6, 725–730 (1996).
Bavry, A. A. et al. Harmful effects of nsaids among patients with hypertension and coronary artery disease. Am. J. Med. 124, 614–620 (2011).
Banik, M., Gopi, S. P., Ganguly, S. & Desiraju, G. R. Cocrystal and salt forms of furosemide: solubility and diffusion variations. Cryst. Growth Des. 16, 5418–5428 (2016).
Goud, N. R. et al. Novel furosemide cocrystals and selection of high solubility drug forms. J. Pharm. Sci. 101, 664–680 (2012).
Harriss, B. I., Vella-Zarb, L., Wilson, C. & Evans, I. R. Furosemide cocrystals: Structures, hydrogen bonding, and implications for properties. Cryst. growth Des. 14, 783–791 (2014).
Karaytuğ, M. O. et al. Piperazine derivatives with potent drug moiety as efficient acetylcholinesterase, butyrylcholinesterase, and glutathione s-transferase inhibitors. J. Biochem. Mol. Toxicol. 37, e23259 (2023).
Deshler, L. & Zuman, P. Polarographic reduction of aldehydes and ketones: Part xviii. ethacrynic acid. Analytica Chim. Acta 73, 337–354 (1974).
Litwin, A., Adams, L. E., Zimmer, H. & Hess, E. V. Immunologic effects of hydralazine in hypertensive patients. Arthritis Rheumatism J. Am. Coll. Rheumatol. 24, 1074–1077 (1981).
Main, B. G. & Tucker, H. 3 recent advances in β-adrenergic blocking agents. Prog. Med. Chem. 22, 121–164 (1985).
Shrivastav, P. S., Buha, S. M. & Sanyal, M. Detection and quantitation of β-blockers in plasma and urine. Bioanalysis 2, 263–276 (2010).
Rampášek, L. et al. Recipe for a general, powerful, scalable graph transformer. Adv. Neural Inf. Process. Syst. 35, 14501–14515 (2022).
Wang, L., Liu, Y., Lin, Y., Liu, H. & Ji, S. Comenet: Towards complete and efficient message passing for 3d molecular graphs. In Koyejo, S. et al. (eds.) Advances in Neural Information Processing Systems, vol. 35, 650–664 (Curran Associates, Inc., 2022).
Gu, Y. et al. Domain-specific language model pretraining for biomedical natural language processing. ACM Trans. Comput. Healthc. (HEALTH) 3, 1–23 (2021).
Chen, T., Kornblith, S., Norouzi, M. & Hinton, G. A simple framework for contrastive learning of visual representations. In International conference on machine learning, 1597–1607 (PMLR, 2020).
Ganin, Y. & Lempitsky, V. Unsupervised domain adaptation by backpropagation. In International conference on machine learning, 1180–1189 (PMLR, 2015).
Wang, H. et al. Multi-modal learning with missing modality via shared-specific feature modelling. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, 15878–15887 (2023).
Wishart, D. S. et al. Drugbank 5.0: a major update to the drugbank database for 2018. Nucleic acids Res. 46, D1074–D1082 (2018).
Wu, Z. et al. Moleculenet: A benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018).
Xiong, Z. et al. Multi-relational contrastive learning graph neural network for drug-drug interaction event prediction. In Proceedings of the Thirty-Seventh AAAI Conference on Artificial Intelligence. 5339–5347 (2023).
Gilson, M. K. et al. Bindingdb in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic acids Res. 44, D1045–D1053 (2016).
Zitnik, M., Sosic, R. & Leskovec, J. Biosnap datasets: Stanford biomedical network dataset collection. http://snap.stanford.edu/biodata Cited by 5 (2018).
Yu, Q. et al. Multimodal molecular pretraining via modality blending. In The Twelfth International Conference on Learning Representations.
Hamilton, W., Ying, Z. & Leskovec, J. Inductive representation learning on large graphs. Advances in neural information processing systems 30, (2017).
Sun, F.-Y., Hoffmann, J., Verma, V. & Tang, J. Infograph: Unsupervised and semi-supervised graph-level representation learning via mutual information maximization. In International Conference on Learning Representations (2020).
Xu, M., Wang, H., Ni, B., Guo, H. & Tang, J. Self-supervised graph-level representation learning with local and global structure. In International Conference on Machine Learning, 11548–11558 (PMLR, 2021).
Rong, Y. et al. Self-supervised graph transformer on large-scale molecular data. Adv. Neural Inf. Process. Syst. 33, 12559–12571 (2020).
Yu, H., Zhao, S. & Shi, J. STNN-DDI: A substructure-aware tensor neural network to predict drug-drug interactions. Brief. Bioinforma. 23, bbac209 (2022).
Nyamabo, A. K., Yu, H. & Shi, J.-Y. SSI-DDI: Substructure-substructure interactions for drug-drug interaction prediction. Brief. Bioinf. https://doi.org/10.1093/bib/bbab133 (2021).
Nyamabo, A. K., Yu, H., Liu, Z. & Shi, J.-Y. Drug-drug interaction prediction with learnable size-adaptive molecular substructures. Brief. Bioinf. https://doi.org/10.1093/bib/bbab441 (2021).
Yang, Z., Zhong, W., Lv, Q. & Chen, C. Y.-C. Learning size-adaptive molecular substructures for explainable drug-drug interaction prediction by substructure-aware graph neural network. Chem. Sci. 13, 8693–8703 (2022).
Zhu, X., Shen, Y. & Lu, W. Molecular Substructure-Aware Network for Drug-Drug Interaction Prediction. In Proceedings of the 31st ACM International Conference on Information & Knowledge Management, 4757–4761 (2022).
Li, Z. et al. DSN-DDI: an accurate and generalized framework for drug-drug interaction prediction by dual-view representation learning. Briefings Bioinformatics24. https://doi.org/10.1093/bib/bbac597 (2023).
He, H., Chen, G. & Yu-Chian Chen, C. 3DGT-DDI: 3D graph and text based neural network for drug-drug interaction prediction. Briefings in Bioinformatics 23, https://doi.org/10.1093/bib/bbac134, https://academic.oup.com/bib/article-pdf/23/3/bbac134/43745041/bbac134.pdf (2022).
Cortes, C. & Vapnik, V. Support-vector networks. Mach. Learn. 20, 273–297 (1995).
Ho, T. K. Random decision forests. In Proceedings of 3rd international conference on document analysis and recognition, vol. 1, 278–282 (IEEE, 1995).
Lee, I., Keum, J. & Nam, H. Deepconv-dti: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 15, e1007129 (2019).
Nguyen, T. et al. Graphdta: Predicting drug–target binding affinity with graph neural networks. Bioinformatics 37, 1140–1147 (2021).
Huang, K., Xiao, C., Glass, L. M. & Sun, J. Moltrans: molecular interaction transformer for drug–target interaction prediction. Bioinformatics 37, 830–836 (2021).
Xiong, Z. Data of multi-to-uni modal knowledge transfer pre-training for molecular representation learning: V1.0, https://doi.org/10.57967/hf/7153 (2025).
Xiong, Z. Source data files for paper “multi-to-uni modal knowledge transfer pre-training for molecular representation learning. https://doi.org/10.5281/zenodo.18219572 (2026).
Xiong, Z. Zhankun-xiong/m2umol: M2umol, https://doi.org/10.5281/zenodo.17798744 (2025).
Irwin, R., Dimitriadis, S., He, J. & Bjerrum, E. J. Chemformer: A pre-trained transformer for computational chemistry. Mach. Learn. Sci. Technol. 3, 015022 (2022).
Yang, M., Chen, T., Liu, Y.-X. & Huang, L. Visualizing set relationships: Evenn’s comprehensive approach to venn diagrams. Imeta 3, e184 (2024).
Acknowledgements
W.Z. is supported by the National Natural Science Foundation of China (62372204, 62072206), National Administration of Traditional Chinese Medicine Science and Technology Project (No. GZY-KJS-2025-003), Huazhong Agricultural University Scientific & Technological Self-innovation Foundation and Fundamental Research Funds for the Central Universities (2662024SZ006). S.L. is supported by the National Natural Science Foundation of China (62472191). P.Z. is not funded by any of the funders.
Author information
Authors and Affiliations
Contributions
Z.X., Z.W., and F.H. contributed equally. Z.X. conceived the research project. Z.X. developed the primary method and code. Z.W. analyzed the baselines in the paper. M.Q., S.F., and L.Y. assisted in analyzing the effectiveness and interpretability of the method. Z.X., Z.W., F.H., P.Z., and W.Z. wrote the paper. All authors, including X.Z. and S.L., read and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Prayag Tiwari, and the other anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, Z., Wang, Z., Huang, F. et al. Multi-to-uni modal knowledge transfer pre-training for molecular representation learning. Nat Commun 17, 3797 (2026). https://doi.org/10.1038/s41467-026-69302-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69302-6
