
Abstract
Icosahedral carboranes, C2B10H12, have long been considered to be aromatic but the extent of conjugation between these clusters and their substituents is still being debated. m- and p-Carboranes are compared with m- and p-phenylenes as conjugated bridges in optical functional chromophores with a donor and an acceptor as substituents here. The absorption and fluorescence data for both carboranes from experimental techniques (including femtosecond transient absorption, time-resolved fluorescence and broadband fluorescence upconversion) show that the absorption and emission processes involve strong intramolecular charge transfer between the donor and acceptor substituents via the carborane cluster. From quantum chemical calculations on these carborane systems, the charge transfer process depends on the relative torsional angles of the donor and acceptor groups where an overlap between the two frontier orbitals exists in the bridging carborane cluster.
Similar content being viewed by others


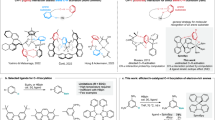
Introduction
Carborane derivatives such as the three closo-C2B10H12 isomers (Fig. 1) and their derivatives possess unique molecular and electronic structures1, which have been exploited in various applications such as in uranyl capture2,3, in medical chemistry4,5,6,7,8,9,10,11,12,13,14, and as luminescent molecules15,16,17,18,19,20,21,22,23,24,25. The skeleton of the carborane cluster is formed by 3c-2e bonds and may be considered as a three-dimensional (3D) aromatic system26. Logically, the cage may act as a conjugated bridge with electron delocalization and charge transfer as found in a phenylene bridge within two-dimensional (2D) π-systems in optoelectronic functional chromophores. However, in o-carboranes with aromatic substituents at the cluster carbon atoms, the overlap between the frontier molecular orbitals (FMOs) of the planar ring and the cage is found to be negligible thus the carborane is considered as a spacer here27,28,29,30,31,32,33. Similarly, m- and p-carboranes behave as spacers due to a lack of FMO overlap according to studies on carborane-bridged luminescent molecules21,34,35,36,37,38,39 and mixed-valence systems40,41,42,43,44.

a Three isomers of closo-C2B10H12 (carborane); b Structures of DA-pCarb, DA-mCarb, DA-pBenz, and DA-mBenz with distances between nitrogen and boron atoms at 10.6, 9.2, 10.2 and 9.0 Å, respectively.
While 2D aromatic bridges are effective in intramolecular charge transfer (ICT) processes, experimental studies of charge transfer processes in 3D carborane bridges remain unexplored. Wade and co-workers revealed that the absorption spectra of donor-acceptor compounds containing phenylene-p-carborane-phenylene bridges are red-shifted. They disclosed that the interaction between carborane and phenylene is antibonding in the HOMO but in-phase in the LUMO45,46. A strong electron acceptor at a carbon atom in a carborane may unlock the potential of carboranes as effective conjugated bridges. This idea led us to design donor-acceptor luminophores with p- and m-carborane bridges, DA-pCarb and DA-mCarb, where the donor is a triarylamine substituent and the acceptor is a dimesitylboron (BMes2) substituent (Fig. 1). In the analogous o-carborane system, the sterics of the bulky substituents present would affect the electronic processes therefore this was not examined here. The phenylene-bridged analogues, DA-pBenz and DA-mBenz, were also synthesized for comparison here. The empty pz orbital at boron makes the BMes2 group a strong π-acceptor and the mesityl groups provide good stability by steric shielding47,48,49,50,51,52,53,54,55,56. Triarylamine is used as the electron donor due to its strong π-electron-donating ability and the low reorganization energy of the amine group supports rapid charge transfer processes51,57,58. The synthesis of all compounds are summarized in the SI (Supplementary Fig. 1–4). Our results demonstrate optically induced intramolecular charge transfer (ICT) through carboranes and confirm the conjugation between the cage and the π-system.
Results
Electrochemistry
To evaluate the donor and acceptor properties of the substituents attached to either carborane or phenylene bridge, cyclic voltammetry for all compounds were measured in THF for reduction and DCM for oxidation (Supplementary Fig. 5, 6). Similar reversible reduction and oxidation potentials against ferrocene/ferrocenium were found for all donor-acceptor systems (dyads) and are listed in Table 1 (see also Supplementary Fig. 6).
The oxidation potentials show that the carborane withdraws electron density from the triarylamine, thus the oxidation potential is increased by 120 mV in DA-pCarb and by 150 mV in DA-mCarb compared to tris(p-tolyl)amine (Eox = +0.34 in DCM)59. In contrast, the phenylene-bridged compounds retain similar oxidation potentials to tris(p-tolyl)amine. On the other hand, the p-carborane substitution leads to a more negative reduction potential than trimesitylborane (Mes3B, Ered = -2.43 V) by 60 mV, while the m-phenylene substitution is more negative than the m-carborane substitution. The band gaps based on the potentials are 22600 cm−1 for DA-pBenz, 23700 cm−1 for DA-pCarb, 23400 cm−1 for DA-mBenz, and 23400 cm−1 for DA-mCarb (Supplementary Fig. 5). These values show that the electronic communication between the donor and the acceptor is stronger in the para-phenylene dyad than in the meta-phenylene analog. However, both carborane dyads display a similar band gap to the meta-phenylene dyad, indicating a similar degree of donor-acceptor interactions in these compounds.
Absorption and fluorescence spectra
Evidence of the donor-acceptor behavior of the dyads was provided by absorption and emission spectra in solvents of different polarities (Fig. 2, Supplementary Fig. 7–13, Table 2). In hexane, DA-pCarb has a broad absorption band at 300 nm with a shoulder at 341 nm (Fig. 2a). Deconvolution analysis of the bands gave two Gaussian bands at around 29200 cm−1 and 33000 cm−1 (Supplementary Fig. 8–11), respectively. While the intense band at 33000 cm−1 of DA-pCarb corresponds to a superposition of the localized (LE) transitions of the triarylamine and the BMes2 group60,61, the weaker one at 29200 cm−1 is assigned the intramolecular charge transfer (ICT) between the amine and BMes2 moieties through the p-carborane cage. The squared transition dipole moment ( = dipole strength) of the ICT transition in hexane for DA-pCarb determined by integrating the ICT band in the absorption spectra is 14.7 D2 (see Table 2)62,63,64, which is about half that of the phenylene bridged DA-pBenz (31.8 D2) where the ICT transition is visible as a separated absorption band at 26,000 cm−1 in hexane (Supplementary Fig. 9). In both compounds, the observed ICT bands in the absorption spectra are little changed when going to a more polar solvent (Supplementary Fig. 12) indicating a small ground state dipole moment46. The 00-energy of the ICT band was estimated by the intersection of a tangent at the low-energy flank with the abscissa where the values of 27,400 cm−1 for DA-pCarb and 23,900 cm−1 for DA-pBenz were obtained. The absorption spectrum for DA-mCarb has one asymmetric broad band (Fig. 2a) where deconvolution analysis showed a strong LE band (32,700 cm−1) and a very weak ICT band (29,200 cm−1) (Supplementary Fig. 10) implying that the ICT transition in DA-mCarb is only weakly allowed. The deconvolution of the absorption spectrum in DA-mBenz also resulted in two intense bands, suggesting that the charge transfer through the meta-position of the phenylene ring is stronger than in the carborane analogue. However, we stress that the deconvolution of the absorption bands of the carborane and phenylene bridged meta-dyads is to some extent arbitrary. The similar ICT transition energies of DA-mBenz and the two carborane dyads are in agreement with similar band gaps measured by cyclic voltammetry for these compounds. The corresponding energies measured for DA-pBenz are significantly smaller.

Absorption (a) and normalized emission (b) spectra of DA-pCarb (excited at 337 nm) and DA-mCarb (excited at 310 nm) in hexane (solid line) and THF (dashed line) at room temperature.
Because the band position and the 00-energy are difficult to estimate from the absorption spectra, we also measured the emission spectra of the four dyads in solvents of different polarities. The emission spectra of all four dyads are strongly solvatochromic (Supplementary Fig. 13). For DA-pCarb and DA-mCarb (Fig. 2b and Supplementary Fig. 13), the fluorescence spectra are broad and featureless in polar solvents, while the bands in the spectra are much sharper in hexane solutions. The Stokes shifts change from 2400 and 2900 cm−1 in hexane to 15,100 and 13,700 cm−1 in acetonitrile (Fig. 3, Supplementary Fig. 13), respectively, indicating the relaxed S1 state has a larger dipole moment than the ground state. Determining the 00-energies (Supplementary Tab. 1) using a tangent to the high energy flank gives similar values of 28,100 cm−1 in hexane for DA-pCarb and DA-mCarb, and 23,000 cm−1 and 22,300 cm−1 in THF for DA-pCarb and DA-mCarb respectively. These values indicate the formation of a relaxed charge transfer state in polar solvents. A fit to the emission spectra (Supplementary Fig. 14) of a large series of solvents with varying polarity yielded a more accurate ΔG00 energy value (the difference in the free energy between the diabatic ground state and the excited state) as well as other reorganization parameters showing that both ΔG00 and the solvent reorganization energy λo vary linearly with solvent polarity f(ε)-0.5 f(n2)65.

Solvents used are from left to right: hexane, toluene, dibutyl ether, THF, DCM, and MeCN, corresponding to the squares/triangles in the picture.
The solvatochromic effects were further investigated with Lippert-Mataga plots66,67,68 (Fig. 3, Supplementary Tab. 2–5), which describe the difference of Stokes shifts (∆ʋ̃) versus orientation polarizability (∆f = f(ε)-f(n2)) in solvents of different polarities. The well-fitted linear correlation together with the large slopes confirms the ICT nature of the emissive S1 state of both carboranes. The slope of DA-pCarb is about twice that of DA-pBenz, indicating a much stronger charge transfer in the former. The dipole moment changes (Δμ) between the excited state and the ground state can be estimated using the effective radius of the spherical Onsager cavity of 7.07 Å and is about 44.9 D for DA-pCarb. Taking the dipole moment in the ground state (μg) to be -1.06 D from DFT calculations (vide infra), we estimate the dipole moment of the relaxed S1 state (μe) to be at ca. 44 D, which corresponds to 86% charge-separation referring to the distance between nitrogen and the three-coordinate boron atom in the excited state. These values are similar to DA-mCarb (μg = 4.36 D, Δμ = 30.6 D) but almost twice that of DA-pBenz (μg = 2.31 D, μe = 18.4 D, 38% charge separation) and DA-mBenz (μg = 1.29 D, Δμ = 14.86 D).
The fluorescence quantum yields (Φf) of the carborane-bridged compounds are strongly dependent on the polarity of the solvent, while the Φf values of DA-pBenz and DA-mBenz are not. The fluorescence quantum yield of DA-pCarb in THF is 64%, which is higher than the meta-carborane (35%). However, the quantum yield of DA-mCarb (5%) is similar to that of DA-pCarb (4%) in hexane. This observation is quite unusual for fluorescence from ICT states. In most donor-acceptor compounds, the ICT state is stabilized by polar solvents and the charges become more localized at the respective moieties. The latter usually leads to a smaller dipole strengths and thus a slow radiative decay rate, while the smaller vertical S1 ← S0 gap in more polar solvents leads to faster nonradiative deactivation, and consequentially to a lower quantum yield69,70. The trend of quantum yield vs. ΔG00 is given in the Supplementary Fig. 15 for both carboranes.
To obtain more insight into the emission dynamics, time-resolved fluorescence measurements were performed by time-correlated single photon counting (TCSPC) (Supplementary Fig. 16). The fluorescence lifetime (τ) of carborane-bridged compounds in hexane are below the time resolution of our TCSPC setup (IRF = 0.9 ns). Indeed, transient absorption measurements with fs time resolution (vide infra) revealed the lifetime of the S1 state in hexane to be only 0.18 ns (DA-pCarb) and 0.76 ns (DA-mCarb) which also explains the low fluorescence quantum yields. However, the fluorescence lifetimes of DA-pCarb (Table 2) in THF (24.2 ns) and DCM (20.4 ns) are two orders of magnitude longer than that in hexane, while lifetimes of DA-pBenz are little affected by solvent polarity (see Table 2). The radiative decay rate constant (kr) of DA-pCarb in THF (2.6 ×107 s−1) is an order of magnitude smaller than that in hexane (2.2 ×108 s−1), while kr of DA-pBenz in hexane is very similar to that in THF. The fast nonradiative decay rate (5.3 ×109 s−1) of DA-pCarb in hexane, which is 400 times higher than that in THF, is responsible for its low quantum yield. The squared transition dipole moment \({\mu }_{{{{{{\rm{fl}}}}}}}^{2}\) ( = dipole strength) of DA-pCarb in hexane (17.5 D2) was determined from the radiative rate constant by the Strickler-Berg equation62 and is slightly larger than that of the absorption. On the other hand, \({\mu }_{{{{{{\rm{fl}}}}}}}^{2}\,\) of DA-pCarb in THF is about one fourth of \({\mu }_{{{{{{\rm{abs}}}}}}}^{2}\) indicating weaker orbital overlap between donor and acceptor moieties in the more polar solvent. In contrast, for DA-pBenz in both solvents \({\mu }_{{{{{{\rm{fl}}}}}}}^{2}\) is larger than \({\mu }_{{{{{{\rm{abs}}}}}}}^{2}\) (Table 2)62,63,64,71.
The comparison of dipole strengths of absorption and emission for DA-pCarb indicates a more local excitation in the non-polar solvent but a CT character in the polar solvent where reduced orbital overlap leads to a smaller dipole strength for the emission. The increasing fluorescence quantum yield in the more polar solvent is, at first sight, contradictory with common observations and the Engelman-Jortner72 gap rule where one would expect a higher nonradiative rate constant for the stabilized CT state in a more polar solvent. Closer investigation of the nonradiative rate constant in an extended series of solvents (Supplementary Tab. 6, Supplementary Fig. 15) indeed manifests the trend that logknr linearly increases with growing ΔG00 of the CT state. This trend which contradicts the Marcus inverted region behavior and the gap rule, is caused by an increasing degree of local excited state admixture and decreasing CT character in the emitting S1 state. While in nonpolar solvents the CT character is less pronounced, it is dominating in more polar solvents but never reaches a full charge separation (vide supra). Indeed, Gould and Farid73 found such a behavior in a series of donor-acceptor exciplexes if the amount of charge transfer is less than ca. 80%.
The room- and low-temperature emissions of DA-mCarb and DA-pCarb in methylcyclohexane were also measured (Supplementary Fig. 18–20). At 77 K, the phosphorescent bands around 450 nm in both compounds with lifetimes of ca. 600 ms are observed along with the fluorescence bands largely unchanged from the fluorescence bands observed at room temperature. The emission spectra and lifetimes of all four compounds in PMMA and solid-state were also determined (Supplementary Fig. 21–23). These photophysical observations are typical of compounds with triphenylamine and BMes2 substituents74.
Transient Absorption Spectroscopy
To gain more insight into the excited state dynamics of both donor-acceptor carborane compounds, femtosecond pump-probe transient absorption spectroscopy (Fig. 4 and Supplementary Fig. 24) measurements in both hexane and THF were performed. The transient map was globally analyzed by a kinetic model involving a sequence of exponential functions. The derived evolution-associated difference spectra (EADS) revealed different behaviors of DA-pCarb, DA-pBenz, DA-mCarb, and DA-mBenz in the two solvents. For DA-pCarb in hexane, the first EADS with τ = 10 ps shows two excited state absorption (ESA) bands (black line, Fig. 4a), one between 350-420 nm and the other one between 600-720 nm, both of which are typical absorption bands of triarylamine radical cations and arylBMes2 radical anions60,75,76. This observation confirms the ICT character of the primarily populated S1 state between the amino group and the BMes2 moiety, and thus, an optically induced charge transfer where the ICT state is directly populated by absorption of light. There is a negative band at around 370 nm, which corresponds to the stimulated emission (SE) from this ICT state of DA-pCarb. Vibrational cooling/solvation was accomplished within 10 ps, giving the relaxed emissive ICT (= S1) state with a lifetime of 0.18 ns (red line, Fig. 4a). The ESA spectrum shows almost no changes during this relaxation process, indicating the solvation effects in hexane are weak as expected for such a nonpolar solvent. In THF, the ESA signals are very similar to those in hexane, suggesting an ICT character of DA-pCarb in both solvents. Vibrational cooling with τ = 2.2 ps in THF is accompanied by a small hypsochromic shift of the ESA band of the first EADS at 680 nm, confirming the strong solvation effect of the CT state in polar solvents57. The cold S1 state has a lifetime of 20.5 ns, which agrees with the fluorescence lifetime of DA-pCarb in THF. Even more significant is the fading of the SE around 410–510 nm. However, because of the strong overlap of ESA and SE, it is difficult to extract specific parameters for the dynamic Stokes shift. Thus, the solvation process in THF was also investigated by broadband fluorescence upconversion spectroscopy (FLUPS) measurements, which show that the fluorescence shifts spectrally from 490 nm to 530 nm with time which is finished within 10 ps (Fig. 4f). The shortest component of the global deconvolution (Supplementary Fig. 24) possesses a time constant of 0.1 ps which is in good agreement with the shortest solvent correlation time of THF77. For DA-mCarb in hexane, the EADS only shows one ESA signal of triarylamine radical cation, and vibrational cooling with τ = 1.17 ps giving a relaxed emissive state with a lifetime of 0.76 ns. In THF, vibrational cooling was finished within 270 fs, then the signal of anionic BMes2 radical moiety was detected which is also accompanied by a hypsochromic shift. The cold S1 state has a lifetime of 31.2 ns, indicating a slower radiative decay of the meta-carborane derivative.

EADS and lifetimes from a global deconvolution of the transient absorption spectra of DA-pCarb in hexane (a) and THF (b), DA-mCarb in hexane (c) and THF (d), at room temperature excited at 340 nm; FLUPS emission map of DA-pCarb in THF excited at 340 nm (e) and fluorescence spectra (f) at indicated delay times.
For DA-pBenz in hexane, the EADS also shows two prominent ESA signals (ca. 480 nm and 610 nm), ground state bleaching (GSB) at 380 nm and SE at 430 nm. While the ESA and GSB signals are invariant with time, the SE signal shifts weakly with τ = 2.6 ps (Supplementary Fig. 24). In THF, the first EADS with τ = 250 fs is very similar to the one in hexane. A series of four EADS follows, which are not associated with different states but serve to model a continuous vibrational cooling/solvent relaxation process expressed by a dynamic Stokes shift. In that way the two ESA signals shift from 480/620 nm to 450/570 nm and the SE from 430 nm to 510 nm. The latter is in very good agreement with the steady-state fluorescence maximum in THF, and the former with the one in hexane. The longest lifetime (τ = 3.4 ns) is again in agreement with the fluorescence lifetime from the TCSPC measurement. Thus, in THF, the primarily populated state has the same characteristics as the one in hexane but unlike that in hexane, it displays a continuous combined molecular/solvent relaxation. The latter is essentially complete within ca. 3 ps. The two principal time constants of τ = 0.25 and 0.73 ps are in very good agreement with the solvent relaxation times of THF77.
Density functional theory (DFT) studies
DFT calculations at B3LYP/6-31 G* in the gas phase were carried out to understand the interaction between the carborane and its substituents (Fig. 5). We used CAM-B3LYP/6-31 G* for time-dependent DFT (TD-DFT) to identify likely transitions in the predicted absorption spectra but the excited states were heavily mixed with no obvious intermolecular charge transfer (ICT) state. B3LYP/6-31 G* on the other hand for TD-DFT provided reasonable results with a clear ICT character of the S1 state. In both carboranes, the HOMO is mainly localized on the triphenylamine moiety with little contribution from the carborane bridge. While the LUMO of DA-pBenz and DA-mBenz is mainly located on the boron center and the biphenylene bridge (see SI), the LUMO of DA-pCarb and DA-mCarb is delocalized over the BMes2 moiety and the entire carborane cage (Supplementary Fig. 25, 26). NAO (natural atomic orbitals) analyses were carried out to describe the fragment contributions to the frontier orbitals (Supplementary Tab. 7, 8)78. In the carborane derivatives, the LUMO consists of the empty boron pz orbital of the BMes2 group (60% for DA-pCarb and 62% for DA-mCarb) and cluster orbitals (15% for p-carborane and 14% for m-carborane), confirming that the carborane cluster is significantly involved in the electronic structure. There is overlap between HOMO and LUMO in both compounds (Supplementary Fig. 25, 26), which is calculated at 0.18 in DA-pCarb and 0.11 in DA-mCarb where a complete overlap would be at 1.

Blue and white indicate the different signs of the wave function. The percentage values are the carborane-contributions to the molecular orbitals.
Because of the spherical structure of the carborane bridge, we anticipated that the rotation of the phenylene ring of the triphenylamine might possess a very shallow potential. To get more insight how the rotation influences the conjugation properties of the carboranes, we rotated the phenylene group of DA-pCarb stepwise by 10° (torsional angle of C33-C32-B23-C1) and optimized all other structural parameters. The resulting potential is depicted in Fig. 6a and indeed shows a quite shallow potential of less than 60 cm−1, which can easily be overcome by thermal energy at room temperature. The minimum structure refers to a parallel orientation of the amino-phenylene ring with the trigonal boron coordination plane whereby the relative orientation of the nitrogen and boron pz orbitals is 39.8°. Taking this geometry as a start, we also optimized the S1 state where this angle widens to 55.5°. On the other hand, the mesityl rings possess a somewhat smaller torsional angle with the trigonal nitrogen plane of 31° in the S1 state compared to 45° in the S0 state. We also calculated the S1 ← S0 transitions at B3LYP/6-31 G* for each fixed torsional step. While the overall transition energy is 23500 cm−1 (425 nm), which is too low in comparison with experiment, the variation with the torsional angle shows a clear trend with a maximum transition energy for the minimum of the S0 potential and a minimal transition energy for the maximum potential. However, the absolute variation is with 80 cm−1 extremely small and would hardly be visible experimentally. A more important point is the variation of the oscillator strength, which goes from almost zero at the maximum to 0.15 at the minimum of the of the S0 potential. Thus, orbital overlap between the phenylene ring and the boron center via the carborane cage clearly has a strong impact on the oscillator strength (Fig. 6, Supplementary Fig. 27, 28). For DA-mCarb, the phenylene plane and the B trigonal plane also are coplanar in the optimized global minimum structure. There is a similar potential barrier of ca. 90 cm−1 for the rotation of the triarylamine in DA-mCarb as in DA-pCarb (Fig. 6). In contrast to DA-pCarb the S1 ← S0 transition energy is minimal in the potential minimum. However, the maximum oscillator strength is much smaller (f = 0.03) in the global minimum structure where the triarylamine phenylene ring and the boron pz orbitals are coplanar. This smaller oscillator strength is in agreement with the lower transition dipole strength of DA-mCarb (Fig. 2).

Relative ground state energy depending on the phenylene-borane-plane torsional angle (blue) and absolute S1 ← S0 transition energies (red), oscillator strength f (green) and HOMO-LUMO overlap % (purple) in the gas phase of a) DA-pCarb and c) DA-mCarb). Optimized global minimum structures of b) DA-pCarb and d) DA-mCarb; blue spheres, pink spheres and gray spheres corresponding to the nitrogen, boron, and carbon atoms, respectively.
Both DA-mCarb and DA-pCarb show torsional-angle-dependent oscillator strengths, despite the spherical shape of the carborane. This reveals that orbital overlap plays a strong role in the electronic transmission, that is, the dihedral angle of N and B plane has a vital influence for the electron communication through the carboranes. The HOMO and LUMO (Fig. 5, Supplementary Fig. 25, 26) at the optimized geometry show some overlap in the carborane region. The transmission of orbital overlap through the meta-carborane is weaker than for para-carborane as is apparent by the smaller oscillator strengths in the former. Both MOs are clearly π-orbitals and the twisting of the groups reduces this overlap and, consequently, the oscillator strength. The effect of carborane orbital contribution to the transmission of charge in the ICT process of DA-pCarb was estimated by a TD-DFT calculation in which the carborane cluster atoms in the global minimum structure were omitted and hydrogen atoms were added to the phenylene and boron. This calculation gave an ICT energy, which is 1500 cm−1 higher but possesses zero oscillator strength instead of 0.15 for DA-pCarb. The contribution of ICT (LUMO ← HOMO) transition to the S1 ← S0 transition is nearly 100% in all four compounds (Supplementary Tab. 9–12, Supplementary Fig. 29–33). The calculated oscillator strength (0.58) in DA-pBenz is much higher than in DA-pCarb and the same is true for DA-mCarb and DA-mBenz, which matches well with the comparatively large transition dipole strength that was estimated from the experimental absorption spectrum (Table 2 and Supplementary Tab. 6).
Discussion
In conclusion, we have synthesized the donor-acceptor systems DA-pCarb and DA-mCarb and studied the communication through carboranes by combining absorption spectroscopy, steady-state and time-resolved fluorescence spectroscopy, fs- transient absorption, FLUPS, cyclic voltammetry, DFT and TD-DFT calculations. Unexpectedly, the fluorescence quantum yield increases with the solvent polarity which is caused by a decrease of nonradiative rate constant tuned by the amount of local excitation to the CT character of the S1 state. The dipole moment of the relaxed S1 state of both carborane derivatives is much larger than that of analogous phenylene-bridged derivatives, indicating a more pronounced charge transfer through the carborane bridge than in the case of phenylene. Femto-second transition absorption measurements and FLUPS of DA-pCarb reveals the spontaneous formation of the charge transfer state with excitation. Thus, the lowest energy absorption and emission band of DA-pCarb correspond to the ICT transition between the amine and the BMes2 moieties through the p-carborane cage with considerable transition dipole strength. A less pronounced optically induced process was found in DA-mCarb. TD-DFT calculations show that the S1 ← S0 transitions of both carborane derivatives are predominantly LUMO ← HOMO transitions that correspond to the migration of electrons from the triphenylamine to the BMes2 group through the carborane bridge. This transition depends on orbital overlap because it is influenced by the dihedral angle of the trigonal N and B plane of the carborane compounds. Maximum oscillator strengths were obtained when the phenylene ring is coplanar with the trigonal boron plane when the HOMO - LUMO overlaps are at maximum. Both experimental and theoretical results confirm the charge transfer through m- and p- carborane bridges, indicating considerable conjugation between the 3-D carborane bridge and the adjacent donor and acceptor 2-D π-systems. Concerning the fluorescence properties (energy, quantum yield, lifetime), meta- and para-substituted carboranes and the meta-phenylene analog are all similar. Our results confirm the potential of developing carborane-bridged fluorescent materials with high quantum yields in polar media.
Methods
Materials
For 11B NMR, 11B{1H} NMR, 1H NMR, 1H{11B} NMR, 13C{1H} NMR, and high-resolution mass spectrometry of compounds in this manuscript and details of the synthetic procedures, see Supplementary Information.
Data availability
All data supporting the findings of this study, including experimental procedures and compounds characterization, the relevent measurement methods, computational details, are avaliable within the paper and Supplementary Information. The coordinates of the optimized structures are present as source data. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Grimes R. N. Carboranes. 7–18 (Elsevier Science, 2016).
Keener, M. et al. Redox-switchable carboranes for uranium capture and release. Nature 577, 652–655 (2020).
Keener, M. et al. Selective electrochemical capture and release of uranyl from aqueous alkali, lanthanide, and actinide mixtures using redox-switchable carboranes. Chem. Sci. 13, 3369–3374 (2022).
Jude, H. et al. Coordination-Driven Self-Assemblies with a Carborane Backbone. J. Am. Chem. Soc. 127, 12131–12139 (2005).
Soloway, A. H. et al. The Chemistry of Neutron Capture Therapy. Chem. Rev. 98, 1515–1562 (1998).
Hawthorne, M. F. The Role of Chemistry in the Development of Boron Neutron Capture Therapy of Cancer. Angew. Chem., Int. Ed. 32, 950–984 (1993).
Stockmann, P., Gozzi, M., Kuhnert, R., Sárosi, M. B. & Hey-Hawkins, E. New keys for old locks: carborane-containing drugs as platforms for mechanism-based therapies. Chem. Soc. Rev. 48, 3497–3512 (2019).
Marfavi, A., Kavianpour, P. & Rendina, L. M. Carboranes in drug discovery, chemical biology and molecular imaging. Nat. Rev. Chem. 6, 486–504 (2022).
Scholz, M. & Hey-Hawkins, E. Carbaboranes as Pharmacophores: Properties, Synthesis, and Application Strategies. Chem. Rev. 111, 7035–7062 (2011).
Ali, F. et al. Boron Chemistry for Medical Applications. Molecules 25, 828–852 (2020).
Hosmane N. S., Eagling R. D. Handbook of Boron Science: With Applications In Organometallics, Catalysis, Materials And Medicine. (World Scientific Publishing Company, 2018).
Endo, Y. Carboranes as Hydrophobic Pharmacophores. In Boron‐Based Compounds, Vol. 1 (eds Hey-Hawkins, E. & Teixidor, C. V.) 1–19 (Wiley, 2018).
Sauerwein W. A. G., Wittig A., Moss R., Nakagawa Y. Neutron Capture Therapy: Principles and Applications. (Springer Berlin Heidelberg, 2012).
Barth R. F., Carpenter D. E., Soloway A. H. Advances in Neutron Capture Therapy. (Springer US, 2012).
Furue, R., Nishimoto, T., Park, I. S., Lee, J. & Yasuda, T. Aggregation-Induced Delayed Fluorescence Based on Donor/Acceptor-Tethered Janus Carborane Triads: Unique Photophysical Properties of Nondoped OLEDs. Angew. Chem., Int. Ed. 55, 7171–7175 (2016).
Wang, Z. et al. Through-Space Charge Transfer: A New Way to Develop a High-Performance Fluorescence Sensing Film towards Opto-Electronically Inert Alkanes. Angew. Chem., Int. Ed. 61, e202207619 (2022).
Shi, C. et al. Variable Photophysical Properties of Phosphorescent Iridium(III) Complexes Triggered by closo- and nido-Carborane Substitution. Angew. Chem., Int. Ed. 52, 13434–13438 (2013).
Wee, K. R., Cho, Y. J., Song, J. K. & Kang, S. O. Multiple photoluminescence from 1,2-dinaphthyl-ortho-carborane. Angew. Chem., Int. Ed. 52, 9682–9685 (2013).
Gao, X. et al. A Portable Fluorescence Sensor with Improved Performance for Aniline Monitoring. Adv. Mater. Interfaces 9, 2201275 (2022).
Huang, R. et al. Single-Fluorophore-Based Organic Crystals with Distinct Conformers Enabling Wide-Range Excitation-Dependent Emissions. Angew. Chem., Int. Ed. 61, e202211106 (2022).
Wee, K. R. et al. Carborane photochemistry triggered by aryl substitution: carborane-based dyads with phenyl carbazoles. Angew. Chem., Int. Ed. 51, 2677–2680 (2012).
Ochi, J., Tanaka, K. & Chujo, Y. Recent Progress in the Development of Solid-State Luminescent o-Carboranes with Stimuli Responsivity. Angew. Chem., Int. Ed. 59, 9841–9855 (2020).
Makarov, N. S. et al. Impact of Electronic Coupling, Symmetry, and Planarization on One- and Two-Photon Properties of Triarylamines with One, Two, or Three Diarylboryl Acceptors. J. Phys. Chem. A 116, 3781–3793 (2012).
Núñez, R., Tarrés, M., Ferrer-Ugalde, A., de Biani, F. F. & Teixidor, F. Electrochemistry and Photoluminescence of Icosahedral Carboranes, Boranes, Metallacarboranes, and Their Derivatives. Chem. Rev. 116, 14307–14378 (2016).
Wei, X. et al. Aggregation-Induced Electrochemiluminescence of Carboranyl Carbazoles in Aqueous Media. Angew. Chem., Int. Ed. 58, 3162–3166 (2019).
King, R. B. Three-dimensional aromaticity in polyhedral boranes and related molecules. Chem. Rev. 101, 1119–1152 (2001).
Yang, X. et al. The Marriage of Carborane with Chalcogen Atoms: Nonconjugation, σ-π Conjugation, and Intramolecular Charge Transfer. Org. Lett. 21, 8285–8289 (2019).
Zhang, C., Wang, J., Su, W., Lin, Z. & Ye, Q. Synthesis, Characterization, and Density Functional Theory Studies of Three-Dimensional Inorganic Analogues of 9,10-Diboraanthracene-A New Class of Lewis Superacids. J. Am. Chem. Soc. 143, 8552–8558 (2021).
Buzsaki, D., Kovacs, M. B., Humpfner, E., Harcsa-Pinter, Z. & Kelemen, Z. Conjugation between 3D and 2D aromaticity: does it really exist? The case of carborane-fused heterocycles. Chem. Sci. 13, 11388–11393 (2022).
Poater, J., Vinas, C., Sola, M. & Teixidor, F. 3D and 2D aromatic units behave like oil and water in the case of benzocarborane derivatives. Nat. Commun. 13, 3844 (2022).
Wright, J. H., Kefalidis, C. E., Tham, F. S., Maron, L. & Lavallo, V. Click-Like Reactions with the Inert HCB11Cl11- Anion Lead to Carborane-Fused Heterocycles with Unusual Aromatic Character. Inorg. Chem. 52, 6223–6229 (2013).
Asay, M. et al. Isolation of a Carborane-Fused Triazole Radical Anion. Angew. Chem., Int. Ed. 52, 11560–11563 (2013).
Chan, T. L. & Xie, Z. Synthesis, structure and aromaticity of carborane-fused carbo- and heterocycles. Chem. Sci. 9, 2284–2289 (2018).
Kwon, S., Wee, K. R., Cho, Y. J. & Kang, S. O. Carborane dyads for photoinduced electron transfer: photophysical studies on carbazole and phenyl-o-carborane molecular assemblies. Chem. Eur. J. 20, 5953–5960 (2014).
Ziessel, R., Ulrich, G., Olivier, J. H., Bura, T. & Sutter, A. Carborane-Bodipy scaffolds for through space energy transfer. Chem. Comm. 46, 7978–7980 (2010).
Indelli, M. T., Bura, T. & Ziessel, R. p-Carborane-Bridged Bipyridine Ligands for Energy Transfer between Two Iridium Centers. Inorg. Chem. 52, 2918–2926 (2013).
Harriman, A., Alamiry, M. A., Hagon, J. P., Hablot, D. & Ziessel, R. Through-space electronic energy transfer across proximal molecular dyads. Angew. Chem., Int. Ed. 52, 6611–6615 (2013).
Hablot, D., Sutter, A., Retailleau, P. & Ziessel, R. Unsymmetrical p-carborane backbone as a linker for donor-acceptor dyads. Chem. Eur. J. 18, 1890–1895 (2012).
Hablot, D., Ziessel, R., Alamiry, M. A. H., Bahraidah, E. & Harriman, A. Nanomechanical properties of molecular-scale bridges as visualised by intramolecular electronic energy transfer. Chem. Sci. 4, 444–453 (2013).
Fox, M. A. et al. Ruthenium complexes of C,C’-bis(ethynyl)carboranes: an investigation of electronic interactions mediated by spherical pseudo-aromatic spacers. J. Am. Chem. Soc. 130, 3566–3578 (2008).
Wedge, T. J. et al. Metal-Metal Communication through Carborane Cages Supporting Electroactive [η5-CpFe(CO)2] Substituents.Organometallics 23, 482–489 (2004).
Brown, N. J. et al. Molybdenum Complexes of C,C-Bis(ethynyl)carboranes: Design, Synthesis, and Study of a Weakly Coupled Mixed-Valence Compound. Organometallics 30, 884–894 (2010).
Bitner, T. W., Wedge, T. J., Hawthorne, M. F. & Zink, J. I. Synthesis and luminescence spectroscopy of a series of [η5-CpFe(CO)2] complexes containing 1,12-dicarba-closo-dodecaboranyl and -ylene ligands. Inorg. Chem 40, 5428–5433 (2001).
Cook, A. R. et al. p-Carborane Conjugation in Radical Anions of Cage-Cage and Cage-Phenyl Compounds. J. Phys. Chem. A 122, 798–810 (2018).
Clegg W. et al. Platinum-catalysed 1,4-diboration of 1,3-dienes. J. Chem. Soc., Dalton Trans. 1431–1438 (1998).
Jakubowski, R. et al. Photophysical Behavior of Self-Organizing Derivatives of 10- and 12-Vertex p-Carboranes, and their Bicyclo[2.2.2]octane and Benzene Analogues. Chem. Eur. J. 29, e202203948 (2023).
Ji, L. et al. Electron Delocalization in Reduced Forms of 2-(BMes2)pyrene and 2,7-Bis(BMes2)pyrene. J. Am. Chem. Soc. 137, 6750–6753 (2015).
Ji, L., Griesbeck, S. & Marder, T. B. Recent developments in and perspectives on three-coordinate boron materials: a bright future. Chem. Sci. 8, 846–863 (2017).
Jäkle, F. Advances in the Synthesis of Organoborane Polymers for Optical, Electronic, and Sensory Applications. Chem. Rev. 110, 3985–4022 (2010).
Kahlert, J. et al. Syntheses and reductions of C-dimesitylboryl-1,2-dicarba-closo-dodecaboranes. Dalton Trans. 44, 9766–9781 (2015).
Heckmann, A. & Lambert, C. Organic Mixed-Valence Compounds: A Playground for Electrons and Holes. Angew. Chem., Int. Ed. 51, 326–392 (2012).
Taniguchi, T. Advances in chemistry of N-heterocyclic carbene boryl radicals. Chem. Soc. Rev. 50, 8995–9021 (2021).
He, J. J., Lyu, P. B., Sun, L. Z., Garcia, A. M. & Nachtigall, P. High temperature spin-polarized semiconductivity with zero magnetization in two-dimensional Janus MXenes. J. Mater. Chem. C. 4, 6500–6509 (2016).
He, J. et al. N-Heterocyclic Olefins as Electron Donors in Combination with Triarylborane Acceptors: Synthesis, Optical and Electronic Properties of D-π-A Compounds. Chem. Eur. J. 25, 13777–13784 (2019).
Yuan, M.-S., Liu, Z.-Q. & Fang, Q. Donor-and-Acceptor Substituted Truxenes as Multifunctional Fluorescent Probes. J. Org. Chem. 72, 7915–7922 (2007).
Zhu, H. et al. Ultrafast Investigation of Intramolecular Charge Transfer and Solvation Dynamics of Tetrahydro[5]-helicene-Based Imide Derivatives. Sci. Rep. 6, 24313 (2016).
Kohler, J. et al. Ultrafast charge-transfer dynamics of donor-substituted truxenones. Phys. Chem. Chem. Phys. 14, 11081–11089 (2012).
Shirota, Y. & Kageyama, H. Charge carrier transporting molecular materials and their applications in devices. Chem. Rev. 107, 953–1010 (2007).
Amthor, S., Noller, B. & Lambert, C. UV/Vis/NIR spectral properties of triarylamines and their corresponding radical cations. Chem. Phys. 316, 141–152 (2005).
Fiedler, J., Zališ, S., Klein, A., Hornung, F. M. & Kaim, W. Electronic Structure of π-Conjugated Redox Systems with Borane/Borataalkene End Groups. Inorg. Chem. 35, 3039–3043 (1996).
Nelson, R. R. & Adams, R. N. Anodic oxidation pathways of substituted triphenylamines. II. Quantitative studies of benzidine formation. J. Am. Chem. Soc. 90, 3925–3930 (2002).
Marciniak, H. et al. Reduction of the Fluorescence Transition Dipole Moment by Excitation Localization in a Vibronically Coupled Squaraine Dimer. J. Phys. Chem. C. 123, 3426–3432 (2019).
Lewis, J. E. & Maroncelli, M. On the (uninteresting) dependence of the absorption and emission transition moments of coumarin 153 on solvent. Chem. Phys. Lett. 282, 197–203 (1998).
Chako, N. Q. Absorption of Light in Organic Compounds. J. Chem. Phys. 2, 644–653 (1934).
Gould, I. R. et al. Radiative and Nonradiative Electron-Transfer in Contact Radical-Ion Pairs. Chem. Phys. 176, 439–456 (1993).
Gould, I. R., Young, R. H., Mueller, L. J., Albrecht, A. C. & Farid, S. Electronic Structures of Exciplexes and Excited Charge-Transfer Complexes. J. Am. Chem. Soc. 116, 8188–8199 (1994).
Herbich, J. & Kapturkiewicz, A. Electronic Structure and Molecular Conformation in the Excited Charge Transfer Singlet States of 9-Acridyl and Other Aryl Derivatives of Aromatic Amines. J. Am. Chem. Soc. 120, 1014–1029 (1998).
Lippert, E. Habilitationsschrift Zur Erlangung Der Lehrberichtigung (Venia Legendi) Fur Das Fach Physikalische Chemie Ander Techischen-Hochschule-Stuttgart - Spektroskopische Bestimmung Des Dipolmomentes Aromatischer Verbindungen Im Ersten Angeregten Singulettzustand. Z. Elektrochem. 61, 962–975 (1957).
Merz, J. et al. Pyrene Molecular Orbital Shuffle-Controlling Excited State and Redox Properties by Changing the Nature of the Frontier Orbitals. Chem. Eur. J. 23, 13164–13180 (2017).
Gong, Y. et al. Photophysical properties of photoactive molecules with conjugated push-pull structures. J. Phys. Chem. A 111, 5806–5812 (2007).
Strickler, S. J. & Berg, R. A. Relationship between Absorption Intensity and Fluorescence Lifetime of Molecules. J. Chem. Phys. 37, 814–822 (1962).
Stahl, R., Lambert, C., Kaiser, C., Wortmann, R. & Jakober, R. Electrochemistry and photophysics of donor-substituted triarylboranes: symmetry breaking in ground and excited state. Chem. Eur. J. 12, 2358–2370 (2006).
Gould, I. R. & Farid, S. Radiationless decay in exciplexes with variable charge transfer. J. Phys. Chem. B 111, 6782–6787 (2007).
Cheng, A. Y. et al. Origin of Red-Shifted Phosphorescence from Triphenylamines: Triplet Excimer or Impurity? Angew. Chem., Int. Ed. 61, e202206366 (2022).
Schmidt, W. & Steckhan, E. Über organische Elektronenüberträgersysteme, I. Elektrochemische und spektroskopische Untersuchung bromsubstituierter Triarylamin‐Redoxsysteme. Chem. Ber. 113, 577–585 (2006).
Maksimenka, R. et al. Femtosecond dynamics of electron transfer in a neutral organic mixed-valence compound. Chem. Phys. 347, 436–445 (2008).
Horng, M. L., Gardecki, J. A., Papazyan, A. & Maroncelli, M. Subpicosecond Measurements of Polar Solvation Dynamics: Coumarin 153 Revisited. J. Phys. Chem. C. 99, 17311–17337 (1995).
Nkungli, N. K., Ghogomu, J. N., Nogheu, L. N. & Gadre, S. R. DFT and TD-DFT Study of Bis[2-(5-Amino-[1,3,4]-Oxadiazol-2-yl) Phenol](Diaqua)M(II) Complexes [M = Cu, Ni and Zn]: Electronic Structures, Properties and Analyses.J. Comput. Chem 3, 29–44 (2015).
Acknowledgements
We are grateful for generous financial support by the Nature Science Foundation of China (Grant No. 62174137), the Key Research and Development Projects of Shaanxi Province (Grant No. 2020GXLH-Z-022), Ningbo Natural Science Foundation (Grant No. 2021J044, 2023J038), the Fundamental Research Funds for the Central Universities, and Northwestern Polytechnical University. L. W. acknowledges generous financial support from the China Scholarship Council. C.L. thanks the Bavarian State Ministry of Education, Culture, Science, and the Arts for funding this work by the SolTech initiative. We gratefully thank Prof. Dr. Dr. h.c. Todd B. Marder for selfless support during the whole project, we also thank the kind help from Dr. Florian Rauch.
Author information
Authors and Affiliations
Contributions
L.J., C.L., M.A.F. and M.F. conceived and directed the project. L.W. and F.P. synthesized and characterized the compounds reported in this paper. L.W., A.S. and P.M. did the photophysical measurements. L.W., M.H. J.S. did the theoretical studies. M.M. did the electrochemistry. L.W., T.N., R.B. did the boron-related NMR studies and M.A.F. analyzed the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kangmoon Lee and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, L., Holzapfel, M., Schmiedel, A. et al. Optically induced charge-transfer in donor-acceptor-substituted p- and m- C2B10H12 carboranes. Nat Commun 15, 3005 (2024). https://doi.org/10.1038/s41467-024-47384-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-47384-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
