
Abstract
In developing an organic light-emitting diode (OLED) panel for a foldable smartphone (specifically, a color filter on encapsulation) aimed at reducing power consumption, the use of a new optically clear adhesive (OCA) that blocks UV light was crucial. However, the incorporation of a UV-blocking agent within the OCA presented a challenge, as it restricted the traditional UV-curing methods commonly used in the manufacturing process. Although a visible-light curing technique for producing UV-blocking OCA was proposed, its slow curing speed posed a barrier to commercialization. Our study introduces a highly efficient photo-initiating system (PIS) for the rapid production of UV-blocking OCAs utilizing visible light. We have carefully selected the photocatalyst (PC) to minimize electron and energy transfer to UV-blocking agents and have chosen co-initiators that allow for faster electron transfer and more rapid PC regeneration compared to previously established amine-based co-initiators. This advancement enabled a tenfold increase in the production speed of UV-blocking OCAs, while maintaining their essential protective, transparent, and flexible properties. When applied to OLED devices, this OCA demonstrated UV protection, suggesting its potential for broader application in the safeguarding of various smart devices.
Similar content being viewed by others


Introduction
Smartphones have revolutionized the way we live, work, and interact with the world, making them an indispensable tool for communication, entertainment, and access to information for both personal and professional use. The recent emergence of smartphones with various form factors has further provided users with improved features such as a better viewing experience, increased durability, and enhanced portability1.
The intricate functionality of a smartphone is achieved through the seamless integration of distinct films, each designed to fulfill specific functions. In this respect, an adhesive is one of the most crucial components of a modern smartphone2,3. With the emergence of smartphones featuring various form factors, adhesives must fulfill multiple requirements beyond connecting the individual element films4,5,6. Specifically, they need to provide stress dissipation properties while maintaining a suitable balance between flexibility and elasticity7. In addition, they should exhibit high optical transparency and exceptional stability under conditions of moisture and heat3. Consequently, a well-designed adhesive can maintain its structural integrity even in extreme operating conditions that involve multiple deformations without exhibiting permanent deformation or failure.
Reducing power consumption is a crucial issue in smartphones for improving battery life, device performance, and lifespan, while minimizing environmental impact during manufacturing and charging. Indeed, recently, a state-of-the-art foldable smartphone has been introduced, featuring innovative technology that reduces the display panel’s power consumption by 25% (i.e., color filter (CF) on encapsulation, Fig. 1a)8. The new panel design implemented in the current foldable display eliminates the requirement for an additional polarizer layer, which is typically employed to mitigate external light reflection. This innovative approach necessitates the modification of existing layers to possess UV-blocking characteristics to replace the polarizer’s function of safeguarding the panel from external UV radiation. To incorporate a UV-blocking ability into the target layer, its fabrication process needs to be compatible with additives which efficiently absorb UV radiation, such as pigments or UV absorbers (UVAs). One approach to introduce these capabilities would involve fine-tuning the pigments in the CF layer, but it would require endeavours to ensure they absorb UV light and visible light simultaneously at desired wavelengths9,10,11. An alternative strategy would be imbedding UVAs into the CF layer. However, since the fabrication of these filters typically relies on UV-light curing12,13, substantial efforts are needed to overcome potential hindrances of UV-light curing associated with their high UV-light absorbance. Optically clear adhesives (OCA) layer could be regarded as the other alternative, but the preparation of UV-blocking adhesive still proves to be a complex task in the presence of UVAs, which impedes the effectiveness of the conventional photocuring method that relies on a UV-photoinitiator.

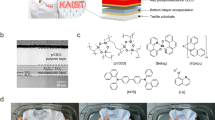
a Device structure of a foldable display with UV-blocking optically clear adhesive (OCA) film; TFT, BM, CF, ToE, TFE, and PDL denote thin film transistor, black matrix, color filter, touch-panel on encapsulation, thin film encapsulation and pixel define layer, respectively. b Schematic illustration of this work; here EnT and ET denote triplet-triplet energy transfer and electron transfer, respectively. Image of the prepared UV-blocking OCA is shown. c Comparison of adhesive film curing rate without or with UV absorbers (UVA). The OCA films were cured by previous photo-initiating system (PIS) with tertiary amines (black line) and a newly designed PIS with HNu 254 and Borate V (orange line). Error bars represent the standard deviation of uncertainty in the reproducibility (n = 5).
A simple approach is to utilize visible light instead of UV light for curing, with the aid of a photosensitizer that efficiently absorbs visible light14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32; while visible-light curing can technically be used to add the UVAs to the CF layer, it is likely inefficient and thus impractical, mainly due to the high absorption of visible light by the pigments12,13. Kwon and colleagues have made noteworthy progress in the development of OCA for foldable displays in a recent study33. They achieved a substantial reduction in the amount of photocatalyst (PC) used in visible-light photocuring by employing a compound that generates long-lived lowest triplet excited states (T1) as a PC34,35,36,37,38,39,40,41,42. This led to the successful production of UV-blocking OCA that exhibits high transparency in the visible-light range while also curing well under visible-light irradiation. However, a major drawback of this system was somehow the substantial reduction in curing speed when UVAs that do not absorb visible light is present (light dosage = 27,000 mJ m−2 for 95.0% conversion), which falls short of the production speed required for its commercialization (Fig. 1c, black line in the plot on the right). This limitation ultimately affects the availability of UV-blocking OCAs on the market.
Here, we have developed a highly efficient PIS that operates under visible-light irradiation, allowing for the production of UV-blocking OCAs at a much faster rate compared to the previously reported PIS33. Through a mechanistic analysis of existing PIS, we found that electron transfer (ET) and triplet-triplet energy transfer (EnT) between the PC and the UVAs predominate over those between the PC and the co-initiator. This reduces the efficiency of previous PIS, consequently impeding the curing rate. To address this issue, we carefully replaced the existing PC (i.e., 4Cz-IPN) with 4DP-IPN, aiming to minimize ET and triplet-triplet EnT efficiency with UVAs (Fig. 1b). Additionally, we introduced co-initiators, i.e., HNu 254 and Borate V, that facilitate faster electron transfer and PC regeneration compared to previously used amine-based co-initiators. With such a PIS, we were able to produce UV-blocking OCAs at a rate fast enough for commercialization under 452 nm visible-light irradiation (light dosage = 2400 mJ cm−2 for 98.1% conversion)43, which is approximately 10 times faster than the previous system (Fig. 1c, orange line in the plot on the right). The resulting film exhibited excellent UV-blocking capabilities (T345 nm ≈ 1.3%), while maintaining high optical transparency (T455 nm ≈ 100.0%) and possessing viscoelastic properties suitable for use in foldable displays as OCA. Finally, to evaluate the practical UV-blocking performance of the prepared film, we conducted a UV-blocking test on an organic light-emitting diode (OLED) device. Compared to the device without the UV-blocking OCA, the OLED covered with our OCA film exhibited less reduction in luminance under UV irradiation (λmax = 368 nm, 70 mW cm−2), demonstrating its excellent UV-blocking capabilities. We believe that the UV-blocking OCA film developed in this study will find extensive practical applications, extending beyond the foldable OLEDs showcased, in providing protection against UV radiation for diverse smart devices with various form factors including epidermal44,45,46, and wearable electronics47,48.
Results
Origin of rate inhibition in the previous PIS
In the previous system, 4Cz-IPN and tertiary amine analogs (i.e., DMAEA and DMAEAc) were selected as a PC and co-initiators, respectively (Fig. 2a)33. Here, the amines are oxidized to transfer an electron to the T1 state of the PC (3PC*), leading to the formation of α-amino radical species that act as initiators for the polymerization; 4Cz-IPN, a cyanoarene-based thermally activated delayed fluorescent (TADF) material, generates both lowest singlet (S1) and T1 excited states with negligible energy differences35,36, and its T1 concentration is approximately 100 times higher, predominantly contributing to ET (Supplementary Fig. 11). To obtain a clear understanding of the reduction in the curing rate resulting from UVAs in the previous system, we conducted a Stern-Volmer relationship experiments by monitoring photoluminescence (PL) decay quenching on 4Cz-IPN, in the presence of each UVA (Fig. 2b and Supplementary Fig. 12). Dimethyl 2-(4-(dimethylamino)benzylidene)malonate (UVA-1) and ethyl 2-cyano-3,3-diphenylacrylate (UVA-2) were used to block the UVA (315–400 nm) and UV-B (280–315 nm) regions, respectively. The Stern-Volmer plots revealed that the quenching rate constants of 4Cz-IPN with UVAs (kq (UVA-1) = 3.2 × 109 M−1 s−1 and kq (UVA-2) = 7.5 × 108 M−1 s−1) are close to the diffusion limit, which is about 10–100 times faster than photoinduced ET (PET) with DMAEAc (kPET = 2.1 × 107 M−1 s−1) (Fig. 2c)33. These observations were consistent with the hindered curing rate of existing PIS in the presence of UVAs. To elucidate the origin of this dissipation of the PC in excited state, our initial hypothesis was that the triplet-triplet EnT played a role in the quenching phenomena. Based on our observations, the conditions for a Dexter-type triplet-triplet EnT49, outlined below, were however not met for the case of PC and UVA-1, being i) EPC (T1) > EUVA (T1), ii) IPPC− EPC (T1) < EAUVA, and iii) EAUVA + EUVA (T1) < IPPC, where IP and EA stands for ionization potential and electron affinity, respectively (Fig. 2d and Supplementary Fig. 15). In contrast, these criteria were satisfied in the case of UVA-2, which is in accordance with previous reports that UVA-2 acts as a triplet quencher50,51. In fact, the feasibility of PET between PC and UVAs can be assessed via the ground and excited state redox potentials of both PC and UVAs (Supplementary Table 3). Careful data analysis indeed suggested that UVA-1 is likely acting as reductant to reduce 3PC* (Eox0 = 0.95 V vs SCE) in the reductive quenching pathway (Fig. 2a). On the contrary, in the case of UVA-2, the less favorable driving force for PET suggests that the primary quenching process would likely be the triplet-triplet EnT process rather than ET. It is anyway noted that a quantitative analysis of the triplet-triplet EnT and ET processes involving UVAs is challenging, and further detailed analysis is currently conducted. To sum up, the main factors contributing to the inhibition in curing rate are thought to be the triplet-triplet EnT and ET between the 3PC* and the UVAs, which happen at a considerably faster rate compared to the ET between 3PC* and tertiary amines.

a Proposed mechanism of the previous PIS33. b, c Stern-Volmer plots for PL quenching of 4Cz-IPN in ethyl acetate along with addition of b UVAs and c DMAEAc at RT33, respectively. Generally, PL quenching experiments were conducted by measurement of the change of PL decay with time-correlated single photon counting (TCSPC) techniques, but they were conducted with UVA-1 by monitoring the change in steady-state PL emission intensity maxima. d Electronic structure of PCs and UVAs obtained by experiments; here, IP and EA denote ionization potential and electron affinity, respectively (see Supplementary Table 2 for more detail). The driving forces (i.e., ∆Eox and ∆Ered) for each step of electron exchange in Dexter-type triplet-triplet EnT between 4Cz-IPN and UVAs are given; initial transfer pathway (orange dashed line) and following transfer pathway (gray dashed line).
Selection of a PC for the advanced PIS
We envisioned that by reducing the rates of triplet-triplet EnT and ET between 3PC* and UVAs, while simultaneously promoting ET between PC and co-initiators, the issue could be potentially resolved. To effectively suppress both triplet-triplet EnT and ET with UVAs concurrently, it becomes necessary to elevate the energy of highest occupied molecular orbital (HOMO) of PC, while maintaining the lowest unoccupied molecular orbital (LUMO) level less changed. This adjustment aims to reduce the driving force for the overall quenching processes involving UVAs. To resolve the issue, we screened the PC candidates established in our previous study, utilizing the systematic molecular catalyst design platform36. This platform enables systematic control over critical catalytic properties, such as redox potentials and the energies of S1 and T1, across a broad range. Among the PC candidates, 4DP-IPN emerged as an outstanding choice, primarily due to its higher HOMO energy, which results from substituting carbazole with the more electron-donating diphenylamine. In fact, the 4DP-IPN compound exhibited triplet energy of 2.38 eV and an excited state reduction potential (Ered* = 0.72 V), both approximately 0.30 eV and 0.75 eV lower than those of 4Cz-IPN, respectively (Fig. 3a). Interestingly, as compared to 4Cz-IPN, 4DP-IPN not only exhibited a longer triplet lifetime but also demonstrated superior T1 generation (Fig. 3b)33, which facilitate the reactive radical generation in the polymerization through the fast rate of ET process52,53,54,55. To fully leverage the excited-state reducing power of 4DP-IPN and maximize ET, we selected an electron-deficient co-initiator, an iodonium salt (i.e., HNu 254)56,57,58. According to the proposed mechanism, HNu 254 accepts electrons from 3PC*, followed by the dissociation of the C−I bond, generating the required radicals for the initiation59,60. Moreover, we chose a borate salt (i.e., Borate V) as an electron-rich co-initiator61,62. This co-initiator facilitated the production of the radicals necessary for initiation while simultaneously promoting the rapid regeneration of the PC by donating electrons to the intermediate PC radical cation (PC•+).

a Proposed mechanism of the newly designed PIS. b Kinetics simulation of relative T1 population of 4DP-IPN (orange line) and 4Cz-IPN (black line) at 10 ppm in ethyl acetate without or with UVAs (UVA-1 (2000 ppm) and UVA-2 (6400 ppm)) under continuous 452 nm irradiation. c, d Stern-Volmer plots for PL quenching of 4DP-IPN in ethyl acetate along with addition of c UVAs and d HNu 254 and Borate V, respectively. Generally, PL quenching experiments were conducted by measurement of the change of PL decay with TCSPC techniques, but they were conducted with UVA-1 by monitoring the change in steady-state PL emission intensity maxima. e Kinetics simulation of radical generation in the newly designed PIS (orange line) and the previous PIS (black line) without or with UVAs mimicking homopolymerization of EHA under continuous 452 nm irradiation (see Supplementary Table 5 for more detail).
Evaluation of newly design PIS
To evaluate the effectiveness of the newly designed PIS, the 3PC* dissipation of 4DP-IPN was first monitored through PL quenching experiments with UVAs (Fig. 3c and Supplementary Fig. 13). A decrease in the order of magnitude was observed for rate constants with UVAs (kq (UVA-1) = 1.8 × 108 M−1 s−1 and kq (UVA-2) = 2.3 × 108 M−1 s−1), as compared to those associated with 4Cz-IPN. We then conducted PL quenching experiments between 4DP-IPN and co-initiators. Based on the Stern-Volmer equation, we evaluated the rate constants as kPET = 2.0 × 109 M−1 s−1 for HNu 254 and kPET = 2.7 × 106 M−1 s−1 for Borate V (Fig. 3d), which correspond well with their redox potentials (Ered0 (HNu 254) = −0.53 V and Eox0 (Borate V) = 0.58 V). Notably, the rate constant between 4DP-IPN and HNu 254 was found to be close to the diffusion limit, and approximately 1000 times faster than that of Borate V. These results suggest that our PIS relies primarily on an oxidative quenching cycle mediated by HNu 254, with Borate V being responsible for PC regeneration. In contrast to the previous system (4Cz-IPN and tertiary amines), the rate of ET is now competitive with the rate of 3PC* quenching by the UVAs, ensuring sufficient curing rates even in the presence of UVAs. Besides rapid ET, the process of radical generation by one-electron oxidized/reduced co-initiators, subsequent to ET, is critical for the curing rate. Upon ET, HNu 254 quickly decomposes into phenyl radicals through concerted ET59,60. Additionally, boranyl radicals generated from Borate V quickly produce alkyl radical species (kdiss. ~ 1011 s−1), rendering back electron transfer (BET) negligible61,62. However, amine-based co-initiators have a much slower rate constant (kamine = 2.3 × 105 M−1 s−1, Supplementary Fig. 18) for the generation of α-amino radical species after PET with PCs. Using the obtained rate constants, we performed simulations to estimate the amount of radical species generated by each PIS over time with the same amounts (i.e., 10 ppm) of PCs (Fig. 3e); as the quantitative analysis of the triplet-triplet EnT and ET processes with UVAs is challenging, to be simplified, triplet-triplet EnT with UVAs was considered as a primary quenching pathway of 3PC* without considering polymerization-factors such as radical propagation and termination. Strikingly, our results indicate that the newly designed PIS generates significantly more initiating radicals, approximately 1000 times more, particularly in the presence of UVAs. This should allow for fast and efficient curing to prepare UV-blocking OCAs.
Preparation of UV-blocking OCAs
Based on the newly designed PIS, we prepared OCA films. Typically, acrylic OCA films are prepared using a two-step photocuring process (Supplementary information for the details)63. First, bulk polymerization of the monomer mixture is performed to obtain the appropriate viscosity for achieving the desired thickness of the final OCA film with high reproducibility. Then, the acrylic syrup, which has the desired viscosity, is cast to the desired thickness, leading to the formation of the ultimate OCA film through photocuring. Unlike conventional photoinitiator-based methods, our approach eliminates the need for additional PC incorporation in post-bulk polymerization. This is because the PC is regenerated through an ET process with a co-initiator in the catalytic cycle33,64. In our current experiments, we synthesized acrylic syrup through bulk polymerization of monomers under 455 nm irradiation. Subsequently, the resulting acrylic syrup was cast to a thickness of approximately 50 μm between two 100 μm-thick silicon-treated release films, followed by irradiation with 452 nm light for film curing.
We first reproduced OCAs using the same conditions as in our previous system, which involves the use of 4Cz-IPN and tertiary amines29. For this purpose, we prepared OCAs using the same combination of monomers, i.e., n-butyl acrylate (BA) and 4-hydroxybutyl acrylate (HBA) as indicated in Table 1 (entries 1 and 2) and Supplementary Table 6. Consequently, the OCA film required a light dosage of 5400 mJ cm−2 to achieve a film conversion of over 95%. However, in the presence of UVAs, the light dosage required for film curing increased to 27,000 mJ cm−2, which is approximately an order of magnitude higher than the light intensity needed for curing conventional OCAs43. Such a high light dosage is impractical for commercialization purposes.
Next, we prepared OCAs using HNu 254 and Borate V as co-initiators (500 ppm and 300 ppm with respect to monomers, respectively), while maintaining the same type and amount of PC (10 ppm of 4Cz-IPN with respect to monomers). Here, we replaced BA (Tg, poly(BA) ≈ −48 °C) with 2-ethylhexyl acrylate (EHA, Tg, poly(EHA) ≈ −58 °C) as the monomer65. This substitution offers the advantage of reducing the glass transition temperature (Tg) and providing significant flexibility even at RT. As a result, it imparts suitable adhesive strength and peel adhesion properties66. Furthermore, in this study, we optimized the process of incorporating UVAs. Previously, acrylic syrup was first prepared through bulk polymerization, followed by the addition of UVAs (0.3 and 1 phr (per hundred resin) for UVA-1 and UVA-2, respectively) and subsequent film formation through photocuring67. However, to enhance reproducibility, in this study, UVAs were added during the bulk polymerization step to produce the acrylic syrup, which was then cured to the film. As anticipated, the use of HNu 254 and Borate V exhibited significantly faster kinetics in film curing (light dosage = 3000 mJ cm−2 for 94.3% conversion in Table 1, entry 3; see Supplementary Table 7 for comprehensive data on the optimization processes) compared to tertiary amines. However, the presence of UVAs inevitably reduced both bulk polymerization and curing processes, with film curing necessitating a substantial amount of light (light dosage = 6000 mJ cm−2 in Table 1, entry 4).
Finally, we prepared OCAs using 4DP-IPN as the PC along with HNu 254 and Borate V as co-initiators (Table 1, entries 5–10; see Supplementary Tables 8−11 for comprehensive data regarding the optimization processes and reproducibility tests). By systematically varying the amount of PC and co-initiators, we identified the optimal conditions, achieving a catalyst reduction to 3 ppm—approximately three times lower than the previous PC, 4Cz-IPN (Table 1, entries 9 and 10). Under these optimized conditions, we achieved a conversion of 97.8% with a remarkably low light dosage of 600 mJ cm−2. Even in the presence of UVAs, a conversion of 97.8% was attained with only 2400 mJ cm−2 of light dosage, which is typically required integrated light intensity for conventional OCA film preparation43. These compelling results indicate the feasibility of mass-production of UV-blocking OCAs using this method. Furthermore, we made an intriguing observation within our system. We observed a strong dependency of monomer conversion on the light intensity employed. Higher irradiation intensity (i.e., 100 mW cm−2) led to accelerated initial kinetics; however, the conversion reached a saturation point without further increase (Supplementary Fig. 21). This behavior can be attributed to the excessive generation of reactive radical species in the early stages, leading to accelerated termination between radical species. To address this challenge, we intentionally chose lower irradiation intensity for both bulk polymerization and film curing processes (i.e., 25 mW cm−2 and 10 mW cm−2, respectively), leading to improved monomer conversion. Under these conditions, we achieved a significant gel content in the resulting OCA films, without requiring additional crosslinkers (see Table 2). This outcome is likely attributed to the involvement of hydroxy groups in the crosslinking reactions between polymer chains in the presence of radical intermediates66,68. We are currently conducting an in-depth investigation into the detailed crosslinking mechanism involving the hydroxy group.
Properties of the resulted OCAs
We tested the optical properties of the prepared OCA film and confirmed that it possesses UV-blocking capabilities comparable to commercial standards. To determine the film’s transmittance, we employed a UV/vis spectrophotometer (Fig. 4c and Supplementary Fig. 23). The UV-blocking OCA exhibited remarkable effectiveness in blocking UV light (T345 nm ≈ 1.3%), while maintaining exceptional transparency in the visible-light range (T455 nm ≈ 100.0%). Given that the UV-blocking ability did not significantly differ across the OCA thickness range of 50–200 μm, we proceeded with a 50 μm thickness for subsequent experiments, aligning with the standard for foldable OCA films (see Supplementary Fig. 23a). We next evaluated the mechanical and adhesive properties of the prepared OCA film (Table 2 and Supplementary Figs. 25−28). As a control, a commercially available OCA for foldable displays (3 M, CEF 3602) was also tested. To verify the applicability as the OCA for foldable displays, we measured the viscoelastic properties through dynamic mechanical analysis (DMA)69. As illustrated in Table 2, entry 5, the UV-blocking OCA prepared in optimized conditions exhibits decent strain recovery, stress relaxation, and maximum stress characteristics that are comparable to those of CEF 3602.

a Schematic illustration of a UV-blocking test; ETL, EML, and HTL denote electron transport layer, emission layer, and hole transport layer, respectively. b Electroluminescence (EL) emission spectra of tested OLED devices. Images of tested OLED devices and their EL emissions under the current (J0 = 100 mA cm−2) are shown (inset). c UV/vis transmittance of prepared OCAs and UV LED emission spectra. The transmittances of non-UV-blocking OCA film (black line) and UV-blocking OCA film (orange line) were measured with quartz plates as a cover glass. d Schematic illustration of the operation of UV-blocking test. During the measurement of luminance levels over time with a 10 min interval, the UV LED was momentarily deactivated for 5 s to prevent the detection of photoluminescent signals originating from the OLEDs due to UV irradiation. e, f Time-dependent changes of e luminance (L0 ~ 3690 nit) and f voltage (V0 ~ 7.20 V) of blue OLED covered by the prepared non-UV-blocking OCA film (black line) and UV-blocking OCA film (orange line) exposed by UV irradiation (λmax = 368 nm, 70 mW cm−2) under constant current (J0 = 100 mA cm−2). Changes in the luminance and voltage of blue OLED were recorded every 10 min.
We then proceeded to evaluate the peel strength of the prepared OCAs on both glass (as an alternative to ultrathin glass, UTG) and colorless polyimide (CPI), commonly used as cover windows in foldable displays (Table 2). The observed peel strength values are generally higher on the CPI than those on the glass, which would be attributed to the presence of phthalic anhydride derivatives70 and benzidine derivatives71 giving an abundant hydrogen bond site and the higher polarity to make interfacial interactions much stronger. In the case of the OCA without UVAs, the peel strength was similar to that of CEF 3602. However, a slight reduction in peel strength was observed in the UV-blocking OCA. This reduction may be attributed to UVAs with high concentrations migrating towards the surface of the acrylic adhesive film72, or acting as defects that hinder adhesive properties73. Nevertheless, it is important to highlight that despite this slight decrease, the peel strength remains sufficient for the effective utilization of the OCA in foldable applications. To assess the high- and low-temperature reliability of the UV-blocking OCA film, we examined the viscoelastic behaviours of the OCA film across various temperatures74. The storage modulus (Gʹ), loss modulus (Gʺ), and damping factor (tan δ) were measured by a rheometer through a dynamic temperature sweep ranging from −50 °C to 90 °C (Supplementary Table 12). Notably, the measured values at all temperatures met commercial OCA requirements. At low temperatures, the storage modulus value (Gʹ = 150.3 kPa at −20 °C) was found to be slightly higher than that of CEF 3602 (Gʹ = 115.0 kPa at −20 °C), yet it still meets the commercial standard where the storage modulus of commercially available OCAs is within the 100–150 kPa range75,76. Furthermore, the Tg of the OCA film, measured at −39.7 °C during a dynamic temperature sweep, is consistent with the observed low Gʹ at −20 °C, as detailed in Supplementary Fig. 30. We also conducted dynamic folding tests to assess the suitability of the prepared UV-blocking OCAs for foldable displays (Supplementary Figs. 31, 32)46,69. The test specimen structure was designed based on an actual foldable smartphone with a fixed radius of curvature of 1.5 mm and a folding cycle of 0.5 Hz. Remarkably, the prepared OCAs demonstrated excellent folding durability within 5 × 104 folding cycles without cracks even at low temperatures77, nearly meeting the specifications for foldable displays with similar values of surface texture (∆Z) to CEF 3602 (Supplementary Fig. 32), thanks to their low storage modulus and high strain recovery at −20 °C (Supplementary Table 12).
UV-blocking capability of the OCA
Finally, we assessed the UV-blocking capability of our OCA film when employed in blue fluorescent OLEDs (Fig. 4 and Supplementary Fig. 4). The choice of blue fluorescent OLEDs for experiments stemmed from the inherent vulnerability of their constituent molecules to UV irradiation, in contrast to their counterparts used in red and green OLEDs78,79,80. OLEDs covered by the OCA with and without UVA were operated at a constant current density of J0 = 100 mA cm−2 during continuous exposure to UV irradiation, exhibiting a peak wavelength of λmax = 368 nm and a high intensity of 70 mW cm−2. We compared the degradation in the optoelectronic performance of these devices with and without UV blockage. Ideally, for UV-blocking testing, all layers, including the CF layer, should be integrated to create an OLED device. However, due to practical difficulties in fabricating CF in the laboratory, we opted for a simplified device structure (Fig. 4a). Specifically, Fig. 4e, f show the temporal evolution of normalized luminance (L) with respect to its initial value (L0) at t = 0, and the voltage rise (i.e., ∆V = V – V0) with V0 as the initial voltage at t = 0, respectively (see Supplementary Fig. 24 for more detail). The UV-blocking OCA effectively shielded the blue OLED from UV exposure for 30 h, maintaining its normalized luminance (L/L0) at 96.3%; On the other hand, the device covered by the non-blocking OCA experienced a significant reduction in its L/L0 to 39.4%. The steeper voltage rise of ∆V = 4.0 V vs 0.8 V for the non-UV-blocked and UV-blocked devices, respectively, indicates that the former device generated more degradation-induced defects due to stimulated molecular dissociation caused by UV exposure. We observed an unusual increase in the luminance during the early stages of UV irradiation, the underlying cause of which is currently under active investigation. The operational stability of OLEDs covered by the UVA-containing OCA clearly demonstrates the successful UV-blocking ability of our OCAs, even under harsh UV irradiation. This suggests that our UV-blocking OCA holds potential for the commercialization, boasting an extended product lifespan.
Discussion
In summary, we have developed a highly efficient PIS that operates under visible-light irradiation. Through a thorough analysis of existing PIS, we identified the triplet-triplet EnT and ET with UVAs as the main factors limiting the curing rate. By employing the PC with higher HOMO energy and incorporating iodonium and borate salts as co-initiators with faster ET and radical generation, we addressed this issue. The resulting PIS allowed us to produce UV-blocking OCAs, achieving a high conversion (98.1%) with a light dosage of 2400 mJ cm−2, approximately 10 times lower than the previous system33. The lower light dosage and fast curing rate in our PIS highlight its economic potentials for the commercialization, making it comparable to the conventional UV-curing process43. The prepared OCAs exhibited excellent UV-blocking capabilities while maintaining high optical transparency and demonstrated fascinating physicochemical properties, including adhesive properties, viscoelasticity, and folding durability, comparable to those of the commercial OCA. Moreover, we successfully demonstrated their practical UV-blocking capabilities by applying them to OLED devices, indicating their potential for various applications that require UV-blocking abilities. We believe that the UV-blocking OCA prepared here will have enormous practical applications beyond the foldable OLEDs, providing protection against UV radiation for diverse smart devices with various form factors. Furthermore, the newly developed PIS is believed to be used as a key PIS not only for the preparation of (functional) OCA film, but also visible-light-based 3D/4D printing81,82,83,84,85, optically clear resins33, soft actuator/robotics86,87, dental resin88,89, and more90.
Methods
General experimental procedures for synthesis of acrylic syrup
A 20 ml vial (glass, Sungho SIGMA) equipped with a stirring bar was charged with monomer mixture, PCs, and co-initiators. Afterwards, the vial was capped with a rubber septum sealed with parafilm and degassed with 99.999% N2 for 30 min. PCs and co-initiators were used after dilution in acrylate monomers for reproducibility. In the case of addition of UVAs, they were added in this reaction solution before the bulk polymerization in newly designed PIS. During the degassing process, the reaction solution was kept in a dark condition to block the room light preventing undesired polymerization. Subsequently, acrylic syrup was synthesized under irradiation of 3 W MR 16 LED (λmax = 455 nm, 25–100 mW cm−2) at RT.
General experimental procedures for film curing
For film curing, two 15 W string-type blue LEDs (λmax = 452 nm) were used, and their intensities were set up as 10 mW cm−2. Film curing was carried out by additionally irradiating visible light to the prepared acrylic syrup. Since the PIS used in this process reuses the PC within the catalyst cycle of the acrylic syrup preparation process, no further additives were added during the film process. Moreover, a crosslinking agent was also not included in this process. For film curing, acrylic syrup was coated between two release films and casted to a thickness of 50 μm using a film casting applicator. The samples for measurements of optical and viscoelastic properties were cured for 40 min for the previous PIS (i.e., 4Cz-IPN and tertiary amines) and 30 min for this PIS (i.e., 4DP-IPN, HNu 254 and Borate V) to obtain fully cured OCAs.
General experimental procedures for UV-blocking test of OLEDs
Both an OLED covered by non-UV-blocking OCA and the same device with UV-blocking OCA underwent simultaneous illumination using a UV LED source (Kessil, PR160L) with a peak wavelength of λmax = 368 nm and an intensity of 70 mW cm−2 at RT. Both devices were consistently operated under a constant current density of J = 25–100 mA cm−2, utilizing a multi-channel source measure unit (Agilent Technologies, U2722A). During the measurement of luminance levels over time with a 10 min interval, we momentarily deactivated the UV LED for 5 s to prevent the detection of photoluminescent signals originating from the OLEDs due to UV irradiation. Upon turning off the UV LED, a waiting period of 3 s was included, followed by a 2 s measurement of electroluminescence from the OLEDs. This measurement involved the averaging of 20 collected signals, each spaced at 0.1 s intervals, in order to minimize detection errors. Degradation of OLEDs’ luminance over time was captured using a photodiode (Thorlabs, SM1PD1A) and subsequently recorded through a data logger (Keysight, DAQ970A). All stages of OLED testing were fully automated through an in-house LabVIEW virtual instruments.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information.
References
Kim, D. W. et al. Fabrication of practical deformable displays: advances and challenges. Light. Sci. Appl. 12, 61 (2023).
Hayashida, S. et al. Impact strength of joints bonded with high-strength pressure-sensitive adhesive. Int. J. Adhes. Adhes. 56, 61–72 (2015).
Abrahamson, T. et al. Optically clear adhesives for OLED. Lumin. - OLED Technol. Appl. (2020).
Han, Z. & Fina, A. Thermal conductivity of carbon nanotubes and their polymer nanocomposites: a review. Prog. Polym. Sci. 36, 914–944 (2011).
Taboada, G. M. et al. Overcoming the translational barriers of tissue adhesives. Nat. Rev. Mater. 5, 310–329 (2020).
Hillmyer, M. A. & Tolman, W. B. Aliphatic polyester block polymers: renewable, degradable, and sustainable. Acc. Chem. Res. 47, 2390–2396 (2014).
Salmon, F. et al. Modeling the mechanical performance of a foldable display panel bonded by 3M optically clear adhesives. SID Symp. Dig. Tech. Pap. 48, 938–941 (2017).
Samsung Display Newsroom. Samsung Display unveils new Eco2 OLEDTM that reduces power consumption, offers enhanced under-panel camera, and features eco-friendly design. https://global.samsungdisplay.com/28402 (2021).
Sakong, C. et al. The synthesis of thermally-stable red dyes for LCD color filters and analysis of their aggregation and spectral properties. Dyes Pigm. 88, 166–173 (2011).
Choi, J. et al. Synthesis and characterization of thermally stable dyes with improved optical properties for dye-based LCD color filters. N. J. Chem. 36, 812–818 (2012).
Namgoong, J. W. et al. Aryloxy- and chloro-substituted zinc(II) phthalocyanine dyes: synthesis, characterization, and application for reducing the thickness of color filters. Dyes Pigm. 154, 128–136 (2018).
Williams, C. et al. Grayscale-to-color: scalable fabrication of custom multispectral filter arrays. ACS Photonics 6, 3132–3141 (2019).
Jiang, L. et al. Microfabrication of a color filter array utilizing colored SU-8 photoresists. Appl. Opt. 59, 137–145 (2020).
Yagci, Y. Wavelength flexibility in photoinitiated cationic polymerization. Macromol. Symp. 215, 267–280 (2004).
Tasdelen, M. A., Lalevée, J. & Yagci, Y. Photoinduced free radical promoted cationic polymerization 40 years after its discovery. Polym. Chem. 11, 1111–1121 (2020).
Lalevée, J. et al. Metal and metal-free photocatalysts: mechanistic approach and application as photoinitiators of photopolymerization. Beilstein J. Org. Chem. 10, 863–876 (2014).
Xiao, P. et al. Visible light sensitive photoinitiating systems: recent progress in cationic and radical photopolymerization reactions under soft conditions. Prog. Polym. Sci. 41, 32–66 (2015).
Dietlin, C. et al. Photopolymerization upon LEDs: new photoinitiating systems and strategies. Polym. Chem. 6, 3895–3912 (2015).
Sun, K., Xiao, P., Dumur, F. & Lalevée, J. Organic dye-based photoinitiating systems for visible-light-induced photopolymerization. J. Polym. Sci. 59, 1338–1389 (2021).
Shao, J., Huang, Y. & Fan, Q. Visible light initiating systems for photopolymerization: status, development and challenges. Polym. Chem. 5, 4195–4210 (2014).
Zhang, G. et al. W. Free radical polymerization initiated and controlled by visible light photocatalysis at ambient temperature. Macromolecules 44, 7594–7599 (2011).
Corrigan, N., Yeow, J., Judzewitsch, P., Xu, J. & Boyer, C. Seeing the light: advancing materials chemistry through photopolymerization. Angew. Chem. Int. Ed. 58, 5170–5189 (2019).
Xu, J., Jung, K., Atme, A., Shanmugam, S. & Boyer, C. A robust and versatile photoinduced living polymerization of conjugated and unconjugated monomers and its oxygen tolerance. J. Am. Chem. Soc. 136, 5508–5519 (2014).
Theriot, J. C. et al. Organocatalyzed atom transfer radical polymerization driven by visible light. Science 352, 1082–1086 (2016).
Tucker, B. S., Coughlin, M. L., Figg, C. A. & Sumerlin, B. S. Grafting-From proteins using metal-free PET-RAFT polymerizations under mild visible-light irradiation. ACS Macro Lett. 6, 452–457 (2017).
Zhou, H. et al. Initiator-activation strategy-enabled organocatalyzed reversible-deactivation radical polymerization driven by light. Angew. Chem. Int. Ed. 62, e202304461 (2023).
Zhao, Y. et al. Controlled radical copolymerization of fluoroalkenes by using light-driven redox-relay catalysis. Nat. Synth. 2, 653–662 (2023).
Stevens, L. M. et al. Counting all photons: efficient optimization of visible light 3D printing. Adv. Mater. Technol. 8, 2300052 (2023).
Chung, K. U. & Page, Z. A. Boron-methylated dipyrrometheneasa green light activated type I photoinitiator for rapid radical polymerizations. J. Am. Chem. Soc. 145, 17912–17918 (2023).
Corakci, B., Hacioglu, S. O., Toppare, L. & Bulut, U. Long wavelength photosensitizers in photoinitiated cationic polymerization: the effect of quinoxaline derivatives on photopolymerization. Polymer 54, 3182–3187 (2013).
Dumur, F. Recent advances on coumarin-based photoinitiators of polymerization. Eur. Polym. J. 163, 110962 (2022).
Dumur, F. Recent advances on photobleachable visible light photoinitiators of polymerization. Eur. Polym. J. 186, 111874 (2023).
Back, J. H. et al. Visible light curable acrylic resins toward UV-light blocking adhesives for foldable displays. Adv. Mater. 35, 2204776 (2023).
Speckmeier, E., Fischer, T. G. & Zeitler, K. A toolbox approach to construct broadly applicable metal-free catalysts for photoredox chemistry: deliberate tuning of redox potentials and importance of halogens in donor-acceptor cyanoarenes. J. Am. Chem. Soc. 140, 15353–15365 (2018).
Bryden, M. A. & Zysman-Colman, E. Organic thermally activated delayed fluorescence (TADF) compounds used in photocatalysis. Chem. Soc. Rev. 50, 7587–7680 (2021).
Singh, V. K. et al. Highly efficient organic photocatalysts discovered via a computer-aided-design strategy for visible-light-driven atom transfer radical polymerization. Nat. Catal. 1, 794–804 (2018).
Lee, Y. & Kwon, M. S. Emerging organic photoredox catalysts for organic transformations. Eur. J. Org. Chem. 38, 6028–6043 (2020).
Wu, C., Liu, W., Lim, C. H., Miyake, G. & Lim, C. H. Rational design of photocatalysts for controlled polymerization: effect of structures on photocatalytic activities. Chem. Rev. 122, 5476–5518 (2022).
Corbin, D. A. & Miyake, G. M. Photoinduced organocatalyzed atom transfer radical polymerization (O-ATRP): precision polymer synthesis using organic photoredox catalysis. Chem. Rev. 122, 1830–1874 (2022).
Lee, Y. et al. A water-soluble organic photocatalyst discovered for highly efficient additive-free visible-light-driven grafting of polymers from a protein at ambient and aqueous enviornments. Adv. Mater. 34, 2108446 (2022).
Kwon, Y. et al. Formation and degradation of strongly reducing cyanoarene-based radical anions towards efficient radical anion-mediated photoredox catalysis. Nat. Commun. 14, 92 (2023).
Uddin, A. et al. Do the twist: efficient heavy-atom-free visible light polymerization facilitated by spin-orbit charge transfer inter-system crossing. Angew. Chem. Int. Ed. 62, e202219140 (2023).
Takahashi, H. & Watanabe, A. (Dexerials Co.), Method of manufactureing image display device. JP Patent 2020-46557 A (2020).
Kwak, M. K., Jeong, H. E. & Suh, K. Y. Rational design and enhanced biocompatibility of a dry adhesive medical skin patch. Adv. Mater. 23, 3949–3953 (2011).
Kim, J. et al. Epidermal electronics with advanced capabilities in near-field communication. Small 11, 906–912 (2015).
Lim, D. et al. Carboxyethyl acrylate incorporated optically clear adhesives with outstanding adhesion strength and immediate strain recoverability for stretchable electronics. Chem. Eng. J. 437, 135390 (2022).
Liu, Y., Pharr, M. & Salvatore, G. A. Lab-on-Skin: a review of flexible and stretchable electronics for wearable health monitoring. ACS Nano 11, 9614–9635 (2017).
Lim, H. R. et al. Advanced soft materials, sensor integrations, and applications of wearable flexible hybrid electronics in healthcare, energy, and environment. Adv. Mater. 32, 1901924 (2020).
Praveen, V. K., Ranjith, C., Bandini, E., Ajayaghosh, A. & Armaroli, N. Oligo(phenylenevinylene) hybrids and self-assemblies: versatile materials for excitation energy transfer. Chem. Soc. Rev. 43, 4222–4242 (2014).
Kikuchi, A., Nakabai, Y., Oguchi-Fujiyama, N., Miyazawa, K. & Yagi, M. Energy-donor phosphorescence quenching study of triplet-triplet energy transfer between UV absorbers. J. Lumin. 166, 203–208 (2015).
Kockler, J., Oelgemöller, M., Robertson, S. & Glass, B. D. Photostability of sunscreens. J. Photochem. Photobiol. C. Photochem. Rev. 13, 91–110 (2012).
Song, Y. et al. Organic photocatalyst for ppm-level visible-light-driven reversible addition-fragmentation chain-transfer (RAFT) polymerization with excellent oxygen tolerance. Macromolecules 52, 5538–5545 (2019).
Back, J. H. et al. Synthesis of solvent-free acrylic pressure-sensitive adhesives: via visible-light-driven photocatalytic radical polymerization without additives. Green. Chem. 22, 8289–8297 (2020).
Back, J. H. et al. Visible-light-curable solvent-free acrylic pressure-sensitive adhesives via photoredox-mediated radical polymerization. Molecules 26, 385–396 (2021).
Ko, H. M., Lee, C. W. & Kwon, M. S. Degradation behavior of donor‐acceptor cyanoarenes‐based organic photocatalysts. ChemCatChem 15, e202300661 (2023).
Al Mousawi, A. et al. Carbazole scaffold based photoinitiator/photoredox catalysts: toward new high performance photoinitiating systems and application in LED projector 3D printing resins. Macromolecules 50, 2747–2758 (2017).
Al Mousawi, A. et al. Carbazole derivatives with thermally activated delayed fluorescence property as photoinitiators/photoredox catalysts for LED 3D printing technology. Macromolecules 50, 4913–4926 (2017).
Timpe, H. et al. Photoinitiated polymerization of acrylates and methacrylates with decahydroacridine-l,8-dione/onium salt initiator systems. Macromolecules 26, 4560–4566 (1993).
Romańczyk, P. P. & Kurek, S. S. The Reduction potential of diphenyliodonium polymerisation photoinitiator is not −0.2 V vs. SCE. A computational study. Electrochim. Acta 255, 482–485 (2017).
Romańczyk, P. P. & Kurek, S. S. Reliable reduction potentials of diaryliodonium cations and aryl radicals in acetonitrile from high-level ab initio computations. Electrochim. Acta 351, 136404 (2020).
Chatterjee, S. et al. Photochemistry of carbocyanine alkyltriphenylborate salts: intra-ion-pair electron transfer and the chemistry of boranyl radicals. J. Am. Chem. Soc. 112, 6329–6338 (1990).
Polykarpov, A. Y., Hassoon, S. & Neckers, D. C. Tetramethylammonium tetraorganylborates as coinitiators with 5,7-diiodo-3-butoxy-6-fluoronein visible light polymerization of acrylates. Macromolecules 29, 8274–8276 (1996).
Behling, E. R. et al. (3M innovative properties company), Acrylic-based flexible assembly layer. US Patent 10640689 B2 (2020).
Zeitler, K. Photoredox catalysis with visible light. Angew. Chem. Int. Ed. 48, 9785–9789 (2009).
BASF. Acrylic & methacrylic monomers. https://chemicals.basf.com/global/en/Petrochemicals/specialty-monomers.html (2021).
Czech, Z. Synthesis and cross-linking of acrylic PSA systems. J. Adhes. Sci. Technol. 21, 625–635 (2007).
Lee, J. H. et al. Stretchable and recoverable acrylate-based pressure sensitive adhesives with high adhesion performance, optical clarity, and metal corrosion resistance. Chem. Eng. J. 406, 126800 (2021).
Ballard, N. & Asua, J. M. Radical polymerization of acrylic monomers: an overview. Prog. Polym. Sci. 79, 40–60 (2018).
Park, H. et al. Tailoring pressure sensitive adhesives with H6XDI-PEG diacrylate for strong adhesive strength and rapid strain recovery. Adv. Funct. Mater. 33, 2305750 (2023).
Abdulhamid, M. A. et al. Plasticization-resistant carboxyl-functionalized 6FDA-polyimide of intrinsic microporosity (PIM-PI) for membrane-based gas separation. Ind. Eng. Chem. Res. 59, 5247–5256 (2020).
Hasegawa, M. et al. Solution-processable colorless polyimides with ultralow coefficis of thermal expansion for optoelectronic applications. Polym. Int. 65, 1063–1073 (2016).
Shin, S. et al. Synthesis and reactivity of novel cinnamonitrile derivatives as reactive UV stabilizers for enhanced light protection and performance of coatings. Polym. Degrad. Stab. 201, 109969 (2022).
Heidarpour, F., Farahani, M. & Ghabezi, P. Experimental investigation of the effects of adhesive defects on the single lap joint strength. Int. J. Adhes. Adhes. 80, 128–132 (2018).
Hwang, J. et al. Ambient air-operated thermo-switchable adhesion of N-isopropylacrylamide-incorporated pressure sensitive adhesives. Mater. Horiz. 10, 2013–2023 (2023).
Campbell, C. J. et al. Optically clear adhesives enabling foldable and flexible OLED displays. SID Symp. Dig. Tech. Pap. 48, 2009–2011 (2017).
Song, K. et al. (Samsung Display Co., LTD.) Display device. US Patent 11502270 B2 (2022).
Samsung display newsroom. Samsung foldable OLED: ‘Folding Test in Extreme Cold’. https://global.samsungdisplay.com/28434/ (2021).
Wen, S. W., Lee, M. T. & Chen, C. H. Recent development of blue fluorescent OLED materials and devices. J. Disp. Technol. 1, 90–99 (2005).
Wong, M. Y. & Zysman-Colman, E. Purely organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Adv. Mater. 29, 1605444 (2017).
Monkman, A. Why do we still need a stable long lifetime deep blue OLED emitter? ACS Appl. Mater. Interfaces 14, 20463–20467 (2021).
Zhang, Z., Corrigan, N., Bagheri, A., Jin, J. & Boyer, C. A Versatile 3D and 4D printing system through photocontrolled RAFT polymerization. Angew. Chem. Int. Ed. 58, 17954–17963 (2019).
Jung, K. et al. Designing with light: advanced 2D, 3D, and 4D Materials. Adv. Mater. 32, 1903850 (2020).
Ahn, D. et al. Rapid high-resolution visible light 3D printing. ACS Cent. Sci. 6, 1555–1563 (2020).
Ahn, D., Stevens, L. M., Zhou, K. & Page, Z. A. Additives for ambient 3D printing with visible light. Adv. Mater. 33, 2104906 (2021).
Ma, Q. et al. How to overcome the light penetration issue in photopolymerization? An example for the preparation of high content iron-containing opaque composites and application in 3D printing. Eur. Polym. J. 165, 111011 (2022).
Roy, D., Cambre, J. N. & Sumerlin, B. S. Future perspectives and recent advances in stimuli-responsive materials. Prog. Polym. Sci. 35, 278–301 (2010).
Roy, D., Brooks, W. L. A. & Sumerlin, B. S. New directions in thermoresponsive polymers. Chem. Soc. Rev. 42, 7214–7243 (2013).
Ferracane, J. L. Resin composite - state of the art. Dent. Mater. 27, 29–38 (2011).
Sprick, E. et al. New hydrogen donors for amine-free photoinitiating systems in dental materials. Dent. Mater. 37, 382–390 (2021).
Figg, C. A. et al. Polymerization-induced thermal self-assembly (PITSA). Chem. Sci. 6, 1230–1236 (2015).
Acknowledgements
This work was supported by the Technology Innovation Program (20011317, Development of an adhesive material capable of morphing more than 50% for flexible devices with a radius of curvature of 1 mm or less) and by the Industrial Strategic Technology Development Program (20011059, Development of inks for emitting layers with high performance and long lifetime), which were funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea). It was also backed by the National Research Foundation of Korea (NRF) grants provided by the Korean government (MSIT) under grant numbers 2021R1A5A1030054 and NRF-2022R1A2C2011627. The work in Madrid was supported by the Spanish Ministerio de Ciencia, Innovación y Universidades, and the European Structural Funds through projects PID2022-138222NB-C21, CEX2020-001039-S, and by the Campus of International Excellence (CEI) UAM + CSIC.
Author information
Authors and Affiliations
Contributions
Y.Kwon, S.L., and M.S.K. were responsible for the initial conception of the project and wrote the initial draft of the manuscript. Y.Kwon and W.J. performed the photophysical measurements and CV measurements. S.L., J.K., and Y.P. synthesized the OCA films and measured the mechanical- and optical properties of OCA films. J.J. fabricated OLED devices and performed the UV-blocking test supported by S.L. and J.K. under supervision from J.L. S.L., Y.P., Y.Kim, and H.J.K. discussed the mechanical properties of OCA films. Y.Kwon, J.G., and M.S.K. were involved in the discussion of the photophysics and photochemical reactions. Y.Kwon performed DFT calculations and the kinetic simulation under supervision from M.S.K. All the authors discussed the results and commented on the manuscript. J.L., Y.Kim, and M.S.K. supervised the project and were responsible for editing the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The patent application (K.R. application number: 1020230026669 and 1020230089201) was filed by M.S.K., Y.Kwon, and S.L. from Seoul National University R&DB Foundation. The patent application covered the preparation of methods for preparing photocured resin and their application into UV-blocking adhesive. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kwon, Y., Lee, S., Kim, J. et al. Ultraviolet light blocking optically clear adhesives for foldable displays via highly efficient visible-light curing. Nat Commun 15, 2829 (2024). https://doi.org/10.1038/s41467-024-47104-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-47104-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
