
Abstract
It is a long-standing scientific controversy to achieve anti-Kasha-type multiple emissions by tuning the structures at a molecular level. Although it is known that some conjugated structures have excitation-dependent multiple emissions, no all-benzenoid molecules have yet been reported, the emissions of which originate from different excited states. Herein, we report the design of two symmetry-breaking heterogeneous carbon bisnanohoops that in solution become multiple fluorescent emitters with unusual anti-Kasha characteristics. This phenomenon can be spectroscopically and theoretically explained and will find applications in a wide range of sensing and imaging technologies.
Similar content being viewed by others


Introduction
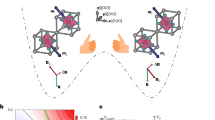
Kasha’s rule, a general principle in basic spectroscopy, states that the emitting level of a given multiplicity is the lowest excited level of that multiplicity, which was formulated by Michael Kasha in 19501. Most fluorescent molecules follow this rule very well, but very few molecules are reported to break this rule, such as azulene and thioketones (Figs. 1a, b)2,3,4,5,6,7. Azulene only shows anti-Kasha S2 fluorescence due to a large energy separation between the S2 and S1 states (ΔE(S2 - S1) ~ 14,000 cm−1), leading to a relatively slow internal conversion of S2 → S1, and S2 radiative emission rates can compete favorably with internal conversion (Fig. 1d)3,4. Some thioketones, such as thiophosgene5 and xanthione (Fig. 1b)6, have a larger oscillator strength of S2 than that of S1 and demonstrate fluorescent emission from the S2 excited state. In those molecules, the S1 → S0 transition is negligible (Fig. 1d)8. The anti-Kasha emission provides possibilities to control the excited-state transformations for a variety of fundamental research and practical applications9,10,11,12,13,14. In the past few decades, a few other molecular systems, such as phenanthroimidazole-type dyes15, tricarbocyanine16, cycl[3.3.3]azines17, and triphenylmethane18,19, have also been reported to show anti-Kasha emissions, but many examples are still under debate20. Moreover, almost all the reported organic molecules that violate Kasha’s rule are non-benzenoid aromatic hydrocarbons or heteroatom-containing organic molecules, regardless of whether they are a definite anti-Kasha molecular system or a controversial molecular system20,21,22,23. To date, no such molecule containing only benzenoid moieties has been reported to have multiple fluorescent emissions with unusual anti-Kasha characteristics.

a Structure of azulene. b Structures of thiophosgene (left) and xanthione (right). c Structures of heterogeneous bisnanohoops SCPP[10,8] (left), SCPP[10,9] (middle) and homogeneous bisnanohoop SCPP[10] (right). d Anti-Kasha emission of azulene. e S2, S1 dual excited-state emissions of SCPPs bisnanohoops.
Benzenoid-type luminogens have the advantages of wide variety, dynamic tunability, rich physical properties, and excellent stability, which are attractive for many applications24,25,26,27,28,29. The conjugated backbones of benzenoid macrocycles support delocalized electronic excitations, resulting in interesting tunable dynamics by changing cyclic geometries and/or symmetries30,31. Conjugated cycloparaphenylene (CPP) nanohoops, representing the shortest sidewall segments of carbon nanotubes, have demonstrated intriguing luminescent and electronic properties31,32,33,34,35. Due to a conservation of orbital symmetry for the centrosymmetric CPPs, the HOMO-LUMO optical transitions are Laporte forbidden36. As a result, the photophysical properties of CPP differ significantly from those of linear conjugated materials. CPPs have virtually identical absorbance maxima and unique fluorescence emissions from spatially localized S1′ states to the ground state S037. In 2019, Jasti and coworkers discovered that breaking the centrosymmetric CPP can alter fluorescent properties, although no multiple emissions were observed38.
Herein, we report a distinctive type of symmetry-breaking heterogeneous carbon bisnanohoops, (1,4)[10,9]cycloparaphenylenophane (SCPP[10,9]) and (1,4)[10,8]cycloparaphenylenophane (SCPP[10,8]) (Fig. 1c), achieving excitation-dependent multiple emissions from the upper and lowest singlet excited states with unusual anti-Kasha characteristics. SCPP[10,9] and SCPP[10,8] have two different CPP-based nanohoops that are fused in a central benzene. Bisnanohoops with heterogeneous CPP-based macrocycle compositions are expected to have extraordinary photophysical phenomena derived from breaking the symmetry of the original centrosymmetric structure. The homogeneous bisnanohoop SCPP[10] was also studied for comparison (Fig. 1c)26. By adjusting the size of the bisnanohoops and tuning the balance between the high-energy excited state and low-energy excited state, the interesting excitation-dependent multicolor luminescence and systematic photophysical properties of bisnanohoops were investigated. This work demonstrates an intriguing example of organic all-benzenoid molecules with S2 and S1 dual excited-state emissions and wide-range emissions spanning over 150 nm (Fig. 1e).
Results
Synthesis and characterizations of SCPPs
The key to the synthesis of heterogeneous bisnanohoops was the use of ridged 1,4-syn-dimethoxy-2,5-cyclohexadienes (1 and 2) as masked benzene rings to react with ditriflate [10]CPP single nanohoop 3. This appropriately substituted cyclohexadiene unit is able to alleviate strain and allow for Suzuki-Miyaura cross-coupling/macrocyclization to bisnanohoops with varying sizes. In the final reductive aromatization step, these cyclohexadiene-containing macrocycles were treated with H2SnCl4 at room temperature to achieve the fully benzenoid bisnanohoops SCPP[10,9] and SCPP[10,8] (Fig. 2). The successful synthesis of heterogeneous bisnanohoops can be supported by different characterization techniques, including various NMR spectra and high-resolution mass spectrometry (HR-MS) (Supplementary Figs. 1–20).

Synthesis strategies of heterogeneous bisnanohoops SCPP[10,9] and SCPP[10,8].
Anti-Kasha emissions of SCPPs in solution
The photophysical properties of SCPP[10,8], SCPP[10,9], and SCPP[10] were investigated in dilute dichloromethane (DCM) solutions. As shown in Fig. 3a–c, the UV-Vis absorption spectra of SCPPs exhibited broad absorption bands in the region of 280–480 nm. The maximum absorption peaks appear at 359 nm, 356 nm, and 351 nm for SCPP[10,8]-SCPP[10], respectively. In addition, there are an obvious shoulder peak at approximately 340 nm and a weak shoulder peak in the longer wavelength region (at ~400 nm) for all SCPPs.

Absorption and emission spectra (under excitation at 360 nm) of SCPPs: (a) SCPP[10]; (b) SCPP[10,9]; and (c) SCPP[10,8]. Fluorescence photographs and emission spectra of SCPPs under 300/365 nm UV light in dilute DCM (c = 5 × 10−6 mol/L): (d) SCPP[10]; (e) SCPP[10,9]; and (f) SCPP[10,8]. Excitation-dependent emissive properties of SCPPs: (g) SCPP[10]; (h) SCPP[10,9]; and (i) SCPP[10,8].
Furthermore, the steady-state photoluminescence (PL) spectra of SCPP[10], SCPP[10,9], and SCPP[10,8] were preliminarily measured under excitation at 360 nm (Fig. 3a–c). Each SCPP has a size-dependent low-energy emission (520 nm for SCPP[10], 528 nm for SCPP[10,9], and 545 nm for SCPP[10,8]) and two high-energy shoulder peaks at around 470 nm and 390 nm.
Unexpectedly, distinctive excitation-dependent multicolor fluorescence under different excitation wavelengths was clearly observed in their photoluminescent spectra and photographs (Fig. 3d–f). Under irradiation at 300 nm UV light, SCPP[10] and SCPP[10,9] displayed green fluorescence, and SCPP[10,8] showed blue fluorescence. Under irradiation at 365 nm UV light, SCPP[10] emitted light yellow fluorescence, SCPP[10,9] emitted yellow fluorescence, and SCPP[10,8] was remarkably orange-emissive. The emission color of SCPPs can be reversibly tuned through UV 365/300 nm switch processes, showing excellent sensitivity and reversibility. These processes were also monitored by photoluminescent spectra.
To further understand these anomalous phenomena, the detailed excitation wavelength-dependent fluorescence spectra of SCPPs were further measured (Fig. 3g–i). When the excitation wavelength ranged from 300 to 400 nm, these SCPPs demonstrated interesting multiple emissions. For instance, when a short excitation wavelength (<340 nm) was applied to SCPP[10,8], two emission bands maximized at 392 nm and 467 nm were observed, while the latter dominated the fluorescence spectra. However, when the excitation wavelengths were slowly changed from 340 to 400 nm, a lower-energy emission band maximized at ~545 nm clearly appeared and gradually became the major emission, accompanied by a decrease in the higher-energy emission bands. The wide-range wavelength shift (155 nm) in the emission spectrum is consistent with the remarkable fluorescent color change of SCPP[10,8] from blue to orange. Interestingly, SCPPs exhibited similar high-energy fluorescence emission band maximized at ~390 nm and significantly different low-energy emission bands, in which the maximum peaks are located at 520 nm for SCPP[10], 528 nm for SCPP[10,9], and 545 nm for SCPP[10,8]. The relative proportions of different emission bands are directly related to the excitation wavelength, with higher energy excitations leading to increased emissions at shorter wavelengths. These results strongly indicated that the emissions come from more than one excited state. Besides, fluorescence quantum yields for the different emission processes were measured in a dilute solution at room temperature (Supplementary Table 1). The quantum yields from the low energy emission of SCPPs decreased as bisnanohoops got smaller, which is similar to the size-dependent emission properties of CPPs.
Solid-state emissions of SCPPs
The solid-state fluorescence properties of SCPPs have also been studied. As observed with the naked eye, the color of SCPPs gradually changed from light yellow to orange as the ring size decreased (Supplementary Fig. 21). All these solid samples exhibited a single and bright fluorescence under a handheld UV light, and their emissions did not change with different excitation wavelengths (Fig. 4a, c and Supplementary Fig. 22). To further verify this phenomenon, the PL spectra of SCPP films were measured, and the results confirmed the independence of excitation (Fig. 4b, d and Supplementary Fig. 23). Therefore, SCPPs exhibit excitation-dependent multiple emission properties in solution, which violates the Kasha’s rule. However, in the solid state, the high energy emissions vanished and only showed the lowest energy emissions, which obeys the Kasha’s rule (Fig. 4e).

Fluorescence photographs of SCPPs powder (a) and PMMA film (b) under 300/365 nm UV light. Emission spectra of SCPPs powder (c) and PMMA film (d). e Fluorescent behavior of SCPP[10,8] in a dilute solution (left and middle) and in solid state (right).
Theoretical calculations of SCPPs
The subtle interactions of geometry, symmetry, and strain plays a significant role in many interesting properties of these SCPPs. Density functional theory (DFT) calculations were performed at the theoretical level of B3LYP/6-31 G(d, p) utilizing the Gaussian 16 program to investigate these basic properties of SCPPs39. Geometrical structure optimization showed that SCPPs feature C2 symmetry, and the symmetry of bisnanohoops is obviously reduced compared with that of D2-symmetric single nanohoop [10]CPP31. The twist angle of the bridging phenyl is 12.74° for SCPP[10,8], 11.53° for SCPP[10,9], and 10.05° for SCPP[10]. As the ring size decreases, the torsion of bridging phenyl gradually increases. Moreover, the strain energies were estimated to be 110.59 kcal/mol for SCPP[10], 118.95 kcal/mol for SCPP[10,9], and 130.98 kcal/mol for SCPP[10,8] (Supplementary Fig. 24 and Supplementary Table 2). Strain is a critical factor that endows nanohoops with unusual photophysical properties31. As the diameter decreases, these SCPPs have increasing strain energies, changing the geometry and optical properties.
Experimental and computational verification of anti-Kasha emission
To further explore the anti-Kasha properties of SCPPs, the absorption spectra were further investigated by theoretical calculation and compared with excitation spectra (Fig. 5a–d). Frontier molecular orbital analyses of SCPPs were performed to provide insight into the slight changes in absorption peaks. Time-dependent density functional theory (TDDFT) calculations revealed that the high-energy absorption bands of SCPPs are assignable to the S0 → S4 (HOMO → LUMO + 3 and HOMO - 3 → LUMO) transition, and the relatively low-energy absorption bands of SCPPs are assignable to the S0 → S3 (HOMO - 1 → LUMO + 1, HOMO - 2 → LUMO, and HOMO → LUMO + 2) transition, and the weak shoulder peak in the longer wavelength region (at ~400 nm) are assignable to S0 → S1 (HOMO → LUMO) (Fig. 5d, Supplementary Figs. 25–28 and Supplementary Tables 3–6). As the ring size increases, the energy level of unoccupied molecular orbitals moves downward, while those of occupied molecular orbitals move upward. The redshift of the absorption spectra in SCPPs can be ascribed to the slight shift of the frontier molecular orbitals. Moreover, the excitation spectra were measured and compared with the absorption spectra (Fig. 5a–c). The excitation spectra of all SCPPs agreed well with the corresponding absorption spectra. For example, according to the excitation spectrum, the high-energy emission at 392 nm of SCPP[10,8] was easily detected under excitation at 320 nm, which probably corresponds to the absorption band at 310 nm. The emission at 467 nm can be detected when the excitation wavelength was 340 nm, which is consistent with the absorption peak at 340 nm, and the emission centered at 545 nm arose primarily when the excitation was ~360 nm, which also agrees well with the absorption peak at 360 nm. The excitation and absorption spectra of SCPP[10] and SCPP[10,9] also showed the same characteristics. These data provide strong evidence for emissions derived from both the upper and lowest excited states.

a Excitation spectra of SCPP[10] from emissions monitored at 390 nm (green dotted line), at 470 nm, and at 520 nm (blue dotted line); absorption spectrum of SCPP[10] (purple solid line). b Excitation spectra of SCPP[10,9] from emissions monitored at 394 nm (green dotted line), at 472 nm, and at 528 nm (blue dotted line); absorption spectrum of SCPP[10,9] (purple solid line). c Excitation spectra of SCPP[10,8] from emissions monitored at 392 nm (green dotted line), at 467 nm, and at 545 nm (blue dotted line); absorption spectrum of SCPP[10,8] (purple solid line). d TDDFT-calculated molecular orbitals and energy diagrams. The f value represents the oscillator strength. e Jablonski diagram showing the calculated photophysical processes of SCPP[10,8]. kIC internal conversion rate, kr radiative transition rate, ∆G energy gap between two excited states.
To provide deeper insight into the fluorescence process, excited-state calculations were performed. The calculation results indicated that the allowed transitions of all SCPPs are S2 → S0 and S1 → S0 (Fig. 5e and Supplementary Figs. 29–31), showing the anti-Kasha emission feature. The high-energy emission (peak at ~390 nm) of these SCPP bisnanohoops comes from the S2 state emission, the lowest energy emission derives from the S1 state emission, and the emission at ~470 nm is probably from the vibronic mixing of the S1 state with higher electronic states. Calculations and experiments have shown that the SCPP bisnanohoops have similar S2 excited states with an emission peak at 384–397 nm and S3 excited states with an emission peak 350–357 nm. However, as the size of the bisnanohoop decreases, the higher strain energy leads to a decrease in the torsional angle and increases the degree of conjugation. As a result, the increased conjugation further enhances vibration coupling and reduces S1-S0 energy, resulting in a gradual redshift of the low energy emission.
The validity of Kasha’s rule is based on the rates of internal conversion, intersystem crossing, and vibrational relaxation from a given excited state to another excited state being much faster than the intrinsic radiative rate of that state8. As mentioned before, prototypical examples with anti-Kasha emission are azulene and thioketones, of which the rates of radiative Sn → S0 decay can compete with internal conversion to the S1 state owing to the large energy gap between the S2 and S1 states. Similarly, the excited states of the SCPP bisnanohoops were characterized by a large energy separation between the S1 and S2 states, and ΔE(S2 - S1) was calculated to be 6299 cm−1 for SCPP[10], 7401 cm−1 for SCPP[10,9], and 7073 cm-1 for SCPP[10,8]. Therefore, S2 fluorescence can compete favorably with internal conversion because of such a large energy gap. The internal conversion rate (kIC) and radiative transition rate (kr) were calculated based on different excited states. The kr of all SCPPs is approximately 108 s−1, which is faster than the internal conversion rate of S2 → S1 (kIC = 1.47 × 106 for SCPP[10,8], kIC = 2.26 × 105 s−1 for SCPP[10,9], and kIC = 5.05 × 107 s−1 for SCPP[10]). Therefore, under high-energy excitation, SCPPs behaved as the anti-Kasha emission of S2 → S0. In addition, the luminescence rate of the S1 state of all SCPPs is ~108 s−1, which is in the range of the fluorescence emission rate (107–1010 s−1), resulting in S1 → S0 emission under low-energy excitation. All these calculation results are consistent with the excitation-dependent multicolor emission phenomenon of SCPP bisnanohoops.
It is worth noting that the S1 → S0 emission of SCPPs is significantly different from that of the azulene system. The radiative decay rate of the S1 state of the latter is very low (kr1 = 1.00 × 105 s−1); therefore, it is difficult to observe the emission of the S1 → S0 state40. Azulene derivatives were constructed to obtain interesting dual fluorescence by adjusting the energy gaps between the electronic states41. As mentioned above, the radiative transition rates of the S2 and S1 excited states for SCPPs are both in the range of the fluorescence emission rate, resulting in more abundant and flexible luminescent properties of SCPPs than the classic azulene system.
Emission lifetimes and TRES for SCPP[10,8]
Furthermore, the time-resolved photoluminescence (TRPL) technique was used to study the excited-state lifetimes of SCPPs in DCM (Fig. 6a–c and Supplementary Figs. 32–33). Emission lifetimes of SCPPs at ~390 nm were measured upon excitation at 315 nm, and emission lifetimes at ~470 nm, 520 nm, 528 nm and 545 nm were measured upon excitation at 375 nm. All plots can be fitted by biexponential functions, and the fitting results are listed in Supplementary Table 7. SCPPs showed a short-lived species and a long-lived species for all monitored emissions, suggesting that two populations are involved in the decay processes. The long S2 state lifetime (τS2 > 10−10 s) also demonstrated that a large ∆E(S2 - S1) energy gap and a high radiative rate constant (kS2→S0 ~ 108 s−1) are responsible for anomalous fluorescence from the S2 state.

a–c Emission lifetimes for SCPP[10,8]. d Time-resolved emission spectrum for SCPP[10,8].
Excited-state decay processes were also studied by time-resolved emission spectroscopy (TRES) (Fig. 6d). The TRES of SCPP[10,8] was measured upon excitation at 375 nm. The time-dependent data of SCPP[10,8] were collected at wavelengths across the emission band ranging from 378 to 730 nm. The TRES showed three distinct emission spectra, which is consistent with the steady-state emission spectrum. At short delay time (4.85 ns), the spectrum showed a narrow emission peak at 390 nm and a broad emission band maximized at 550 nm with a small shoulder. At a longer delay time, the high-energy emission peak at 390 nm (S2 emission) decreased. The emission intensity of the low-energy emission peak at 550 nm (S1 emission) and the shoulder increased, and two comparable emission bands were detected at 5.31 ns. These observations indicated that the medium emission band (maximized at ~475 nm) and S1 emission are partly from the vibrational relaxation of the S2 excited state. As the delay time increased, the low-energy emission decreased faster, and the high-energy emission decreased relatively slower. In addition, the emission maximized at 475 nm became dominant, while the emission band maximized at 550 nm appeared only as a small shoulder. The results of TRES were matched with emission lifetimes of different excited states.
Discussion
Overall, we can conclude that all-benzenoid heterogeneous bisnanohoops demonstrated anomalous excitation-dependent multiple emissions with anti-Kasha characteristics. The spectroscopic results confirmed that the emission processes of bisnanohoops include the participation of both the upper excited states and the lowest excited state. Furthermore, the excited-state calculations indicated that there are large energy gaps between the S2 and S1 excited states in these bisnanohoops, resulting in anti-Kasha emission. Compared with the S2 dominant emission of azulene, our present heterogeneous bisnanohoops exhibit obvious S2 and S1 dual excited-state emission. Heterogeneous bisnanohoops have the advantages of an interesting symmetry-breaking structure, unique dual excited-state emission, and dynamic adjustability and are attractive organic luminescent materials for future potential applications.
Methods
Materials and general information
NMR spectra were recorded on a Bruker BioSpin (1H 400 MHz, 13C 100 MHz) spectrometer or a JEOL JNM-ECZ600R (1H 600 MHz, 13C 150 MHz) NMR spectrometer. Chemical shifts were reported as the delta scale in ppm relative to tetramethylsilane (δ = 0.00 ppm), CDCl3 (δ = 7.26 ppm) for 1H NMR and CDCl3 (δ = 77.0 ppm) for 13C NMR. Data were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), coupling constant (Hz), and integration. High resolution mass spectrometry (HR-MS) analyses were carried out using MALDI-TOF-MS techniques. All solvents for syntheses were dried by distillation under nitrogen prior to use (tetrahydrofuran and 1,4-dioxane were distilled after reflux with sodium under nitrogen). Other chemicals were obtained from commercial suppliers (Innochem or Acros) and the purity of chemicals are greater than 99%. Air-sensitive reactions were all carried out under argon.
Synthesis of compound 1
The synthesis of compound 142 was carried out using a modified procedure: To a degassed solution of chlorine substituted precursor (2.8 g, 3.24 mmol) and bis(pinacolato)diboron (3.28 g, 12.94 mmol) in 100 mL of dry 1,4-dioxane was added KOAc (3.18 g, 32.34 mmol) under argon, then the mixture was degassed for 15 min. Pd(OAc)2 (85 mg, 0.38 mmol) and S-Phos (313 mg, 0.76 mmol, 24 mol%) were added to the mixture. The mixture was degassed 10 min, after which it was heated with stirring under argon atmosphere at 90 °C for 48 h. After the reaction mixture was cooled to room temperature, the solvent was removed under reduced pressure and the residue was extracted with dichloromethane. The combined organic layer was washed with brine and dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. Then the resulting product was dissolved in petroleum ether/CH2Cl2 (50:50 mL) and passed through a short silica gel column. After removal of volatiles under reduced pressure to afford a crude product, further purification by an ultrasound wash in cold ethanol to afford pure compound 1 (3 g, 2.85 mmol, 89% yield) as a white solid. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.76 (dd, J = 7.8, 6.1 Hz, 4H), 7.55–7.50 (m, 4H), 7.45 (dd, J = 7.2, 3.2 Hz, 4H), 7.44–7.38 (m, 4H), 7.36 (s, 4H), 6.11 (dd, J = 13.2, 10.3 Hz, 12H), 3.47–3.40 (m, 18H), 1.33 (t, J = 6.7 Hz, 24H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 146.37, 146.35, 142.73, 142.62, 142.42, 142.33, 139.98, 139.91, 134.87, 134.85, 133.34, 133.32, 133.27, 133.20, 127.08, 126.33, 126.02, 126.00, 125.27, 125.23, 83.70, 74.87, 74.86, 74.62, 74.60, 74.58, 51.95, 51.91, 24.80, 24.78. The characterization matches well with the reported data42.
Synthesis of compound 2
The synthesis of compound 243 was carried out using a modified procedure: 1,1’-(1,4-dimethoxy-2,5-cyclohexadiene-1,4-diyl)bis[4-[4-(4-bromophenyl)-1,4-dimethoxy-2,5-cyclohexadien-1-yl]benzene] (1.0 g, 1.14 mmol) was dissolved in THF (15 ml) and cooled to −78 °C. To this solution was added a 2.5 M solution of n-BuLi in hexanes (1.0 mL, 2.50 mmol) over 2 min. Then, 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2dioxaborolane (0.6 mL, 2.89 mmol) was added rapidly and the solution was stirred for 20 min. Water (10 mL) was added to the solution and the mixture was allowed to stir for 15 min at room temperature. The aqueous layer was extracted with CH2Cl2 and concentrated under reduced pressure. The crude product was purified by column chromatography (hexane/ethyl acetate = 1/1, v/v) to afford compound 2 (0.9 g, 85% yield) as a white solid. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.78 (dd, J = 17.6, 8.3 Hz, 4H), 7.58–7.48 (m, 2H), 7.40 (d, J = 8.3 Hz, 4H), 7.34 (s, 6H), 6.09 (dt, J = 5.9, 3.3 Hz, 12H), 3.42 (dd, J = 4.6, 2.6 Hz, 15H), 3.28–3.25 (m, 3H), 1.33 (d, J = 2.5 Hz, 24H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 146.37, 146.34, 142.65, 142.62, 142.60, 142.57, 142.51, 134.81, 134.78, 133.90, 133.85, 133.64, 133.57, 133.29, 133.19, 133.03, 128.26, 125.91, 125.83, 125.22, 125.17, 83.62, 74.74, 74.49, 74.45, 74.21, 51.84, 51.83, 24.74. The characterization matches well with the reported data43.
Synthesis of compound 3
The synthesis of compound 326 was carried out using a modified procedure: To a mixture of benzyloxy substituted [10]cycloparaphenylene (44 mg, 0.045 mmol), 1,2,4,5-tetramethylbenzene (60 mg, 0.45 mmol) under N2 atmosphere was added anhydrous dichloromethane (20 mL). Boron trichloride solution (0.22 ml, 1 M in hexane, 0.22 mmol) was added dropwise to the reaction mixture at −78 °C. The mixture was stirred at −78 °C for 30 min. After warmed up to room temperature, the mixture was quenched with H2O. The organic and aqueous layer was then separated, and the aqueous layer was extracted with CH2Cl2. The organic layers dried with anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was used in the next step without further purification. To the above crude product was added anhydrous dichloromethane (20 mL) under N2 atmosphere. The mixture was added pyridine (0.1 mL, 1.25 mmol) and trifluoromethanesulfonic anhydride (Tf2O, 0.1 mL, 0.60 mmol) at 0 °C. The mixture was then warmed to room temperature and stirred for 12 h. The reaction was quenched with H2O. The organic and aqueous layer was then separated, and the aqueous layer was extracted with CH2Cl2. The organic layers dried with anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (hexane/CH2Cl2 = 3/1, v/v) to afford compound 3 (30 mg, ~55% over two steps) as a yellow solid. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.65–7.52 (m, 32H), 7.48 (d, J = 8.7 Hz, 4H), 7.35 (s, 2H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 145.31, 141.26, 138.64, 138.42, 138.21, 138.19, 138.14, 137.96, 137.81, 134.78, 131.98, 129.57, 127.85, 127.47, 127.42, 127.35, 125.43. The characterization matches well with the reported data26.
Synthesis of SCPP[10, 9]
To a mixture of 1 (42 mg, 40 μmol), 3 (42 mg, 40 μmol) and K2CO3 (66 mg, 0.48 mmol) in a round-bottomed flask (500 mL) were added THF (150 mL), H2O (30 mL), and Pd(PPh3)4 (10 mg, 9 μmol) under an argon atmosphere. Thereafter, the solution was heated at 75 °C for 48 h. After cooling to room temperature, the solvent was removed under vacuum and the remaining aqueous fraction was extracted with CH2Cl2. The combined organic layer was dried over anhydrous MgSO4 and concentrated under reduced pressure to afford crude product as a yellow solid that was used in the next step without further purification. To a 50-mL round-bottom flask containing a magnetic stirring bar were added SnCl2·2H2O (110 mg, 0.49 mmol), THF (10 mL) and concentrated HCl/H2O (0.1 ml, 12 mol/L) were added, and the resultant mixture was further stirred at room temperature for 30 min. The solution of H2SnCl4/THF was added dropwise to a solution containing the above crude product in 2 mL of THF. After stirring the mixture at room temperature for 2 h, the mixture was quenched with aqueous sodium hydroxide, extracted with CH2Cl2, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography (hexane/CH2Cl2 = 2/1, v/v) to afford SCPP[10, 9] (10 mg, ~19% over two steps) as a yellow solid. 1H NMR (CDCl3, 600 MHz): δ (ppm) 7.75 (s, 2H), 7.73 (d, J = 3.8 Hz, 4H), 7.71 (s, 4H), 7.61 (dd, J = 8.6, 4.5 Hz, 8H), 7.56 (dd, J = 7.9, 4.7 Hz, 24H), 7.53 (s, 8H), 7.52 (d, J = 8.7 Hz, 4H), 7.45 (d, J = 8.6 Hz, 4H), 7.43 (d, J = 8.0 Hz, 4H), 7.40 (d, J = 8.3 Hz, 4H), 7.36 (d, J = 8.2 Hz, 4H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 141.90, 141.86, 141.79, 141.66, 141.60, 141.25, 141.14, 141.11, 141.00, 140.96, 140.88, 140.86, 140.73, 140.66, 138.24, 137.67, 132.41, 132.27, 132.16, 131.07, 130.81, 130.63, 130.57, 130.50, 130.44, 130.33, 130.27, 130.14. HR-MS (MALDI-TOF) m/z calcd. for C108H70 [M]+: 1367.5320, found: 1367.5511.
Synthesis of SCPP[10, 8]
To a mixture of 2 (39 mg, 40 μmol), 3 (42 mg, 40 μmol) and K2CO3 (66 mg, 0.48 mmol) in a round-bottomed flask (500 mL) were added THF (150 mL), H2O (30 mL), and Pd(PPh3)4 (10 mg, 9 μmol) under an argon atmosphere. Thereafter, the solution was heated at 75 °C for 48 h. After cooling to room temperature, the solvent was removed under vacuum and the remaining aqueous fraction was extracted with CH2Cl2. The combined organic layer was dried over anhydrous MgSO4 and concentrated under reduced pressure to afford crude product as a yellow solid that was used in the next step without further purification. To a 50-mL round-bottom flask containing a magnetic stirring bar were added SnCl2·2H2O (110 mg, 0.49 mmol), THF (10 mL) and concentrated HCl/H2O (0.1 ml, 12 mol/L) were added, and the resultant mixture was further stirred at room temperature for 30 min. The solution of H2SnCl4/THF was added dropwise to a solution containing the above crude product in 2 mL of THF. After stirring the mixture at room temperature for 2 h, the mixture was quenched with aqueous sodium hydroxide, extracted with CH2Cl2, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography (hexane/CH2Cl2 = 2/1, v/v) to afford SCPP[10, 8] (26 mg, ~51% over two steps) as an orange solid. 1H NMR (CDCl3, 600 MHz): δ (ppm) 7.78 (d, J = 8.4 Hz, 4H), 7.74 (d, J = 8.6 Hz, 4H), 7.72 (s, 2H), 7.61 (dd, J = 8.4, 5.8 Hz, 8H), 7.56 (dd, J = 12.7, 4.2 Hz, 20H), 7.52 (s, 8H), 7.51 (d, J = 8.3 Hz, 4H), 7.46 (d, J = 8.5 Hz, 4H), 7.43 (d, J = 8.1 Hz, 4H), 7.40 (d, J = 7.5 Hz, 4H), 7.34 (d, J = 6.5 Hz, 4H). 13C NMR (CDCl3, 150 MHz): δ (ppm) 141.85, 141.77, 141.62, 141.35, 141.31, 141.22, 141.15, 141.07, 141.00, 140.97, 140.85, 140.73, 140.54, 140.50, 140.16, 137.98, 137.51, 133.10, 132.17, 131.60, 130.95, 130.74, 130.59, 130.51, 130.28, 130.16. HR-MS (MALDI-TOF) m/z calcd. for C102H66 [M]+: 1291.5578, found: 1291.5198.
Spectroscopy analysis
UV-Vis absorption spectra were performed on a UNIC-3802 spectrophotometer. Steady-state fluorescence spectra were obtained with a Horiba FluoroMax-4 compact spectrofluorometer equipped with an ozone-free xenon lamp optics source. The relative quantum yields were measured using anthracene and quinine sulfate as the references. The time-resolved spectra were recorded using a PL lifetime spectrometer (Edinburgh Instruments, LifeSpec-II) based on a time-correlated single photon counting (TCSPC) technique with excitations of 375 nm and 315 nm picosecond laser. SCPPs was measured in DCM with a concentration of 5.0 × 10−6 M.
Computational details
Theoretical calculations were performed using the Gaussian 16 software package39. All calculations were carried out using the density functional theory (DFT) method. Geometrical structure optimization of three compounds were optimized at the B3LYP/6-31 G(d) level, and the energies were calculated at the CAM-B3LYP/6-31 G(d) level. The strain energy was calculated using the reported computational methods33,44. The molecular orbitals were generated by the Multiwfn softwar (A Multifunctional Wavefunction Analyzer Version 3.8 dev)45. The photophysical properties were calculated by using the photophysics module of the Molecular Materials Property Prediction Package (MOMAP Version 2021 A 2.3.1)46,47,48,49,50,51,52. The Gaussian 16 software package was used to perform geometry optimization and frequency calculations on the initial and final states. The bi-exponential decays indicate that there are two possible populations with different radiative lifetimes involved in the S2 and S1 emissions. These two transitions probably originate from the different configurations of the molecule in the excited state, with one configuration being the dominant component. Due to the complexity of excited state configuration, it is difficult to determine which specific configurations are involved in the luminescence process. In order to simplify the calculation process, the minimum energy geometry in the excited state was chosen as the only configuration for the calculation of the radiation rate and internal conversion rate. The transition dipole moment and the transition electric field between the two states were calculated and used for the kr calculation. The non-adiabatic coupling matrix element (NACME) was obtained by Gaussian 16 for the kIC calculation.
Data availability
Characterization studies, 1H NMR spectra, 13C NMR spectra, mass spectrometry data and useful information are available in the Supplementary Information. Coordinates of the optimized structures are provided in the source data file. Additional data that support the findings of this study are available from the corresponding author upon request. Source data are provided with this paper.
Code availability
The Molecular Material Property Prediction Package (MOMAP) workshop is commercially available at http://www.momap.net.cn/. The Multifunctional Wavefunction Analyzer (Multiwfn) program is free access at http://sobereva.com/multiwfn.
References
Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 9, 14–19 (1950).
Beer, M. & Longuet‐Higgins, H. C. Anomalous light emission of azulene. J. Chem. Phys. 23, 1390–1391 (1955).
Viswanath, G. & Kasha, M. Confirmation of the anomalous fluorescence of azulene. J. Chem. Phys. 24, 574–577 (1956).
Huppert, D., Jortner, J. & Rentzepis, P. M. S2 → S1 emission of azulene in solution. Chem. Phys. Lett. 13, 225–228 (1972).
Levine, S. Z., Knight, A. R. & Steer, R. P. Fluorescence from the second excited singlet state of thiophosgene vapour. Chem. Phys. Lett. 29, 73–76 (1974).
Maciejewski, A. & Steer, R. P. The photophysics, physical photochemistry, and related spectroscopy of thiocarbonyls. Chem. Rev. 93, 67–98 (1993).
Wu, Y. H. et al. Multiple‐state emissions from neat, single‐component molecular solids: suppression of Kasha’s rule. Angew. Chem. Int. Ed. 59, 10173–10178 (2020).
Itoh, T. Fluorescence and phosphorescence from higher excited states of organic molecules. Chem. Rev. 112, 4541–4568 (2012).
Dong, J.-X. & Zhang, H.-L. Azulene-based organic functional molecules for optoelectronics. Chin. Chem. Lett. 27, 1097–1104 (2016).
Xin, H. & Gao, X. Application of azulene in constructing organic optoelectronic materials: new tricks for an old dog. ChemPlusChem 82, 945–956 (2017).
Xin, H., Hou, B. & Gao, X. Azulene-based π-functional materials: design, synthesis, and applications. Acc. Chem. Res. 54, 1737–1753 (2021).
Chen, C. et al. Pushing the length limit of dihydrodiboraacenes: Synthesis and characterizations of boron‐embedded heptacene and nonacene. Angew. Chem. Int. Ed. 61, e202200779 (2022).
Wang, H. et al. Breaching Kasha’s rule for dual emission: mechanisms, materials and applications. J. Mater. Chem. C 9, 10154–10172 (2021).
Shi, L. et al. De novo strategy with engineering anti-Kasha/Kasha fluorophores enables reliable ratiometric quantification of biomolecules. Nat. Commun. 11, 793 (2020).
Jiang, Q. et al. Stable high-energy excited states observed in a conjugated molecule with hindered internal conversion processes. J. Phys. Chem. C 123, 6190–6196 (2019).
Das, D. K., Makhal, K. & Goswami, D. Solvent effect on multiple emission and ultrafast dynamics of higher excited states. Chem. Phys. Lett. 706, 375–379 (2018).
Leupin, W., Berens, S. J. & Magde, D. Picosecond fluorescence from the second excited singlet state of cycl[3.3.3]azine, a bridge 12π-perimeter annulene. Spectra and kinetics for S2 → S0 and S2 → S1 transitions. J. Phys. Chem. 88, 1376–1379 (1984).
Janowski, A. & Rzeszotarska, J. Anomalous (S2 → S0) luminescence of some derivatives of triphenylmethane dyes and their complexes with rare earth metals. J. Lumin. 21, 409–416 (1980).
Nakayama, A. & Taketsugu, T. Ultrafast nonradiative decay of electronically excited states of malachite green: Ab initio calculations. J. Phys. Chem. A 115, 8808–8815 (2011).
Del Valle, J. C. & Catalán, J. Kasha’s rule: a reappraisal. Phys. Chem. Chem. Phys. 21, 10061–10069 (2019).
Turro, N. J., Ramamurthy, V., Cherry, W. & Farneth, W. The effect of wavelength on organic photoreactions in solution. Reactions from upper excited states. Chem. Rev. 78, 125–145 (1978).
Demchenko, A. P., Tomin, V. I. & Chou, P.-T. Breaking the Kasha rule for more efficient photochemistry. Chem. Rev. 117, 13353–13381 (2017).
Behera, S. K., Park, S. Y. & Gierschner, J. Dual emission: classes, mechanisms, and conditions. Angew. Chem. Int. Ed. 60, 22624–22638 (2021).
Golder, M. R. & Jasti, R. Syntheses of the smallest carbon nanohoops and the emergence of unique physical phenomena. Acc. Chem. Res. 48, 557–566 (2015).
Wang, J., Zhang, X., Jia, H., Wang, S. & Du, P. Large π-extended and curved carbon nanorings as carbon nanotube segments. Acc. Chem. Res. 54, 4178–4190 (2021).
Zhang, X. et al. A highly strained all‐phenylene conjoined bismacrocycle. Angew. Chem. Int. Ed. 60, 17368–17372 (2021).
Zhang, X., Liu, H., Zhuang, G., Yang, S. & Du, P. An unexpected dual-emissive luminogen with tunable aggregation-induced emission and enhanced chiroptical property. Nat. Commun. 13, 3543 (2022).
Leonhardt, E. J. & Jasti, R. Emerging applications of carbon nanohoops. Nat. Rev. Chem. 3, 672–686 (2019).
Guo, Q.-H., Qiu, Y., Wang, M.-X. & Stoddart, F. Aromatic hydrocarbon belts. Nat. Chem. 13, 402–419 (2021).
Omachi, H., Segawa, Y. & Itami, K. Synthesis of cycloparaphenylenes and related carbon nanorings: a step toward the controlled synthesis of carbon nanotubes. Acc. Chem. Res. 45, 1378–1389 (2012).
Darzi, E. R. & Jasti, R. The dynamic, size-dependent properties of [5]-[12]cycloparaphenylenes. Chem. Soc. Rev. 44, 6401–6410 (2015).
Jasti, R., Bhattacharjee, J., Neaton, J. B. & Bertozzi, C. R. Synthesis, characterization, and theory of [9]-, [12]-, and [18]cycloparaphenylene: Carbon nanohoop structures. J. Am. Chem. Soc. 130, 17646–17647 (2008).
Takaba, H., Omachi, H., Yamamoto, Y., Bouffard, J. & Itami, K. Selective synthesis of [12]cycloparaphenylene. Angew. Chem. Int. Ed. 48, 6112–6116 (2009).
Yamago, S., Watanabe, Y. & Iwamoto, T. Synthesis of [8]cycloparaphenylene from a square‐shaped tetranuclear platinum complex. Angew. Chem. Int. Ed. 49, 757–759 (2010).
Lewis, S. E. Cycloparaphenylenes and related nanohoops. Chem. Soc. Rev. 44, 2221–2304 (2015).
Segawa, Y. et al. Combined experimental and theoretical studies on the photophysical properties of cycloparaphenylenes. Org. Biomol. Chem. 10, 5979–5984 (2012).
Adamska, L. et al. Self-trapping of excitons, violation of Condon approximation, and efficient fluorescence in conjugated cycloparaphenylenes. Nat. Lett. 14, 6539–6546 (2014).
Lovell, T. C., Colwell, C. E., Zakharov, L. N. & Jasti, R. Symmetry breaking and the turn-on fluorescence of small, highly strained carbon nanohoops. Chem. Sci. 10, 3786–3790 (2019).
Frisch, M. J. et al. Gaussian 16 Revision A.03 (Gaussian, Inc., Wallingford, CT 2016).
Bearpark, M. J. et al. The azulene S1 state decays via a conical intersection: a CASSCF study with MMVB dynamics. J. Am. Chem. Soc. 118, 169–175 (1996).
Eber, G., Grüneis, F., Schneider, S. & Dörr, F. Dual fluorescence emission of azulene derivatives in solution. Chem. Phys. Lett. 29, 397–404 (1974).
Wang, J. et al. Tuning the (chir)optical properties and squeezing out the inherent chirality in polyphenylene‐locked helical carbon nanorings. Chem. Eur. J. 28, e202103828 (2022).
Wang, S. et al. Precise membrane separation of nanoparticles using a microporous polymer containing radially π-conjugated molecular carbocycles. Chem. Commun. 57, 11867–11870 (2021).
Segawa, Y., Omachi, H. & Itami, K. Theoretical studies on the structures and strain energies of cycloparaphenylenes. Org. Lett. 12, 2262–2265 (2010).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Shuai, Z. & Peng, Q. Organic light-emitting diodes: theoretical understanding of highly efficient materials and development of computational methodology. Nat. Sci. Rev. 4, 224–239 (2017).
Niu, Y. et al. Molecular Materials Property Prediction Package (MOMAP) 1.0: a software package for predicting the luminescent properties and mobility of organic functional materials. Mol. Phys. 116, 1078–1090 (2018).
Peng, Q., Yi, Y., Shuai, Z. & Shao, J. Toward quantitative prediction of molecular fluorescence quantum efficiency: role of Duschinsky. rotation. J. Am. Chem. Soc. 129, 9333–9339 (2007).
Niu, Y., Peng, Q. & Shuai, Z. Promoting-mode free formalism for excited state radiationless decay process with Duschinsky rotation effect. Sci. China B Chem. 51, 1153–1158 (2008).
Shuai, Z. & Peng, Q. Excited states structure and processes: Understanding organic light-emitting diodes at the molecular level. Phys. Rep. 537, 123–156 (2014).
Shuai, Z. Thermal vibration correlation function formalism for molecular excited state decay rates. Chin. J. Chem 38, 1223–1232 (2020).
Niu, Y., Peng, Q., Deng, C., Gao, X. & Shuai, Z. Theory of excited state decays and optical spectra: application to polyatomic molecules. J. Phys. Chem. A 114, 7817–7831 (2010).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22225108, P.D.; 21971229, P.D.), and Hefei National Research Center for Physical Sciences at the Microscale. This work was partially carried out at the Instruments Center for Physical Science, University of Science and Technology of China.
Author information
Authors and Affiliations
Contributions
P.D. conceived and designed this research. X.Z. synthesized all the compounds, conducted all characterizations, and physical measurements. C.C., G.Z. and X.W. did all the calculation studies. X.Z., W.Z., N.Y., B.Y., G.Z., X.W. and P.D. co-wrote the paper, and all the authors commented on it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Chen, C., Zhang, W. et al. Anomalous anti-Kasha excited-state luminescence from symmetry-breaking heterogeneous carbon bisnanohoops. Nat Commun 15, 2684 (2024). https://doi.org/10.1038/s41467-024-46848-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-46848-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
