
Abstract
Herein, we report an electroreduction of unactivated alkyl alkenes enabled by [Fe]-H, which is provided through the combination of anodic iron salts and the silane generated in situ via cathodic reduction, using H2O as an H-source. The catalytic amounts of Si-additive work as an H-carrier from H2O to generate a highly active silane species in situ under continuous electrochemical conditions. This approach shows a broad substrate scope and good functional group compatibility. In addition to hydrogenation, the use of D2O instead of H2O provides the desired deuterated products in good yields with excellent D-incorporation (up to >99%). Further late-stage hydrogenation of complex molecules and drug derivatives demonstrate potential application in the pharmaceutical industry. Mechanistic studies are performed and provide support for the proposed mechanistic pathway.
Similar content being viewed by others


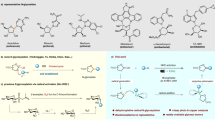
Introduction
Electrochemistry, employing electrons as intrinsically safe and sustainable redox reagents represents an eco-friendly strategy in organic synthesis1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18. In this regime, electrochemical hydrogenation has gained increasing attraction due to the availability of renewable electrical energy, and cost savings obtained from replacing thermal, stoichiometric chemical reductants, and high pressure of H2 inputs. In recent years, elegant work has been reported that low-cost and user-friendly NH3, H2O, alcohol, and DMSO have been employed as H-sources in the electrochemical hydrogenation of carbon-carbon double bonds. However, most of these works are limited to activated olefins, such as styrene derivatives and/or acrylates with more positive reduction potentials19,20,21,22,23,24,25,26,27,28,29,30,31. Contrastingly, as it is more difficult to directly reduce unactivated alkyl alkenes (E1/2 < −3.0 V vs. SCE) than activated olefins (for instance: methyl acrylate, E1/2 = −2.10 V vs. SCE; styrene, E1/2 = −2.58 V) due to their more negative reductive potentials32, strategies for the electrochemical hydrogenation of unactivated alkyl alkenes have proven elusive33,34. Therefore, the development of a general strategy for electrochemical hydrogenation of unactivated alkyl alkenes is still highly desirable and remains challenging.
Transfer hydrogenation (TH) has emerged as a promising and powerful strategy due to its simple operation and is much safer than direct hydrogenation using hydrogen gas, since gas containment or pressure vessel is required35,36,37,38,39. And various reagents including silanes, boranes, acids and alcohols etc. have been used as hydrogen sources in the TH processes of alkenes40,41,42,43,44,45,46. However, there are a few examples using low-cost, non-toxic and eco-friendly water as the hydrogen donor. Formidable challenges remain in the transformations: high bond dissociation energy in H2O (118 kacl mol−1)47; delicate metal-complexes are necessary (Fig. 1A). And it allows easy isotopic labeling of products by employing economical D2O in replacement of H2O as deuterium source during the TH process of alkenes, which could be a straightforward approach to acquire α,β-deuterated alkyl compounds with much cost saving. And the resulting deuterated products are crucial in the field of pharmaceutical science and mechanistic studies48,49,50,51,52,53. Elegant works of H2O activation, including oxidative addition activation and coordination-induced bond weakening, using transition metal complexes or main group elements with empty d or p orbitals have been disclosed54. However, direct proton transfer (PT) from water for further hydrogenation still remains challenging (Fig. 1B). Notably, thiol mediated PT/HAT using water as H-source have been investigated in the field of photocatalysis55, which limiting its application in metal catalyzed transfer hydrogenation might due to the poison effect on metal catalysts. Hence, the development of methodologies for the highly efficient hydrogenation employing H2O/D2O as H/D-source of alkyl alkenes remains critical.

A Transfer hydrogenation (TH) of alkenes. B State-of-the-art in H2O activation modes for hydrogenation. C Our design: Si-mediator as a H/D-carrier from H2O/D2O. D This work: Si-mediated electroreduction of unactivated alkyl alkenes.
Meanwhile, silane is a highly active H-source and is widely used in organic synthesis, especially in combination with transition-metal catalysts in the TH reactions56,57,58. However, in most cases, excess amounts of silane are required, which limits its practical application. Chlorosilane could be activated under electroreductive conditions through two rounds of single-electron transfer to give silyl anions for further transformation59,60. Inspired by the electroreductive events of chlorosilane and our continuous interest in electroreductive transformations61,62,63,64,65, we envisioned that chlorosilane could be used as an H/D-carrier under electroreductive conditions, which could in situ generate a key silane intermediate with H2O/D2O. Then, the Si-species could then engage in the next catalytic cycle after TH with transition metal under the electrochemical conditions (Fig. 1C).
Herein, we report an electroreduction of unactivated alkyl alkenes promoted by a catalytic amount of simple chlorosilane using H2O/D2O as the H/D-source (Fig. 1D). Notable features of this strategy include: (a) using a simple and low-cost catalytic amount of chlorosilane as an H-carrier in the reaction system; (b) using H2O/D2O as the economical H/D-source; (c) noble transition-metal free, low-cost Fe performing dual function as the anode and in situ generation of metal hydride; (d) high chemoselectivity and regioselectivity; (e) late-stage hydrogenation of naturally occurring compounds and drug derivatives. Detailed mechanistic insights provided strong support for the pivotal role of a H-carrier in the transformation.
Results
Optimization of electroreductive hydrogenation
Initially, we employed 4-(pent-4-en-1-yloxy)−1,1′-biphenyl (1) as the model substrate and examined various reaction conditions under a constant current for the electroreductive hydrogenation of unactivated alkyl alkenes. After careful optimization, hydrogenation product 2 was obtained in 92% NMR yield under a constant current (6 mA) in DMF with Ph3SiCl as a catalyst, H2O as an H-source, TBABF4 as a supporting electrolyte, Fe plate as the anode and Ni foam as the cathode (Table 1, entry 1). The control experiment showed that Ph3SiCl was essential for this transformation as its omission led to very low efficiency (entry 2). Several anodic materials were investigated (entries 3−5), while Fe(+) was proven to be optimal. Different cathodic materials, such as Fe plate or GF, could also be used, affording the desired product in 90% and 75% yield, respectively (entry 6). Next, when TMSCl or TMSBr was used as H-carriers, the desired product was provided in good yields as well (entry 7). It was found that (TMS)2O could also promote this transformation with high efficiency (entry 8). When Ph3SiH was employed as an additive in the reaction, it gave the product in 73% yield (entry 9). In addition, the effect of the solvent on the transformation was investigated, including DMF, DMA, NMP, and CH3CN. As a result, DMF was found to be optimal for this transformation (entry 10). Employing MeOH or EtOH as the H-source provided the desired product in high yield (entry 11), however, we chose low-cost and readily available H2O as the preferred H-donor in the transformation. Inspired by previous elegant reports that involved the use of 3d transition metals, such as Co or Ni, for carbon-halogen bond activation via an SET process66,67,68,69,70,71,72, we envisaged that catalytic amounts of Co- or Ni- complexes could behave as a mediator to promote the cleavage of the Si–Cl bond. Indeed, when CoBr2.dtbpy or NiBr2.dtbpy was added to the reaction system, the unactivated alkene substrate could be consumed completely, and the transformation worked in very high efficiency at 5 mA giving the product with 98% isolated yield (entries 12–14). Finally, the control experiment demonstrated the essential role of electricity, as no product could be detected in its absence (entry 15). We chose entry 14 as the optimized electrochemical hydrogenation condition.
Substrate scope
With the optimized reaction conditions established, we next explored the scope and generality of the reaction, as shown in Fig. 2. Initially, various representative terminal alkenes were proven suitable, affording the corresponding hydrogenation products in good to high yields. To our delight, the length of the alkene chain showed no significant influence on the efficiency of the transformation (from 4C to 10C) except only a slight decrease in the yield when the chain became longer (2−8, 90–98%). Next, terminal alkenes derived from phenols bearing different substituents were examined, including those with electron-rich or deficient groups, which all worked smoothly under the standard conditions and furnished the corresponding hydrogenation products in good to high yields (10−21). Simple alkenes without O-linkage in the alkene chain gave an almost quantitative product (9, 98%). Then, several N-containing substrates were tested, and to our delight, the carbazole, indole, and aniline motifs were all impregnable under the electrochemical conditions (22−25). Furthermore, terminal alkenes containing ester linkages derived from simple acids or alcohols proceeded well in the transformation (26−30). Notably, free carboxyl and hydroxyl groups remained intact under the standard conditions, demonstrating good functional group compatibility in this electroreduction process. (31 and 32).

Reaction conditions: aFe as anode, Ni Foam as cathode, constant current = 5 mA, unactivated alkene (0.3 mmol, 1.0 equiv.), NiBr2.dtbpy (5 mol%), Ph3SiCl (20 mol%), H2O (3.0 equiv.), TBABF4 (1.0 equiv.) in DMF (4.0 mL), room temperature, 10 h, under Ar atmosphere. Isolated yields. FE faradaic efficiency. bPh3SiCl (10 mol%), PhSiH3 (30 mol%). c8 mA. dYields were determined by GC.
More importantly, this hydrogenation process showed excellent regioselectivity under standard conditions. When two types of carbon-carbon double bonds were present in the same molecule, the reaction occurred preferentially on the monosubstituted terminal alkene, as demonstrated with products 33 and 34 in Fig. 2. Polysubstituted alkenes showed no conversion under the standard conditions, which might be attributed to more steric hindrance, so in situ generated Ph3SiH suppresses the hydrogenation process. Therefore, we speculated that a smaller silane could promote the hydrogenation process. Conceptually, the addition of catalytic amounts of PhSiH3 (30 mol%) improved the efficiency of the transformation with polysubstituted alkenes to a large extent, including branch alkenes (35 and 38), internal alkenes (36 and 37) and cycloalkenes (39 and 40). To further demonstrate the general application of this methodology, simple commercially available straight-chain aliphatic alkenes were treated under our electroreductive conditions and gave the desired hydrogenation product in moderate to good yields (41−44). Moreover, this protocol could enable the hydrogenation of simple styrenes in moderate to high yields, but conjugated diene worked in very low efficiency under the standard conditions (for details, please see the Supplementary Information).
The electrochemical hydrogenation was further expanded to the late-stage application of complex compounds (Fig. 3). Alkenes derived from drugs (including ibuprofen, flurbiprofen, menthol), α-amino acid derivatives, (including L-valine, L-methionine, and L-phenylalanine), natural compounds, (such as progesterone, estrone and cholesterol), and carbohydrates, (such as fructose, and galactose) were all amenable to the electroreductive hydrogenation transformation, providing the desired products in moderate to good yields (45−58). These results indicated that this strategy offered a potential synthetic method for the selective hydrogenation of complex compounds and drug molecules under mild conditions.

Reaction conditions: aFe as anode, Ni Foam as cathode, constant current = 5 mA, unactivated alkene (0.3 mmol, 1.0 equiv.), NiBr2.dtbpy (5 mol%), Ph3SiCl (20 mol%), H2O (3.0 equiv.), TBABF4 (1.0 equiv) in DMF (4.0 mL), room temperature, 10 h, under Ar atmosphere. Isolated yields. FE faradaic efficiency. b50 °C.
To our delight, this strategy could enable the electroreductive deuteration of alkyl alkenes using D2O as the economical deuterium source under slightly modified reaction conditions, affording the deuterated products in good yields with excellent D-incorporation (up to >99%, Fig. 4). Simple terminal alkenes derived from phenols bearing different substituents worked smoothly under the electroreductive deuteration conditions and gave the desired products in moderate to good yields with excellent D-incorporation (59−70). N-containing substrates, such as carbazole, indole and aniline derivatives, were also tolerant under the electroreductive conditions providing the desired products in moderate to excellent D-incorporation (71−74). These preliminary results demonstrated that this strategy could offer a potential synthetic method for the convenient introduction of deuterium to organic compounds under mild conditions.

Reaction conditions: a Fe as anode, Cu as cathode, constant current = 10 mA, unactivated alkenes (0.3 mmol, 1.0 equiv.), NiBr2.dtbpy (5 mol%), Ph3SiCl (20 mol%), D2O (20.0 equiv.), TBABF4 (1.0 equiv.) in DMF (4.0 mL), room temperature, 10 h, under Ar atmosphere. Isolated yield. FE faradaic efficiency. D-inc. % determined by 1H NMR.
Mechanistic studies
To gain insight into the mechanism of this transformation, several mechanistic experiments were conducted. When substrate 1 was treated under the standard reaction conditions, GC-MS analysis detected the presence of Ph3SiH (Fig. 5a). Control experiments suggested that Ph3SiH could promote the electroreductive hydrogenation process in good efficiency (Fig. 5b). Thus, we speculated that Ph3SiH might be an active intermediate in the catalytic cycle. Moreover, alkyl silicon compound (75) was synthesized and treated under the standard conditions and no hydrogenation product through C−Si bond cleavage was detected (Fig. 5c). In addition, a control experiment using a catalytic amount of 75 in replacement of Ph3SiCl was conducted, and no hydrogenation product was detected (Fig. 5d). These results showed that the activation of alkenes through the formation of C−Si bond in the transformation could be ruled out.

A Investigation on the role of chlorosilane. B Deuteration experiments. C Ring-opening experiments. D 19F NMR spectroscopic evidence for the formation of fluorosilanes. E CV experiments, using glass carbon as the working electrode, Pt plate and Ag/Ag+ as the counter and reference electrodes. Scan rate: 100 mV s−1. DMF/nBu4NBF4 (0.1 M). [Ni] = NiBr2.dme, L = 4,4′-di-tert-butyl-2,2′-bipyridine. F Proposed mechanism.
Next, the deuteration experiments of S3 with 5.0 equivalents of PhMe2SiD with/without Ni-complex gave desired product 3-D in 90% and 88% yields, respectively, with D-incorporation (α/β: 85%/79% and 56%/85%) (Fig. 5e). The deuterated ratio was inverse in the two experiments, which may be attributed to the regioselectivity of different metal hydrides (Ni–H vs. Fe–H). And DFT calculations indicated that the NiH complex preferred to form the branch intermediate with alkenes, which then underwent protonation (Supplementary Fig. 9). For the FeH complex, the regioselectivity was diametrically opposed (Supplementary Fig. 10). These results of DFT calculations were in line with our experimental studies. These results illustrated that Ni-catalyzed cycle was involved in this transformation as well. Combined with the conclusion drawn from Fig. 5a, the catalytic amount of Si-catalyst could work as an H-carrier from H2O/D2O to alkenes. Moreover, when internal alkenes (S36 and S37) were treated with 20 equiv. of D2O, it gave α/β/γ- and α/β/γ/δ-position deuteration products 36-D and 37-D in 53% and 35% yields, respectively (Fig. 5f, g). According to these results, we presumed that the Ni-complex could promote the chain-walking process to provide the terminal alkenes and then hydrogenation occurred, as no product could be detected in its absence.
Thereafter, a commonly used substrate for ring-opening experiments, ((1R,2S)−2-vinylcyclopropyl)benzene (76), was employed in the electroreductive hydrogenation system in the absence of the Ni-complex. The ring-opening product (77) and hydrogenation product (78) were detected by 1H NMR spectroscopy (Fig. 5h). According to literature reports73,74,75,76,77,78, 3d metal salts could react with silane to generate [M]-hydride species, which have a tendency to undergo migration insertion processes with alkenes (providing the intermediate I or I’). We speculated that the ring-opening product was formed through β-carbon elimination of intermediate I, which was followed by protonation to give product 77 (Fig. 5i). Intermediate I’ could then transform into hydrogenation to give product 78.
In order to gain further insight into the reaction mechanism, several cyclic voltammetry (CV) experiments were performed. CV experiments on Ph3SiCl gave the reductive peaks at Ep/2 = −2.4 V vs. Ag/Ag+ in DMF under Ar atmosphere (Supplementary Fig. 16). As shown in Fig. 5D, we systemically investigated the behaviors of TMSCl and TMSBr via 19F NMR tests. We found TMSCl/TMSBr could be mostly transformed into TMSF in very short time even without electricity, which was attributed to the reactions between TMSCl/TMSBr and TBABF4. The characteristic 19F NMR signal of TMSF could be detected in 3 min in the reaction system (13% for TMSCl, 14% for TMSBr vs. 20%, 4-fluoroanisole as internal standard), and there was no significant decline after stirred for 12 h (14% for TMSCl, 14% for TMSBr). These results indicated highly active chlorosilane/bromosilane could exist in the reaction system in the form of fluorosilane. And fluorosilane could efficiently promote the transformation as well (Supplementary Table 2, entry 4). As shown in Fig. 5E, the mixture of nickel catalyst and ligand exhibited two reduction peaks at –1.82 V and –2.55 V vs. Ag/Ag+ (Fig. 5E, red line), corresponding to the reduction of NiII/NiI and NiI/Ni0, respectively. When CV experiments of the nickel catalyst and ligand were conducted in the presence of increasing equivalents of Ph3SiCl, it was observed that the reduction current of NiII/NiI is positively correlated with the amount of Ph3SiCl, and the sign for the generation of Ni0 complex is significantly enhanced. These results suggested that both the NiII-ligand complex and Ph3SiCl underwent reduction reactions at the cathode and the in-situ generated Ni0 complex appears to be the active catalyst79.
Based on the mechanistic experiments and CV studies, a plausible mechanism for this electrochemical hydrogenation of unactivated alkenes was proposed (Fig. 5F). Iron salts were continuously generated at the anode, and the catalytic cycle was initiated by cathodic reduction of chlorosilane to provide silyl radical intermediate A. Then, intermediate A underwent the second one-electron reduction to give silyl anion intermediate B. Upon protonation of B with H2O, a silane intermediate C is formed. The generated iron species Fe-INT1A on the anode react with silane through a σ-bond metathesis reaction, resulting in the formation of hydrogenated iron species Fe-INT2A, and regenerating the halosilane to complete the silicon catalytic cycle. Subsequently, intermediate Fe-INT2A undergoes a migration insertion process with alkene substrate 1, leading to the formation of linear intermediate Fe-INT3A and branched intermediate Fe-INT3B. Both intermediates further undergo protonation with water, resulting in the production of hydrogenation products 2 and the generation of hydroxy iron species Fe-INT4A. Following a σ-bond metathesis reaction between Fe-INT4A and silicon hydride in the system, silanol D is formed, which remains stable in the system and can undergo gradual fluorination to re-enter the silicon catalytic cycle80. Notably, employing D2O instead of H2O could provide the electroreductive deuteration product. According to the mechanistic studies, a Ni-catalyzed cycle was involved in the transformation as well. And we have elucidated the Ni-catalyzed catalytic cycle as a minor pathway in the Supplementary Information (Supplementary Figs. 8, 9 and 11).
As showed in Table 1, Zn(+) or Mg(+) could not enable this transformation under the standard conditions, which was in line with the proposed [Fe]-H involved mechanism. However, when Zn(+) or Mg(+) was employed combined with the deposited Ni Foam cathode (coated by Fe particles after the first round electrolysis) under the standard conditions, provided the hydrogenation products in 83% and 37% yields respectively (Supplementary Table 11, entries 4 and 7). These results showed that the heterogeneous electrochemical hydrogenation process proceeded on the coated Ni Foam cathode might be involved in the transformation as well. (For more details about the heterogeneous process, please see the Supplementary Information)
Discussion
In conclusion, we report an efficient and facile electroreductive hydrogenation/deuteration of unactivated alkyl alkenes promoted by a catalytic amount of simple chlorosilane using H2O/D2O as the H/D source. In this protocol, low-cost Fe performed dual function: Fe worked as anode and the iron salts generated in situ behaved as the high active [Fe]-catalyst to promote the hydrogenation process. The Si-additive worked as an H-carrier from water to in situ generate a highly active silane under electroreductive conditions, which then combined with [Fe]-catalyst to form the [Fe]–H species for further hydrogenation. This approach showed broad substrate scope and good functional group compatibility. In addition to hydrogenation, employing D2O instead of H2O could provide the electroreductive deuterated products with good yields and excellent D-incorporation (up to >99%). Further late-stage carboxylation of biorelevant molecules demonstrated its potential application in the pharmaceutical industry. Mechanistic studies provided strong support for the key role of chlorosilane as a H-carrier. Further applications of the electroreductive hydrogenation strategy are currently underway in our laboratory.
Methods
General procedure of electroreductive hydrogenation of unactivated alkenes
The electrolysis process was carried out in an undivided cell with a Fe plate anode (10 mm × 20 mm × 0.10 mm) and a Ni Foam cathode (10 mm × 20 mm × 0.30 mm). To a 15 mL oven-dried undivided electrochemical cell equipped with a magnetic bar was added unactivated alkene (0.30 mmol, 1.0 equiv.), NiBr2.dtbpy (7.3 mg, 0.015 mmol, 5 mol%), Ph3SiCl (17.7 mg, 0.06 mmol, 20 mol%), H2O (16.2 mg, 0.9 mmol, 3.0 equiv.) and nBu4NBF4 (98.8 mg, 0.30 mmol, 1.0 equiv.) under Ar atmosphere. The electrolysis process was performed at 5.0 mA of constant current for 10 h at room temperature. After that, the electrodes were washed with EtOAc (3 × 5 mL) in an ultrasonic bath. H2O (20 mL) was added to the organic system, and the resulting mixture was extracted with EtOAc (3 × 20 mL) and the combined organic phase was washed with brine, dried by anhydrous MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography to furnish the desired product.
General procedure of electroreductive deuteration of unactivated alkenes
The electrolysis process was carried out in an undivided cell with a Fe plate anode (10 mm × 20 mm × 0.30 mm) and a Cu plate cathode (10 mm × 20 mm × 0.30 mm). To a 15 mL oven-dried undivided electrochemical cell equipped with a magnetic bar was added unactivated alkene (0.30 mmol, 1.0 equiv), NiBr2.dtbpy (7.3 mg, 0.015 mmol, 5 mol%), Ph3SiCl (17.7 mg, 0.06 mmol, 20 mol%), D2O (120 mg, 6 mmol, 20 equiv.) and nBu4NBF4 (98.8 mg, 0.30 mmol, 1.0 equiv.) under Ar atmosphere. The electrolysis process was performed at 5.0 mA of constant current for 10 h at room temperature. After that, the electrodes were washed with EtOAc (3 ×5 mL) in an ultrasonic bath. H2O (20 mL) was added to the organic system, and the resulting mixture was extracted with EtOAc (3 × 20 mL) and the combined organic phase was washed with brine, dried by anhydrous MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography to furnish the desired product.
Gram-scale synthesis of 2
The electrolysis process was carried out in an undivided cell with a Fe plate anode (5 cm × 2.5 cm × 0.5 mm) and a Ni Foam cathode (5 cm × 2.5 cm × 0.5 mm). To a 200 mL oven-dried undivided electrochemical cell equipped with a magnetic bar was added unactivated alkene (10 mmol, 1.0 equiv), NiBr2.dtbpy (242 mg, 0.5 mmol, 5 mol%), Ph3SiCl (600 mg, 2 mmol, 20 mol%), H2O (540 mg, 30 mmol, 3.0 equiv.) and nBu4NBF4 (3.29 g, 10 mmol, 1.0 equiv.) under Ar atmosphere. The electrolysis process was performed at 40 mA of constant current for 26 h at room temperature. After that, the electrodes were washed with EtOAc in an ultrasonic bath. H2O (20 mL) was added to the organic system, and 20 mL of 1 N HCl aq. was carefully added after the completion of the reaction to help the removal of the excess scrap iron from the reaction system, and the resulting mixture was extracted with EtOAc and the combined organic phase was washed with brine, dried by anhydrous MgSO4, filtered, and concentrated in vacuum. The crude product was purified by column chromatography to furnish the desired product (2) in 81% (1.94 g) yield.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. Extra data are available from the author upon request. Source data are provided with this paper.
References
Yoshida, J., Kataoka, K., Horcajada, R. & Nagaki, A. Modern strategies in electroorganic synthesis. Chem. Rev. 108, 2265–2299 (2008).
Francke, R. & Little, R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Waldvogel, S. R., Lips, S., Selt, M., Riehl, B. & Kampf, C. J. Electrochemical arylation reaction. Chem. Rev. 118, 6706–6765 (2018).
Yuan, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution reactions. Acc. Chem. Res. 52, 3309–3324 (2019).
Xiong, P. & Xu, H.-C. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 52, 3339–3350 (2019).
Jiao, K.-J., Xing, Y.-K., Yang, Q.-L., Qiu, H. & Mei, T.-S. Site-selective C–H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 53, 300–310 (2020).
Siu, J. C., Fu, N. & Lin, S. Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc. Chem. Res. 53, 547–560 (2020).
Shi, S.-H., Liang, Y. & Jiao, N. Electrochemical oxidation induced selective C–C bond cleavage. Chem. Rev. 121, 485–505 (2021).
Michael, K. H. et al. Pairing of aqueous and nonaqueous electrosynthetic reactions enabled by a redox reservoir electrode. J. Am. Chem. Soc. 144, 22641–22650 (2022).
Shen, T. & Lambert, T. H. Electrophotocatalytic diamination of vicinal C–H bonds. Science 371, 620–626 (2021).
Zeng, L. et al. Asymmetric-waveform alternating current-promoted silver catalysis for C–H phosphorylation. Nat. Synth. 2, 172–181 (2023).
Liu, Y., Li, P., Wang, Y. & Qiu, Y. Electroreductive cross-electrophile coupling (eXEC) reactions. Angew. Chem. Int. Ed. 62, e202306679 (2023).
Cai, C.-Y. et al. Photoelectrochemical asymmetric catalysis enables site- and enantioselective cyanation of benzylic C–H bonds. Nat. Catal. 5, 943–951 (2022).
Wang, M.-R. et al. Room temperature construction of vicinal amino alcohols via electroreductive cross-coupling of N-heteroarenes and carbonyls. J. Am. Chem. Soc. 145, 10967–10973 (2023).
Park, S. H. et al. Electrocatalytic access to azetidines via intramolecular allylic hydroamination: scrutinizing key oxidation steps through electrochemical kinetic analysis. J. Am. Chem. Soc. 145, 15360–15369 (2023).
Liang, K., Zhang, Q. & Guo, C. Enantioselective nickel-catalysed electrochemical cross-dehydrogenative amination. Nat. Synth. 2, 1184–1193 (2023).
Zhu, C., Chen, H., Yue, H. & Rueping, M. Electrochemical chemo- and regioselective arylalkylation, dialkylation and hydro(deutero)alkylation of 1,3-enynes. Nat. Synth. 2, 1068–1081 (2023).
Xu, Y. & Zhang, B. Recent advances in electrochemical hydrogen production from water assisted by alternative oxidation reactions. ChemElectroChem 6, 3214–3226 (2019).
Kondo, M., Tatewaki, H. & Masaoka, S. Design of molecular water oxidation catalysts with earth-abundant metal ions. Chem. Soc. Rev. 50, 6790–6831 (2021).
Liu, C., Wu, Y., Zhao, B. & Zhang, B. Designed nanomaterials for electrocatalytic organic hydrogenation using water as the hydrogen source. Acc. Chem. Res. 56, 1872–1883 (2023).
Shi, Z. et al. Recent advances in the electrochemical hydrogenation of unsaturated hydrocarbons. Curr. Opin. Electrochem. 28, 100713 (2021).
Yang, J., Qin, H., Yan, K., Cheng, X. & Wen, J. Advances in electrochemical hydrogenation since 2010. Adv. Synth. Catal. 363, 5407–5416 (2021).
Tomida, S., Tsuda, R., Furukawa, Saito, S. M. & Tajima, T. Electroreductive hydrogenation of activated olefins using the concept of site isolation. Electrochem. Commun. 73, 46–49 (2016).
Huang, B., Li, Y., Yang, C. & Xia, W. Electrochemical 1,4-reduction of α,β-unsaturated ketones with methanol and ammonium chloride as hydrogen Sources. Chem. Commun. 55, 6731–6734 (2019).
Li, J., He, L., Liu, X., Cheng, X. & Li, G. Electrochemical hydrogenation with gaseous ammonia. Angew. Chem. Int. Ed. 58, 1759–1763 (2019).
Wu, T., Nguyen, B. H., Daugherty, M. C. & Moeller, K. D. Paired electrochemical reactions and the on-site generation of a chemical reagent. Angew. Chem. Int. Ed. 58, 3562–3565 (2019).
Liu, X., Qiu, J., Cheng, X. & Li, G. Chemical-reductant-free electrochemical deuteration reaction using deuterium oxide. Angew. Chem. Int. Ed. 59, 13962–13967 (2020).
Yang, K., Feng, T. & Qiu, Y. Organo-mediator enabled electrochemical deuteration of styrenes. Angew. Chem. Int. Ed. 62, e202312803 (2023).
Zhang, X., Jiang, R. & Cheng, X. Electrochemical tandem olefination and hydrogenation reaction with ammonia. J. Org. Chem. 86, 16016–16025 (2021).
Kolb, S. & Werz, D. B. Site-selective hydrogenation/deuteration of benzylic olefins enabled by electroreduction using water. Chem. Eur. J. 29, e202300849 (2023).
Zhang, W. et al. Electroreductive dicarboxylation of unactivated skipped dienes with CO2. Angew. Chem. Int. Ed. 62, e202301892 (2023).
Kurimoto, A., Sherbo, R. S., Cao, Y., Loo, N. W. X. & Berlinguette, C. P. Electrolytic deuteration of unsaturated bonds without using D2. Nat. Catal. 3, 719–726 (2020).
Russo, C. et al. eHydrogenation: electrochemical hydrogen-free hydrogenation. Angew. Chem. Int. Ed. 62, e202309563 (2023).
Wang, C., Wu, X. & Xiao, J. Broader, greener, and more efficient: recent advances in asymmetric transfer hydrogenation. Chem. Asian J. 3, 1750–1770 (2008).
Wang, D. & Astruc, D. The golden age of transfer hydrogenation. Chem. Rev., 115, 6621–6686 (2015).
Werkmeister, S., Neumann, J., Junge, K. & Beller, M. Pincer-type complexes for catalytic (de)hydrogenation and transfer (de)hydrogenation reactions: recent progress. Chem. Eur. J. 21, 12226–12250 (2015).
Ai, W., Zhong, R., Liu, X. & Liu, Q. Hydride transfer reactions catalyzed by cobalt complexes. Chem. Rev. 119, 2876–2953 (2019).
Zhu, S.-F. Catalytic hydrogen transfer reactions. Chin. J. Chem. 39, 3211–3218 (2021).
Brunel, J. M. Scope, limitations and mechanistic aspects in the selective homogeneous palladium-catalyzed reduction of alkenes under transfer hydrogen conditions. Tetrahedron 63, 3899–3906 (2007).
Broggi, J. et al. The isolation of [Pd{OC(O)H}(H)(NHC)(PR3)] (NHC = N-Heterocyclic Carbene) and its role in alkene and alkyne reductions using formic acid. J. Am. Chem. Soc. 135, 4588–4591 (2013).
Wang, Y. et al. Transfer hydrogenation of alkenes using ethanol catalyzed by a NCP pincer iridium complex: scope and mechanism. J. Am. Chem. Soc. 140, 4417–4429 (2018).
Horn, S. & Albrecht, M. Transfer hydrogenation of unfunctionalised alkenes using N-heterocyclic carbeneruthenium catalyst precursors. Chem. Commun. 47, 8802–8804 (2011).
Zhang, G., Yin, Z. & Tan, J. Cobalt(ii)-catalysed transfer hydrogenation of olefins. RSC Adv. 6, 22419–22423 (2016).
Mazloomi, Z., Pretorius, R., Pàmies, O., Albrecht, M. & Diéguez, M. Triazolylidene iridium complexes for highly efficient and versatile transfer hydrogenation of C=O, C=N, and C=C bonds and for acceptorless alcohol oxidation. Inorg. Chem. 56, 11282–11298 (2017).
Dong, H. & Berke, H. A mild and efficient rhenium-catalyzed transfer hydrogenation of terminal olefins using alcoholysis of amine–borane adducts as a reducing system. J. Organomet. Chem. 696, 1803–1808 (2011).
Zhang, J., Mück-Lichtenfeld, C. & Studer, A. Photocatalytic phosphine-mediated water activation for radical hydrogenation. Nature 619, 506–513 (2023).
Kushner, D. J., Baker, A. & Dunstall, T. G. C. Pharmacological uses and perspectives of heavy water and deuterated compounds. J. Physiol. Pharmacol. 77, 79–88 (1999).
Wiberga, K. B. The deuterium isotope effect. Chem. Rev. 55, 713–743 (1995).
Gómez-Gallego, M. & Sierra, M. A. Kinetic isotope effects in the study of organometallic reaction mechanisms. Chem. Rev. 111, 4857–4963 (2011).
Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276–5297 (2019).
Li, N., Li, Y., Wu, X., Zhua, C. & Xie, J. Radical deuteration. Chem. Soc. Rev. 51, 6291–6306 (2022).
Norcott, P. L. Current electrochemical approaches to selective deuteration. Chem. Commun. 58, 2944–2953 (2022).
Boekell, N. G. & Flowers, R. A. Coordination-induced bond weakening. Chem. Rev. 122, 13447–13477 (2022).
Dong, J. et al. Formyl-selective deuteration of aldehydes with D2O via synergistic organic and photoredox catalysis. Chem. Sci. 11, 1026–1031 (2020).
Mirza-Aghayan, M., Boukherroub, R., Bolourtchian, M. & Hosseini, M. Palladium-catalyzed reduction of olefins with triethylsilane. Tetrahedron Lett. 44, 4579–4580 (2003).
Bart, S., Lobkovsky, E. & Chirik, P. Preparation and molecular and electronic structures of iron(0) dinitrogen and silane complexes and their application to catalytic hydrogenation and hydrosilation. J. Am. Chem. Soc. 126, 13794–13807 (2004).
Mirza-Aghayan, M., Boukherroub, R. & Bolourtchian, M. A mild and efficient palladium–triethylsilane system for reduction of olefins and carbon–carbon double bond isomerization. Appl. Organometal. Chem. 20, 214–219 (2006).
Lu, L., Siu, J. C., Lai, Y. & Lin, S. An electroreductive approach to radical silylation via the activation of strong Si–Cl bond. J. Am. Chem. Soc. 142, 21272–21278 (2020).
Guan, W. et al. An electrochemical strategy to synthesize disilanes and oligosilanes from chlorosilanes. Angew. Chem. Int. Ed. 62, e202303592 (2023).
Wang, Y. et al. Electrocarboxylation of aryl epoxides with CO2 for the facile and selective synthesis of β-hydroxy acids. Angew. Chem. Int. Ed. 61, e202207746 (2022).
Li, P. et al. Facile and general electrochemical deuteration of unactivated alkyl halides. Nat. Commun. 13, 3774 (2022).
Wang, Y. et al. Metal-free electrochemical carboxylation of organic halides in the presence of catalytic amounts of an organomediator. Angew. Chem. Int. Ed. 61, e202210201 (2022).
Zhao, Z. et al. Site-selective electrochemical C−H carboxylation of arenes with CO2. Angew. Chem. Int. Ed. 62, e202214710 (2023).
Yang, G., Wang, Y. & Qiu, Y. Electroreductive cross-electrophile coupling of aziridines and aryl bromides. Chem. Eur. J. 29, e202300959 (2023).
Knappke, C. E. et al. Reductive cross-coupling reactions between two electrophiles. Chem. Eur. J. 20, 6828–6842 (2014).
Weix, D. J. Methods and mechanisms for cross-electrophile coupling of Csp2 halides with alkyl electrophiles. Acc. Chem. Res. 48, 1767–1775 (2015).
Zhang, P., Le, C. C. & MacMillan, D. W. C. Silyl radical activation of alkyl halides in metallaphotoredox catalysis: a unique pathway for cross-electrophile coupling. J. Am. Chem. Soc. 138, 8084–8087 (2016).
Lucas, E. L. & Jarvo, E. R. Stereospecific and stereoconvergent cross-couplings between alkyl electrophiles. Nat. Rev. Chem. 1, 0065–0072 (2017).
Liu, J., Ye, Y., Sessler, J. L. & Gong, H. Cross-electrophile couplings of activated and sterically hindered halides and alcohol derivatives. Acc. Chem. Res. 53, 1833–1845 (2020).
Fu, H. et al. An asymmetric sp3–sp3 cross-electrophile coupling using ‘ene’-reductases. Nature 610, 302–307 (2022).
Zhang, B. et al. Ni-electrocatalytic Csp3–Csp3 doubly decarboxylative coupling. Nature 606, 313–318 (2022).
Wang, Y., He, Y. & Zhu, S. NiH-catalyzed functionalization of remote and proximal olefins: new reactions and innovative strategies. Acc. Chem. Res. 55, 3519–3536 (2022).
Lo, J. C., Gui, J., Yabe, Y., Pan, C.-M. & Baran, P. S. Functionalized olefin cross-coupling to construct carbon–carbon bonds. Nature 516, 343–348 (2014).
Lo, J. C. et al. Fe-catalyzed C–C bond construction from olefins via radicals. J. Am. Chem. Soc. 139, 2484–2503 (2017).
Kim, D., Rahaman, S. M. W., Mercado, B. Q., Poli, R. & Holland, P. L. Roles of iron complexes in catalytic radical alkene cross-coupling: a computational and mechanistic study. J. Am. Chem. Soc. 141, 7473–7485 (2019).
Kattamuri, P. V. & West, J. G. Hydrogenation of alkenes via cooperative hydrogen atom transfer. J. Am. Chem. Soc. 142, 19316–19326 (2020).
Tong, X., Yang, Z.-P., Aguilar, C. E. D. A. & Fu, G. C. Iron-catalyzed reductive cross-coupling of alkyl electrophiles with olefins. Angew. Chem. Int. Ed. 62, e202306663 (2023).
Jiao, K.-J. et al. Nickel-catalyzed electrochemical reductive relay cross-coupling of alkyl halides to aryl halides. Angew. Chem. Int. Ed. 59, 6520–6524 (2020).
Oguri, N., Takeda, N. & Unno, M. Facile synthesis of cyclic fluorosiloxanes. Chem. Lett. 44, 1506–1508 (2015).
Acknowledgements
Financial support from National Key R&D Program of China (2022YFA1503200) (Y.Q.), National Natural Science Foundation of China (Grant No. 22371149, 22188101) (Y.Q.) and (Grant No. 22301144) (Y.W.), the Fundamental Research Funds for the Central Universities (No. 63223015) (Y.Q.), Frontiers Science Center for New Organic Matter, Nankai University (Grant No. 63181206) (Y.Q.) and Nankai University are gratefully acknowledged
Author information
Authors and Affiliations
Contributions
Y.Q. supervised the project, and provided guidance on the project. Y.Q. and Y.W. conceived and designed the study and wrote the manuscript. Y.W., Q.W. and K.J. performed the experiments, mechanistic studies and revised the manuscript, L.W. and M.W. conducted the DFT calculation and revised the manuscript. Y.W., Q.W. and L.W. contributed equally to this work. All authors contributed to the analysis and interpretation of the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks Lingxiang Lu, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Wang, Q., Wu, L. et al. Electroreduction of unactivated alkenes using water as hydrogen source. Nat Commun 15, 2780 (2024). https://doi.org/10.1038/s41467-024-47168-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-47168-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
