
Abstract
The coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has evoked a worldwide pandemic. As the emergence of variants has hampered the neutralization capacity of currently available vaccines, developing effective antiviral therapeutics against SARS-CoV-2 and its variants becomes a significant challenge. The main protease (Mpro) of SARS-CoV-2 has received increased attention as an attractive pharmaceutical target because of its pivotal role in viral replication and proliferation. Here, we generated a de novo Mpro-inhibitor screening platform to evaluate the efficacies of Mpro inhibitors based on Mpro cleavage site-embedded amyloid peptide (MCAP)-coated gold nanoparticles (MCAP-AuNPs). We fabricated MCAPs comprising an amyloid-forming sequence and Mpro-cleavage sequence, mimicking in vivo viral replication process mediated by Mpro. By measuring the proteolytic activity of Mpro and the inhibitory efficacies of various drugs, we confirmed that the MCAP-AuNP-based platform was suitable for rapid screening potential of Mpro inhibitors. These results demonstrated that our MCAP-AuNP-based platform has great potential for discovering Mpro inhibitors and may accelerate the development of therapeutics against COVID-19.
Similar content being viewed by others
Introduction
The emergence of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has caused a global pandemic. The virus has exhibited a high rate of infectivity and the ability to spread rapidly across all age groups1. According to the World Health Organization (WHO), as of November 2023, there have been over 771 million confirmed infections and over 6.9 million cumulative deaths caused by the SARS-CoV-22. Although vaccines against SARS-CoV-2 are being deployed worldwide, coronavirus disease 2019 (COVID-19) cases have increased because of the emergence of SARS-CoV-2 variants, such as the Delta and Omicron variants, which can break through the protective barrier of vaccination3,4. Various therapeutics have been developed targeting the spike proteins or viral RNA to treat COVID-195,6. However, these drugs have some limitations, such as low efficacy7, various side effects due to frequent variations in the spiked protein7, and mitochondrial RNA dysfunction in humans8. Therefore, there is an urgent need to identify and develop effective antiviral drugs against SARS-CoV-2 to combat this fatal disease.
Viral proteolytic enzymes, including human immunodeficiency virus-1 (HIV-1) and hepatitis C virus (HCV) NS3/4A proteases, play essential roles in viral proliferation and assembly, thereby making them promising potential therapeutic targets9,10. Among these, the main protease (Mpro), a cysteine protease containing a His41–Cys145 catalytic dyad11, is an indispensable enzyme for SARS-CoV-2 replication and proliferation. The Mpro-mediated proteolytic post-processing of the SARS-CoV-2 replicase polyprotein, which cleaves at least 11 conserved sites, is crucial for viral assembly and maturation11. For example, Mpro generates amyloidogenic proteins (e.g., spike protein and non-structural protein 11) in SARS-CoV-2 proteosomes with multiple aggregation-prone regions, facilitating viral self-assembly12. Considering the proven success of protease inhibitors in treating HIV-1 and HCV infections, strategies that specifically target Mpro possess significant potential to thwart viral proliferation10. Importantly, as Mpro has no known homologs in the human proteome, it is feasible to develop effective and selective Mpro inhibitors without eliciting severe side effects11. Furthermore, the genetic similarity between the Mpro of SARS-CoV-2 and SARS coronavirus (SARS-CoV, discovered in 2002) is 90%, whereas their genomes overall have only 79% similarity1. Given its critical role in the viral life cycle and the absence of closely related homologs in the human proteome, Mpro is one of the most attractive targets for antiviral therapeutics to combat COVID-19. For instance, Paxlovid, an oral therapeutic that combines an Mpro inhibitor (PF-07321332) with ritonavir, has garnered FDA approval for the treatment of moderate to severe COVID-19 cases13. Moreover, compounds like Pfizer’s PF-07304814 and Simcere’s SIM0417, designed to inhibit Mpro, are currently undergoing clinical trials as potential oral treatments against SARS-CoV-214,15,16.
Recently, drug screening methods have been developed to discover Mpro inhibitors, such as virtual screening assays, fluorescence resonance energy transfer (FRET) assays, and cell-based assays17,18. However, screening for Mpro inhibitors has been hampered by a high screening cost19, poor reproducibility20, and long screening cycles20. Although structure-based virtual screenings have been highlighted for repurposing and designing pharmaceutical drugs targeting Mpro, the practical use of these in silico methods is hindered by high false-positives due to the conformational flexibility of Mpro and its protein-ligand dynamics21. Therefore, the development of highly rapid, cost-effective, and accurate drug screening methods is necessary to effectively identify medications against COVID-19.
Amyloids are misfolded proteins with a stable, unbranched, fibrous quaternary structure composed of repeating units of β-strands from protein or peptide monomers, and prone to easily self-assemble, forming highly organized fibrillar structure by intermolecular backbone hydrogen bonding22. Interestingly, owing to the well-organized supramolecular structure and exceptional physicochemical traits of amyloid proteins, they have been exploited in synthesizing functional hybrid nanocomposites containing gold nanoparticles (AuNPs), Quantum Dot, and graphene23,24. In particular, there has been some progress in developing protease-responsive nanomaterials that capitalize on the degradation of amyloid aggregates or fibrils. For example, Lee et al. synthesized amyloid corona by combining amyloid proteins and AuNPs as a drug screening platform for amyloid-β oligomer-degrading drugs25. In addition, Li et al. also reported the fabrication of highly conductive, biodegradable nanocomposites for biosensing applications to quantify enzymatic activity using complex structures composed of alternating layers of graphene and amyloid26. Therefore, considering the self-assembly properties of amyloid proteins, we speculated that a hybrid nanocomposite for drug screening can be synthesized with a combination of AuNPs and an amyloid peptide containing both Mpro cleavage and amyloid-forming sequences. By leveraging both the catalytic activity and the localized surface plasmon resonance (LSPR) property of AuNPs, this hybrid nanocomposite is applicable to a high-throughput screening platform for Mpro inhibitor candidates23,25,27.
Here, we designed a bioengineered amyloid peptide containing both amyloid-forming sequences from prion protein (GNNQQY)28 and the Mpro cleavage site (LQS)29 to develop a drug screening platform for discovering Mpro inhibitors (Supplementary Fig. S1). We named our amyloid protein Mpro cleavage site-embedded amyloid peptide (MCAP). We characterized the amyloid properties of MCAP using a thioflavin T (ThT) assay and atomic force microscopy (AFM). By combining MCAPs and AuNPs, we fabricated protease-sensitive nanocomposites, termed MCAP-AuNPs, wherein AuNPs were coated with the MCAP amyloid corona. The amyloid coronas on the surface of MCAP-AuNPs were easily degraded by Mpro, inducing the destabilization and aggregation of AuNPs. The aggregation of AuNPs caused a color change in an aqueous solution due to the intrinsic LSPR of AuNPs. This colorimetric change was used to evaluate the proteolytic activity of Mpro. By measuring the colorimetric response of MCAP-AuNPs, we evaluated the proteolytic activity of Mpro and monitored the efficacy of various Mpro inhibitors. Based on these investigations, we suggest that the MCAP-AuNP-based colorimetric screening system can be used to efficiently discover Mpro inhibitors for combating COVID-19 and the infection of related coronaviruses (COVID-X) in the future.
Results
Fabrication and engineering of the Mpro cleavage site-embedded amyloid peptide (MCAP)
Figure 1a shows the endosomal entry and replication process of SARS-CoV-230. This process is initiated by binding between human angiotensin-converting enzyme-2 (hACE-2) receptor and spike protein (S protein) of SARS-CoV-2. Following these interactions, the S protein undergoes proteolytic cleavage by cathepsin L, leading to the conversion of the protein into a metastable state, subsequently triggering the fusion of the host cell membrane with the virus31. Subsequently, the genomic RNA of the virus is released into the cytoplasm, initiating the translation of co-terminal polyproteins (pp1a/ab). These polyproteins are then cleaved into non-structural proteins (nsps) by Mpro. The nsps product interacts with nsp12 to assemble the replicase-transcriptase complex, which is required for viral genome replication and the transcription of sub-genomic RNAs. Thus, Mpro is essential for the survival of SARS-CoV-2.

a Schematic illustration of the replication of SARS-CoV-2 at the cellular level. Mpro inhibitors have the potential to prevent viral replication. The schematic illustration was created using BioRender.com. b Schematic illustration of MCAP comprising an amyloid sequence and an Mpro cleavage sequence. c ThT fluorescence measurement of MCAP and repeated Mpro cleavage sequence (Re, LQS-LQA-LQG-LQSS) with varying incubation time. The solid lines and error bars indicate the mean values and standard deviations derived from three independent experiments (n = 3). d Circular dichroism spectra of the MCAP solution upon incubation for over 96 h. e Height analysis of MCAP fibril using AFM. The bottom graph represents the cross-section height of the section indicated with the white dashed line. Scale bar: 2 μm. f Height analysis of MCAP fibrils incubated with Mpro for 12 h. Scale bar: 2 μm. The bottom graph represents the cross-section height of the section indicated with the white dashed line. The images of AFM in (e) and (f) were measured at least three times.
Viral infection and replication in vivo involve polyproteins that are cleaved by Mpro at L-Q ↓ (S, A, G) sequences, where ↓ marks the cleavage site29. Inspired by the interaction between Mpro and polyproteins, we designed the MCAP (Fig. 1b). Specifically, Mpro cleavage sequence (LQS) is inserted into the sequence of amyloid-forming peptides (GNNQQY). We speculate that MCAP may have amyloid-like properties and could be cleaved by Mpro, enabling us to analyze the proteolytic activity of Mpro and the inhibitory effects of various drug candidates.
To confirm the amyloid-like properties of MCAP, the solution with MCAP monomers was incubated at 37 °C for 120 h. The results showed that MCAP fibrils were synthesized within five days, confirming the fibril-forming ability of MCAP (Supplementary Fig. S2). We analyzed the persistence length of each MCAP fibril using transmission electron microscopy (TEM) image and Image J software. The analysis showed that the persistence length of MCAP fibrils was 229.95 ± 94.22 nm, shorter than that of the original amyloid sequence (GNNQQY, 1.9 ± 1.3 µm)28. In addition, the height of MCAP fibrils (2.64 ± 0.74 nm, Supplementary Fig. S3) considerably decreased compared to that of the fibrils with original amyloid sequence (43 ± 24 nm)28. These results support that the Mpro cleavage sequences counterbalance the amyloid properties of MCAP.
To investigate if the MCAP fibrils had a β-sheet structure, we first incubated MCAP monomers and monitored the kinetics of fibril formation by recording the fluorescence signal of ThT as they forms the β-sheet structure of amyloid fibrils (Fig. 1c). We observed a lag time of approximately 40 h, after which the growth phase was observed on the sigmoidal form of the ThT curve, strongly supporting the β-sheet formation of MCAP fibrils. In contrast, the peptide with repeated Mpro cleavage sequences (LQS-LQA-LQG-LQSS) only did not show any fluorescence signal for ThT compared to MCAP. Next, we performed circular dichroism (CD) analysis to monitor the β-sheet formation properties of MCAP depending on the incubation time (Fig. 1d). The results showed that the β-sheet peak gradually increased over time, validating the successful synthesis of MCAP fibrils.
Next, we incubated MCAP fibrils in the absence and presence of Mpro (0.1 mg mL-1) and performed AFM analysis to determine the proteolytic ability of Mpro against MCAP fibrils. Without Mpro, the bundles of MCAP protofibrils remain intact (Fig. 1e). In contrast, we observed that Mpro degraded MCAP fibrils into short fragments and small aggregates (Fig. 1f), confirming that Mpro successfully recognizes the Mpro cleavage sites in MCAP. The Fourier transform infrared (FT-IR) and CD analysis also revealed considerable reductions in β-sheet peaks of MCAP fibrils in the presence of Mpro, providing further evidence for the enzymatic degradation of MCAP fibrils by Mpro (Supplementary Fig. S4). Taken together, these results suggest that the bioengineered MCAP we generated possesses both amyloidogenic properties and enzymatic degradability due to the proteolytic activity of Mpro.
Furthermore, we designed engineered amyloid sequences MCAP2 (LQGNLQANQQNY) and MCAP3 (LQGNLQGNQQNY) to explore the availability of different Mpro cleavage sequences (Supplementary Table S1). The AFM, TEM, and CD analysis confirmed the amyloidogenicity of MCAP2, ensuring β-sheet-rich fibril structures (Supplementary Fig. S5), while the MCAP3 showed no such structure (Supplementary Fig. S6). Subsequently, the ThT analysis was conducted to confirm the degree of proteolytic activity of Mpro at the MCAP, the MCAP2, and the MCAP3, respectively (Supplementary Figs. S10a–c). The analysis showed a 42% reduction in the ThT fluorescence intensity for the MCAP fibrils and a 22% reduction for the MCAP2 fibrils by Mpro. In contrast, the MCAP3 aggregates exhibited no significant signal change after reacting with Mpro. These results suggest that both variants of GNNQQY combined with LQS and LQA exhibit amyloidogenicity, and in particular, the MCAP containing LQS has the highest degradability to Mpro.
Interestingly, other types of amyloid peptides can also be used as basic amyloid-forming sequences instead of prion sequences. To explore the availability of other amyloid-forming sequences, we designed various engineered sequences derived from amyloid-forming sequences such as islet amyloid polypeptide (IAPP, SNNFGAIL)32 and amyloid-β (KLVFFAE, GGVVIA)33. The IAPP-derived engineered amyloid sequence is SNLQSNFGAIL (i.e., IAPP MCAP), and the amyloid-β-derived sequences are KLLQSVFFAE (i.e., Aβ MCAP1) and GGLQSVVIA (i.e., Aβ MCAP2) (Supplementary Table. S1). To confirm the amyloidogenicity of each engineered peptide, we conducted AFM, TEM, and CD analysis (Supplementary Figs. S7–S9). The analyses showed that each engineered sequence formed fibrous aggregates with β-sheet structures. To assess the reactivity between Mpro and fibrils synthesized with each amyloid sequence, a ThT assay was conducted (Supplementary Fig. S10). The results showed that after reacting with Mpro, the ThT fluorescence intensity of the MCAP, IAPP MCAP, Aβ MCAP1, and Aβ MCAP2 fibrils decreased by 42%, 26%, 17%, and 23%, respectively. These results indicate that the MCAP possesses superior sensitivity in assessing the proteolytic activity of Mpro compared to other engineered amyloid sequences.
To investigate whether the core sequence of MCAP is cleaved by Mpro in fibrils, we strategically repositioned the Mpro-cleavage sequence and designed an engineered sequence (Repositioned MCAP, LQGNNQQNYLQG) (Supplementary Table S1). The AFM, TEM, and CD analysis revealed that the Repositioned MCAP formed fibrous aggregates with β-sheet structure (Supplementary Fig. S11). The reactivity test between Mpro and Repositioned MCAP fibrils, assessed through AFM analysis, revealed that the fibrillar structure remained unaffected by Mpro treatment (Supplementary Fig. S12). In addition, the CD spectra of Repositioned MCAP fibrils treated with Mpro exhibited negligible peak change (~10% reduction in ThT fluorescence) of the β-sheet structures, compared to the untreated fibrils. Based on these results, we confirmed that Mpro actively degrades only the MCAP fibrils that contain the Mpro-cleavage sequence at the core region of the engineered sequence.
Fabrication of MCAP amyloid corona using AuNPs
To synthesize MCAP amyloid corona with uniform size and morphology, we used AuNPs as the nucleation site of MCAP and fabricated MCAP-AuNP comprising MCAP amyloid corona (Fig. 2a). The negatively charged surface of AuNPs attracted the positively charged N-terminal of MCAP, leading to an increasing local concentration of MCAPs on the AuNP surface23. The increased concentration promotes the events of the formation of β-sheet structure and facilitates the nucleation of MCAPs on AuNPs. After 24 h incubation, amyloid corona was synthesized on AuNPs (MCAP-AuNP). Amyloid coronas on AuNPs predominantly tend to be hard protein coronas because they are interwoven with each other and irreversibly bind to the surface of AuNPs25,34. The amyloid corona can provide several properties to AuNPs, including steric stabilization35,36, antifouling37, and high salt resistance23,25.

a Schematic illustration of the fabrication process of MCAP-AuNPs. b TEM image of MCAP-AuNPs. The white scale bar is 50 nm in size. TEM images were measured at least three times. c UV-vis spectra of MCAP-AuNPs and bare AuNPs. The spectra peak was shifted 3 nm from bare AuNPs to MCAP-AuNPs. d XPS analysis of MCAP-AuNPs and bare AuNPs. The N 1 s peak was only measured in MCAP-AuNPs. e ζ-potential of the bare AuNPs, the MCAP-AuNPs, and the MCAP fibrils Data are presented as mean values ± standard deviation (SD) from n = 3:4:4 independent experiments. The hydrodynamic diameter of bare AuNPs and MCAP-AuNPs (f) before and (g) after freezing. Data are presented as mean values ± SD from n = 4 independent experiments for bare AuNPs and n = 3 for MCAP-AuNPs, respectively. The inserted pictures are actual photographs of each sample.
We optimized the stability of the MCAP-AuNPs by varying the initial concentration of MCAP monomer (Supplementary Fig. S13). At a low concentration of MCAP monomer (<4.04 μM), the ultraviolet-visible (UV-vis) spectra of MCAP-AuNPs shifted from purple to red, because the MCAP monomers cannot fully cover the surface of AuNPs, inducing only a partial aggregation of AuNPs. Similarly, the extent of AuNP aggregation increased at higher concentrations of MCAP monomer (>4.04 μM). Thus, we determined that the optimal concentration of MCAP monomer was 4.04 μM because it was the concentration at which MCAP fully covered the surface of the AuNPs with a high yield (~98%, Supplementary Fig. S14). Moreover, good steric stability was observed in that setup for several weeks. Therefore, we confirmed that the MCAP amyloid corona was homogeneously coated on the surface of each AuNP, revealing long-term steric stability (Fig. 2b and Supplementary Fig. S14). In addition, the FT-IR analysis of the MCAP-AuNPs showed that the MCAP aggregates on the surface of AuNPs have β-sheet-rich conformation (Supplementary Fig. S15).
Physicochemical characteristics of the fabricated MCAP-AuNPs
The LSPR peak in the UV spectra of bare AuNPs and MCAP-AuNPs shifted from ~520 nm to ~523 nm (Fig. 2c). This result indicates that the MCAP amyloid corona covered the surface of the AuNPs, partially increasing the local refractive index of the MCAP-AuNPs38. X-ray photoelectron spectroscopy (XPS) also confirmed the encapsulation of the MCAP amyloid corona on AuNPs because, when compared to the spectrum of bare AuNPs, only the MCAP-AuNP spectrum showed N 1 s peaks (~400 eV) (Fig. 2d). The zeta potential of the bare AuNPs, MCAP-AuNPs, and MCAP fibrils were −53.92 mV, −36.54 mV, and −30.96 mV, respectively. The results indicate that the MCAP-AuNPs are more similar in zeta potential with the MCAP fibrils as compared to the bare AuNPs, thereby supporting successful MCAP-coating on the surface of AuNPs (Fig. 2e).
To verify the stability of the MCAP-AuNPs, we conducted a temperature-dependent stability test and a freeze-thaw test of the MCAP-AuNPs. The temperature-dependent stability test showed that the MCAP-AuNPs were stable at 40 °C but unsteady at 60 °C, thereby inducing particle aggregation due to thermal denaturation (Supplementary Fig. S16). Before freezing, the hydrodynamic diameters of the bare AuNPs and MCAP-AuNPs were 19.02 ± 0.53 nm and 23.18 ± 0.29 nm, respectively, which were consistent with the result in Fig. 2c and Supplementary Fig. S14 (Fig. 2f). The MCAP-AuNPs were ~4 nm larger than the bare AuNPs, corresponding to the TEM image in Fig. 2b. After freeze-thawing, the bare AuNPs aggregated, and their hydrodynamic diameters were not observed (Fig. 2g). In contrast, the MCAP-AuNPs retained their initial hydrodynamic diameters after the freeze-thaw process and were durable up to the fourth cycle of the freeze-thaw test (Supplementary Fig. S17), indicating that the MCAPs were irreversibly bound to AuNP surface as a hard corona25,39. These results also suggest that the MCAP-AuNPs have good cryopreservation storage capacity that prevents the denaturation of MCAPs.
Proteolytic activity of Mpro as measured via MCAP-AuNPs
Figure 3a shows our MCAP-AuNP-based strategy for monitoring the proteolytic activity of Mpro. When Mpro is added to an MCAP-AuNP solution, Mpro degrades the MCAPs on the AuNP surface, exposing the surface of bare AuNPs and promoting their aggregation under physiological conditions40. The aggregation of AuNPs leads to an LSPR shift in its spectrum, causing a color change from red to purple. However, inactivated Mpro cannot degrade the MCAP on the surface of AuNPs. Thus, MCAP-AuNPs remain intact in the solution, whose color does not change. Based on this strategy, we quantitatively measured the proteolytic activity of Mpro.

a Schematic illustration of the principle of measuring the proteolytic activity of Mpro using MCAP-AuNPs. Mpro degrades the MCAP molecules on the AuNP, inducing particle aggregation. However, denatured Mpro cannot induce particle aggregation because it does not have proteolytic activity. b UV–vis spectra of MCAP-AuNPs reacting with various concentrations (1–7: 0.19, 0.56, 0.93, 1.85, 5.55, 9.25, and 18.5 nM) of intact Mpro (upper) and denatured Mpro (bottom). c Plots fitted using the sigmoidal dose-response curve as a function of Mpro concentration. d TEM image of MCAP-AuNPs that reacted with Mpro (left) and denatured Mpro (right). The size of white scale bar is 50 nm. e Selectivity test of MCAP-AuNP. The relative ratio (A650/A525) of MACP-AuNPs was measured after reacting with Mpro, denatured Mpro, HSA, glucose, IgG, and BSA for 24 h. f Concentration-dependent Mpro activity test using MCAP-AuNP platform and FRET-based assay. All data in (c), (e), and (f) are presented as mean values ± SD from n = 3 independent experiments. Error bars are not shown where they are smaller than the circle symbols.
To validate our strategy by measuring the proteolytic activity of Mpro, we quantified the degree of MCAP degradation by intact and denatured Mpro using UV-vis spectra (Fig. 3b). The results revealed that the MCAP-AuNP solution exhibited an LSPR shift from red to purple depending on the Mpro concentration (0.19, 0.56, 0.93, 1.85, 5.55, 9.25, or 18.5 nM). A higher concentration of Mpro induced a larger red shift in the LSPR, indicating that the MCAP-AuNPs aggregated actively at high Mpro concentrations. In contrast, for denatured Mpro, which was incubated at 90 °C for 4 h, the UV-vis spectra of the MCAP-AuNPs remained unchanged (Fig. 3b). This indicates that denatured Mpro cannot degrade the MCAP-AuNPs, resulting in no particle aggregation of them.
For the UV-vis spectra of the MCAP-AuNP solution, the absorbances at A650 and A525 represent the degrees of MCAP-AuNP aggregation and dispersion, respectively. Thus, the relative absorbance ratio A650/A525 was adopted to quantify the degree of MCAP-AuNP aggregation. We found that the A650/A525 ratio of the MCAP-AuNP solution increased depending on the Mpro concentration (Fig. 3c). In contrast, a concentration-dependent A650/A525 ratio change was not observed using denatured Mpro. These results correspond with the TEM images in Fig. 3d. The TEM images showed that an increased aggregation of MCAP-AuNPs took place at high concentrations of Mpro and no aggregation at denatured Mpro. To quantitatively represent the proteolytic activity of Mpro, we analyzed the A650/A525 ratio of MCAP-AuNP solution as a function of Mpro concentration using a sigmoidal dose-response model, shown in Eq. (1) below.
From this equation, the half-maximal effective concentration (EC50) was 4.297 nM and the maximal efficacy was 1.215 (a.u.). This suggests that 4.297 nM of Mpro is sufficient to mediate the massive aggregation of MCAP-AuNPs. We also investigated the concentration- and time-dependent proteolytic activity of Mpro induced by the MCAP-AuNPs (Supplementary Fig. S18). A red shift in the UV spectra was observed at higher Mpro concentrations and reaction times. The selectivity tests of MCAP-AuNPs were conducted with various interfering biomolecules abundant in physiological conditions, such as bovine serum albumin (BSA), immunoglobulin, glucose, and human serum albumin (HSA). The results showed that the MCAP-AuNPs were not affected by these biomolecules and selectively reacted with Mpro (Fig. 3e and Supplementary Fig. S19). Thus, we concluded that MCAP-AuNPs are a suitable platform for measuring the proteolytic activity of Mpro.
We also measured Mpro activity using the commercialized FRET substrate MCA-AVLQSGFR-Lys(Dnp)-Lys-NH2 trifluoroacetate (Sigma Aldrich, USA) (Fig. 3f). We obtained the EC50 values of the FRET system from the sigmoidal dose-response curve as a function of Mpro concentration. The EC50 of the FRET system was estimated to be 211 nM, which is approximately 50 times higher compared to that of our platform (4.4 nM). Furthermore, we observed that the MCAP-AuNP-based system displayed a much lower variance for each data point, compared to the FRET-based system. These results suggest that our MCAP-AuNP-based system offers superior sensitivity and accuracy in measuring Mpro activity as compared to the FRET-based system. Additionally, it requires fewer amounts of expensive enzymes, such as Mpro, for screening.
In our MCAP-AuNP platform, a very small portion of the MCAPs exists in the form of free MCAPs in the supernatant of the MCAP-AuNP solution. These free MCAPs may affect Mpro activity. To scrutinize the effect of free MCAPs, we conducted a Mpro activity test of the MCAP-AuNP solutions with and without free MCAPs (Supplementary Fig. S20). Firstly, we ensured that the free MCAPs were fully removed after centrifugation of the MCAP-AuNP solution. Then, the Mpro activity test was conducted. The results showed that the as slightly shifted from 3.5 nM to 2.1 nM in the presence of the free MCAPs. This behavior indicates that the majority of MCAPs are irreversibly bound to the surface of AuNPs as hard corona. The existence of a small amount of free MCAPs slightly changes the apparent activity of Mpro. For more precise screening, it is recommended to eliminate the free MCAPs prior to drug screening.
Molecular docking analysis of Mpro and its potential inhibitors
Before screening potential inhibitors with MCAP-AuNPs, we conducted molecular docking to evaluate their interaction with the active sites of Mpro. The active site cleft between domains I and II houses the catalytic dyad His41-Cys145, which is known to play a critical role in the proteolytic activity of Mpro41. Based on our analysis, we selected four competitive inhibitors with high binding affinities that directly bind to this crucial catalytic active site for further experiments. We compared their pharmacokinetic properties obtained through molecular docking simulations and the results from the MCAP-AuNPs-based screening platform.
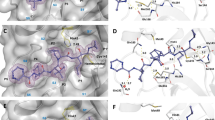
Figure 4 shows the interacting residues and molecular structures of the binding pocket of Mpro with ebselen, leupeptin, hesperetin, and lopinavir. Ebselen is known for its antioxidative, anti-inflammatory, and cytoprotective properties against COVID-19 and has also been investigated for other diseases, such as hearing loss and bipolar disorders42. Figure 4a shows the molecular docking of ebselen and Mpro. At the catalytic site of Mpro, the carbonyl oxygen of ebselen interacts with the Asn142 and Gln189 side chains of Mpro through hydrogen bonding. In addition, hydrophobic contacts between ebselen and the His41, Cys145, Met165, Pro168, Met49, and His164 of Mpro can be made43. The interaction between the catalytic dyad of Mpro and ebselen inhibits the proteolytic activity of Mpro.

Molecular structure of the binding pockets of Mpro (PDB ID: 6LU7) with (a) ebselen, (b) leupeptin, (c) hesperetin, and (d) lopinavir. Orange, gray, blue, red, and black balls indicate selenium, carbon, nitrogen, oxygen, and hydrogen atoms, respectively. Hydrogen atoms bonded to carbon atoms are omitted for clarity.
For leupeptin, a well-known covalent inhibitor of threonine, cysteine, and serine proteases44, its C-terminal aldehyde group reacts with the Cys145 of Mpro to form a hemithioacetal45 (Fig. 4b). In addition, leupeptin forms a water-mediated interaction with the side chain of Glu189 and hydrogen bonds with the main chain of His164 and Cys145 in Mpro. These interactions can reduce the stability of the Mpro-leupeptin complex45. Similarly, hesperetin, which has neuroprotective effects against neurodegenerative diseases, forms a single hydrogen bond with the His41, Leu141, Cys145, Glu166, Arg188, and Thr190 of Mpro (Fig. 4c)46. Lopinavir, which is used to treat HIV infection, interacts with the His41, Cys145, Gln189, Met164, Met49, Glu166, and Leu27 residues of Mpro (Fig. 4d). Thus, the interaction between the inhibitors and residues of Mpro, especially the active sites His41 and Cys145, has the possibility to reduce the proteolytic activity of Mpro.
Next, we evaluated the docking binding free energy (ΔGDock) between four potential inhibitors and the active sites of Mpro to characterize the protein-ligand association. The results revealed that hesperetin (−7.3 kcal/mol) has the highest binding affinity, followed by ebselen (−6.6 kcal/mol), lopinavir (−6.4 kcal/mol), and leupeptin (−6.0 kcal/mol). These values are consistent with those reported in previous studies47,48,49,50. Inhibition constants (\({K}_{i}^{{MD}}\)), representing the potency of each inhibitor, were calculated as below;
where, R is the gas constant (1.987 × 10–3 kcal K-1 mol−1) and T is the temperature in Kelvin (298.15 K). The \({K}_{i}^{{MD}}\) values of hesperetin, ebselen, lopinavir, and leupeptin were determined to be 4.45 μM, 14.51 μM, 20.34 μM, and 39.95 μM, respectively. The ΔGDock and \({K}_{i}^{{MD}}\) values obtained from the docking of selected potential inhibitors against Mpro are summarized in Supplementary Table S2.
MCAP-AuNP-based platform for Mpro inhibitor screening
To validate the efficacy of Mpro inhibitors, we designed an MCAP-AuNP-based screening platform and measured the inhibition efficacy of ebselen, leupeptin, hesperetin, and lopinavir against Mpro using MCAP-AuNPs. Figure 5a shows the working principle of the MCAP-AuNP-based Mpro inhibitor screening platform. In detail, the Mpro-induced proteolysis of MCAP encapsulated on the AuNP surface caused the sterically stabilized MCAP-AuNPs to become unstable and prone to aggregation. Inhibitors that bind to the proteolytic active site of Mpro hamper the Mpro-induced degradation of MCAPs on the surface of AuNPs. The degree of MCAP-AuNP aggregation depended on the state of Mpro inactivation, which was proportional to the concentration of Mpro inhibitors. To investigate the efficacy of the inhibitors, we measured the degree of MCAP-AuNP aggregation using UV-vis spectroscopy (Fig. 5b). In our drug screening platform, the concentration of Mpro was kept constant (18.5 nM), and each Mpro solution was pre-incubated with various concentrations of inhibitors for 20 min before reacting them with MCAP-AuNPs.

a Schematic illustration of the principle of measuring the inhibition efficacy of drugs using MCAP-AuNP. The degree of MCAP-AuNP aggregation was measured as the inhibitor-controlled Mpro activity. b Schematic illustration of measuring the efficacy of Mpro inhibitors using the colorimetric change in MCAP-AuNP solutions. The colorimetric change of the solution represents the inhibition efficacy of the drug against Mpro. The bottom image shows the actual photos of MCAP-AuNP solutions mixed with various concentrations of hesperetin (0, 4.6, 23.1, 46.3, 92.5, 138.8, 161.9, and 185 μM). The dose-dependent inhibition ratio (%) curves of the MCAP-AuNP solution depending on various concentrations of Mpro inhibitors: (c) ebselen, (d) leupeptin, (e) hesperetin, and (f) lopinavir (n = 3 for each). Michaelis-Menten plots of MCAP-AuNP solutions treated with various Mpro inhibitors: (g) ebselen, (h) leupeptin, (i) hesperetin, and (j) lopinavir. All data are presented as mean values ± SD from n = 3 independent experiments. Error bars are not shown where they are smaller than the circle symbols. The bottom table contains the fitting values for each graph.
The LSPR shifts of the MCAP-AuNP solutions decreased depending on the concentration of various inhibitors (Supplementary Fig. S21). Because each inhibitor has different efficacies, we defined the inhibition ratio (%) to quantify the inhibition efficacy of each inhibitor as follows:
where A650 and A525 are the absorption spectra at 650 and 525 nm, respectively, and the maximum \({A}_{525}/{A}_{650}\) is the absorbance when the MCAP-AuNPs are not aggregated.
We also analyzed the inhibition ratio as a function of inhibitor concentration using a sigmoidal dose-response model (Fig. 5c–f). The equations generated for each inhibitor are listed in Supplementary Table S3. From these equations, we determined the half-maximal inhibitory concentration (IC50) values of ebselen, leupeptin, hesperetin, and lopinavir, which were 0.39 μM, 28.57 μM, 43.14 μM, and 10.7 μM, respectively (Fig. 5h). Among these four clinical trial drug candidates, ebselen exhibited the strongest Mpro inhibition activity (IC50 = 0.39 μM), consistent with previous studies11. In addition, we also compared the inhibitory efficacy of two promising antiviral flavonoids, hesperetin and hesperidin, using our platform. Hesperidin, a glycoside containing rutinose (α-L-rhamnopyranosyl-[1→6]-β-D-glucopyranose) linked to the OH-7 of hesperetin, exhibited an IC50 value of 369.4 μM, which was much higher than that of hesperetin (43.14 μM) (Supplementary Fig. S22). This is consistent with previous studies showing that hesperetin has a more substantial inhibitory effect on Mpro than hesperidin51. From these results, we verified that our MCAP-AuNP-based screening platform has excellent capability for validating and quantifying the drug efficacy of Mpro inhibition.
The enzyme kinetics of Mpro was controlled by its inhibitors, so the reaction rate between Mpro and MCAP-AuNP was evaluated depending on the MCAP-AuNP concentration (Fig. 5g–j). This can be represented by Michaelis-Menten model as follows:
where V0 is the initial velocity of the reaction, Vmax is the maximum velocity of the reaction, KM is the Michaelis constant, and [S] is the MCAP-AuNP concentration.
Here, the Vmax and KM were determined depending on the inhibitor concentration (Fig. 5k). We found that the reaction rate tended to decrease at higher inhibitor concentrations. At a high concentration of each inhibitor (2.27 μM ebselen, 81.78 μM leupeptin, 13.6 μM hesperetin, and 27.26 μM lopinavir), the resulting Vmax was approximately 1.18 ~ 1.2-fold lower than that of non-inhibited Mpro. As for ebselen, a relatively low concentration (1.36 μM) decreased the reaction rate, consistent with its relatively low IC50 value (0.39 μM). The TEM image also confirmed the inhibitory effects of each inhibitor in Supplementary Fig. S23. These results indicate that our MCAP-AuNP-based system can be used to evaluate the proteolytic activity of Mpro as controlled by its inhibitors.
We determined the values of experimental inhibition constant \(({K}_{i}^{{Exp}})\) for the four potential inhibitors utilizing the IC50 and KM values obtained from the results with our screening platform. The four inhibitors are competitive and directly interact with the active site of Mpro. Therefore, we calculated \({K}_{i}^{{Exp}}\) values using IC50 and KM values in the following equation52.
Where IC50 is the half-maximal inhibitory concentration, KM is the Michaelis constant, and [S] is the MCAP-AuNP concentration. The \({K}_{i}^{{Exp}}\) values of ebselen, leupeptin, hesperetin, and lopinavir were determined to be 0.23 μM, 16.69 μM, 25.19 μM, and 6.25 μM, respectively. In contrast to the results of molecular docking simulations, ebselen exhibited the lowest \({K}_{i}^{{Exp}}\) value, indicating the highest binding affinity to Mpro in the experiment. The \({K}_{i}^{{Exp}}\) values, along with \({K}_{i}^{{MD}}\) values obtained from the docking simulation for selected potential inhibitors against Mpro, are summarized in Supplementary Table S2. The results revealed that \({K}_{i}^{{Exp}}\) values differed by about 0.17 to 63.71 times compared to each \({K}_{i}^{{MD}}\) value, demonstrating that the theoretically calculated binding affinity cannot perfectly reflect the experimental value21. Our MCAP-AuNP-based screening platform can overcome the limitations of molecular docking by providing precise experimental validation for pharmacokinetic analysis. Taken together, we confirmed that our platform provides a potential means for precisely evaluating both the Mpro activity in the presence of an inhibitor and its drug efficacy in a fast and label-free manner.
Discussion
Despite the rollout of various COVID-19 vaccines, accumulating evidence indicates that the efficacy of these vaccines has diminished against new SARS-CoV-2 variants (e.g., Delta and Omicron) owing to frequent mutations in the S protein53. Therefore, there is an urgent need to develop antiviral drugs to combat COVID-19 and complement the therapeutic effect of currently available preventive vaccines. Mpro, an essential enzyme for viral replication and proliferation, is an outstanding pharmaceutical target for SARS-CoV-2 because it is highly conserved among all coronaviruses6.
Recent studies have revealed that small molecules that bind to the catalytic dyad (His41 and Cys145) and inhibit the proteolytic activity of Mpro are desirable as antiviral therapeutics. Various strategies have been employed to identify Mpro inhibitors that can bind to its catalytic dyad, including in silico drug discovery tools such as molecular docking54,55,56. Despite the high performance of structure-based virtual screening techniques, these methods are limited by several inherent drawbacks. Specifically, the limited consideration of protein flexibility and conformational changes poses significant challenges in predicting the accurate pharmacokinetic properties of Mpro inhibitors21. Therefore, experimental validation of the docking results is crucial for developing antiviral drugs with adequate pharmacokinetic analysis. Despite numerous papers on structure-based virtual screening of potential Mpro inhibitors, only few studies have reported experimental confirmation to validate their docking results11,57. The lack of empirical evaluation is mainly due to the limited access to biological facilities and the lack of in vitro screening techniques. Moreover, SARS-CoV-2 infection and propagation in cell culture require qualified professionals and must be performed in biosafety level-3 (BSL-3) facilities58. Given these critical bottlenecks in the conventional methods, a novel strategy is necessary to develop a rapid, simple, and label-free drug screening platform.
In this study, we developed an MCAP-AuNP-based colorimetric screening platform to discover Mpro inhibitors. Our platform allows rapid (<2 h), label-free, real-time monitoring of drug efficacy, simultaneously across hundreds of compounds with minimal enzyme usage (<10 nM). To optimize our high-throughput screening platform, we considered three crucial factors. First, it should be simple and robust during fabrication and assay processes. We designed the MCAP sequence using the convergence of amyloidogenic and Mpro-cleavable sequences. We confirmed that the bioengineered MCAP sequence facilitates the formation of amyloid fibrils through self-assembly and that Mpro can readily degrade these self-assembled MCAP sequences. Furthermore, our findings demonstrated that the MCAP exhibits superior performance compared to other engineered sequences derived from various types of amyloid sequences. AuNPs provide a nucleation site for MCAP assembly and colorimetric responses via the proteolytic activity of Mpro. Second, the homogeneity and stability of MCAP-AuNPs are necessary for highly reproducible high-throughput drug screening. We optimized the size and stability of MCAP-AuNPs to maximize the reproducibility of our platform. Specifically, we optimized the concentration of MCAP monomers to fabricate a homogeneously coated amyloid corona. These MCAP-AuNPs maintained stability in a high-salt buffer solution even after repeated freeze-thaw cycles, which is attributed to the amyloid hard corona. Third, our platform necessitates high sensitivity to accurately monitor the proteolytic activity of Mpro. We employed MCAP-AuNPs as a protease-sensitive nanomaterial to dynamically track the activity of Mpro over time. Active Mpro progressively cleaved the MCAP, resulting in the loss of the amyloid corona and aggregation of MCAP-AuNPs. These proteolytic reactions triggered colorimetric responses in MCAP-AuNPs, achieving a sensitivity that is remarkably 50 times lower than that of the commercialized FRET method. Furthermore, it exhibited selectivity only for Mpro. These results underscore the performance of our platform in monitoring Mpro’s proteolytic activity, rendering it optimal for assessing the efficacy of Mpro inhibitors.
Finally, various drug candidates including four reagents (ebselen, hesperetin, leupeptin, and lopinavir) as Mpro inhibitors were applied to our MCAP-AuNP-based drug screening platform. From the experiments, we demonstrated that this strategy with MCAP-AuNPs precisely identifies effective drugs that inhibit the proteolytic activity of Mpro. Furthermore, we successfully measured the IC50, Vmax, and Km values of each drug candidate using our drug screening platform, and these values were comparable to those reported previously11,42,45,59,60,61. From the pharmacokinetic results, we also determined the \({K}_{i}^{{Exp}}\) values and compared them to the \({K}_{i}^{{MD}}\) values to assess the discrepancy between experimental and simulated values. Although further validation, such as immunoassays or cellular assays, is required for drug candidates that tested positive, our platform can dramatically reduce the time and cost associated with the drug discovery process and by extent the subsequent validation steps62. In addition, this simple and rapid screening system can be applied to drug screening for COVID-X in the future because of the high genetic similarity of Mpro within the coronavirus family.
In this study, we designed an Mpro inhibitor screening platform based on colorimetric changes in MCAP-AuNP solutions. This rapid, label-free screening platform capable of real-time monitoring may provide a highly efficient and specific drug discovery system for COVID-19. This MCAP-AuNP-based strategy uses a bioengineered peptide, MCAP, which comprises an Mpro cleavage site and an amyloid-forming sequence. This mimics the in vivo process wherein Mpro cleaves the LQS sequence of polyproteins into nsps that are essential for viral replication. In addition, using the self-assembly property of the amyloid sequence, MCAP proteins can be coated on the surface of each AuNP to form an amyloid corona. The MCAP-AuNPs allowed the quantitative measurement of the proteolytic activity of Mpro via the colorimetric response due to the intrinsic plasmonic properties of AuNPs. We also confirmed that this screening platform was suitable for Mpro inhibitor screening by evaluating the inhibition efficacy of Mpro inhibitors (e.g., ebselen, leupeptin, hesperetin, and lopinavir). We anticipate that our MCAP-AuNP-based platform can rapidly screen drug candidates in a chemical database to find antiviral therapeutics for COVID-19 and forthcoming COVID-X.
Methods
Reagents
Lyophilized SARS-CoV-2 main protease (Mpro) was purchased from Biosynth Carbosynth (UK). Mpro cleavage site-embedded amyloid peptides (MCAP; LQGNLQSNQQNY, MCAP2; LQGNLQANQQNY, MCAP3; LQGNLQGNQQNY, IAPP MCAP; SNLQSNFGAIL, Aβ MCAP1; KLLQSVFFAE, Aβ MCAP2; GGLQSVVIA) were commercially synthesized from Peptron (South Korea). Ebselen, hesperidin, hesperetin, lopinavir, leupeptin, chloroauric acid trihydrate (HauCl4 ∙ 3H2O), hydrochloric acid (HCl), thioflavin T (ThT), bovine serum albumin (BSA), immunoglobulin G (IgG), glucose, human serum albumin (HSA), trisodium citrate, and MCA-AVLQSGFR-Lys(Dnp)-Lys-NH2 trifluoroacetate were purchased from Sigma-Aldrich (USA). Distilled water (DW) and phosphate-buffered saline (PBS) were purchased from Gibco (USA).
Preparation of engineered amyloid peptides (MCAP, MCAP2, MCAP3, IAPP MCAP, Aβ MCAP1, Aβ MCAP2, Repositioned MCAP) in vitro and fabrication of engineered amyloid fibrils
The monomers of each engineered amyloid peptide were purified as an acetate salt to prevent reacting to other agents. To aliquot the engineered amyloid peptides, lyophilized monomers were dissolved in DW. The monomer solutions (50 μL; 1 mg mL−1) were distributed into 1.7-mL microcentrifuge tubes and stored at -20 °C in a freezer before the further experiment. To fabricate fibrils of each engineered amyloid peptides, 150 μL of DW (pH 2) was added to 50 μL of monomer solutions, and the mixture was incubated in a shaking incubator (Eppendorf, Germany) at 37 °C with shaking at 1000 Hz for 5 days.
ThT fluorescence measurement for monitoring MCAP fibrillation
To investigate amyloid fibrillation kinetics, ThT fluorescence assays were performed using a microplate reader (Synergy H1 Multi-Mode Reader, BioTek, USA) at an excitation wavelength of 440 nm and an emission wavelength of 485 nm. First, 1 mg mL-1 of MCAP solution was prepared by diluting the peptide in Milli-Q water. Subsequently, DW (pH 2) and a 1 mM ThT solution were added to the peptide solution until the final concentrations of the peptide and ThT were 250 µg mL-1 and 20 µM, respectively. ThT intensity was recorded at steps of 30 min and performed at 37 °C for 99 h with continuous orbital shaking (807 CPM).
ThT analysis of engineered amyloid fibrils
Before the ThT intensity measurement, each type of engineered amyloid fibrils was treated with Mpro for 4 h. After that, each 100 μM solution of engineered amyloid fibrils was treated with 20 μM of ThT molecules for 1 h. Finally, the ThT fluorescence intensities were measured by excitation at 444 nm and emission at 510 nm using a microplate reader (HIDEX, Japan).
Synthesis of AuNPs
To remove chloroauric acid residues, a round beaker was soaked in aqua regia (a 1:3 mixture of HNO3 and HCl) and rinsed with DW. The citrate reduction method was used to synthesize AuNPs. In detail, 2.5 mL of 38.8 mM HAuCl4 solution was mixed with 45 mL of Millipore water in a round beaker, and the mixture was heated to 100 °C with stirring at 1,200 rpm. After boiling, 1 mL of 80 mM sodium citrate solution was added to the mixture, and the final solution was boiled again for 1 h under stirring at 1,200 rpm to make AuNP solution. The final AuNP solution was cooled to room temperature (25 °C) and stored at 4 °C. AuNP synthesis was confirmed by measuring their hydrodynamic diameter and polydispersity index (PDI) using Zetasizer Nano S90 (Malvern Instruments, UK).
Synthesis of MCAP-AuNP
A 10:1 mixture of DW and PBS was added to the aliquoted MCAP to make a 0.1 mg mL-1 MCAP solution. And then, for 1000 μL of AuNP solution, 960 μL of supernatant was removed via centrifugation for 20 min at 6720 × g. Next, 40 μL of the AuNP solution and 110 μL of DW were added to 50 μL of the MCAP solution. The final 200 μL solution was incubated for 24 h (37 °C with shaking at 1000 Hz) in a thermomixer (Eppendorf, Germany) to fabricate MCAP-AuNP solution. Successful MCAP-AuNP synthesis was confirmed using FE-TEM (JEM-2100F, Japan) imaging and hydrodynamic diameter measurements.
Atomic force microscopy (AFM) analysis
Before topological conformation and height analysis using AFM, a silicon wafer was rinsed with a piranha solution (a 1:1 mixture of H2SO4 and H2O2). Fifty microliters of the MCAP monomer and fibril solution were deposited on the silicon wafer at room temperature for 20 min, washed with DW, and then dried for 12 h in a fume hood. AFM analysis was performed using an NX10 system (Park Systems, South Korea) with non-contact cantilever probes (NCHR, Park Systems, South Korea). AFM measurements were conducted in tapping mode at a scanning rate of 0.4-Hz and an image size of 5 × 5 μm. Image flattening and topological analysis were conducted using Park Systems’ Smart Scan software.
Circular dichroism (CD) analysis
Before the conformational analysis of MCAP fibrils using CD, MCAP fibrils were synthesized with varying incubation times. Briefly, 150 μL of DW (pH 2) was added to 50 μL aliquots of MCAP solution, and each mixture was incubated in a shaking incubator at 37 °C with 1000-Hz shaking for 12, 24, 36, 48, 60, and 96 h. Each sample was then deposited in a quartz glass cuvette (Aireka Cells, USA) with a 1 mm path length and 10 mm internal width. The CD spectra of each sample were measured using a J-815 system (Jasco, Japan), at a detection range of 190–300 nm and a scanning rate of 10 nm min-1. The spectra had a resolution of 8 nm and were analyzed using CDTool software (Birkbeck College, UK).
Fourier transform infrared (FT-IR) analysis
To perform conformation analysis of MCAP fibrils using FT-IR, 150 μL of DW (pH 2) was added to 50 μL of MCAP solution, and the mixture was incubated in a shaking incubator at 37 °C for 5 days to make MCAP fibrils. The MCAP fibril solution was then centrifuged at 20,000 × g for 1 h and the supernatant was removed. The resulting pellet was deposited on a silicon wafer and dried for 12 h in a fume hood. FT-IR spectra were measured using a Cary 630 FTIR Spectrometer (Agilent Technologies, USA) with a scanning range of 1600–1700 cm-1. The spectra had a resolution of 4 nm and were analyzed using Agilent MicroLab software.
X-ray photoelectron spectroscopy (XPS) analysis
The supernatants of the MCAP-AuNP and bare AuNP solutions were removed via centrifugation (20 min, 6720 × g). The pellets obtained after centrifuging each solution (<40 μL) were deposited on a silicon wafer and dried for 24 h in a fume hood. For XPS measurements, a K-alpha instrument (Thermo VG, UK) was used with monochromatic X-ray source (Al Kα line:1486.6 eV) at an ultra-high vacuum condition (4.8 × 10−9 mbar). Elemental scans of Au and N peaks were detected using a pass energy of 40 eV, a step size of 0.1 eV, and scanning a range of 0–800 eV.
UV-vis absorbance measurement
The UV-vis absorption spectra of each sample were measured using a spectrophotometer (PerkinElmer, USA), at a scan range of 400–800 nm, and a scan rate of 600 nm min−1. For the US-vis absorbance spectra of AuNPs smaller than 20 nm, which have a dispersed state, the absorbance spectra peaks were detected at 525 nm (A525). When the AuNPs aggregate, which leads to a color change in solution, the absorbance peak shifts near 650 nm (A650). Thus, the degree of AuNP aggregation could be quantified by calculating the A650/ A525 ratio.
Freeze-thaw performance
One milliliter of MCAP-AuNP solution and bare AuNP solution in microcentrifuge tubes were frozen at −80 °C for 2 h. The frozen solutions were defrosted at room temperature for 2 h. This process was performed 8 times for repeated freeze-thaw tests. The hydrodynamic diameter of the MCAP-AuNP solution and bare AuNP solution before and after the freeze-thaw performance was measured using a Zetasizer. In addition, the UV-vis absorbance wavelength ranging from 400 to 800 nm of each solution before and after the freeze-thaw process was measured using a spectrophotometer. Each solution was photographed using a Galaxy Note 20.
Molecular docking studies
To perform the molecular docking analysis, we followed a three-step protocol using three different software tools: Biovia Discovery Studio 2021 Client, UCSF Chimera version 1.14, and PyRx version 0.8.
Firstly, we retrieved the crystal structure of Mpro (PDB ID: 6LU7) from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) in.pdb format. We used Biovia Discovery Studio 2021 Client to remove unnecessary molecules, such as ions, inhibitors, and water molecules, and then loaded the processed structure of Mpro into UCSF Chimera version 1.14. We performed Dock Prep to add hydrogen atoms and converted the processed structure of Mpro into.pdbqt format using PyRx version 0.8.
Secondly, we retrieved the 3D structures of potential inhibitors of Mpro, ebselen (CID: 3194), hesperetin (CID: 72281), leupeptin (CID: 72429), and lopinavir (CID: 92727), from the PubChem database in.sdf format. We imported all four ligands into PyRx software, conducted energy minimization and geometrical confirmation using OpenBabel toolbox, and then converted the ligands into.pdbqt format.
Finally, we performed molecular docking analysis using AutoDock Vina inbuilt PyRx software version 0.8, based on the grid box approach. The grid box dimensions were set to 25 × 25 × 25 Å along X-, Y- and Z-axes, with center coordinates of X = −12.05, Y = 19.03, Z = 70.50. To ensure accuracy, the catalytic active site residues, including His41 and Cys145, and substrate binding site residues were manually inspected and confirmed to be properly confined within the rectangular grid box during the grid generation process. We evaluated the binding free energies (kcal/mol) of the Mpro and potential inhibitors complex after the docking analysis. In specific, the chain of 6LU7 was considered a rigid body and docked with flexible potential inhibitor ligands. Following the docking process, each ligand produced multiple docking poses, with up to nine poses based on root mean square deviation values. The first pose of each potential inhibitor, possessing the highest score, was selected for comparison of the docked ligand structures and binding affinities with Mpro. The docked complex files were then subjected to interaction studies, where we analyzed different types of interactions, such as covalent, carbon-hydrogen, hydrophobic interactions, and Van der Waals attractions using Biovia Discovery Studio.
Kinetic analysis of Mpro by using MCAP-AuNP
Before reaction with MCAP-AuNPs, various concentrations of Mpro (0.925–18.5 nM) were dissolved in PBS for 20 min. Afterward, MCAP-AuNP solutions were added to each Mpro solution to make a total volume of 1 mL. The mixtures of MCAP-AuNP and Mpro solutions were then incubated at 37 °C for 1 h. After incubation, the UV-vis absorption spectra were measured using a spectrophotometer. The degree of particle aggregation (A650/A525) with respect to Mpro concentration was represented using a sigmoidal dose–response curve.
Monitoring Mpro activity using FRET-based assay
Each 16 μM solution of MCA-AVLQSGFR-Lys(Dnp)-Lys-NH2 trifluoroacetate (Sigma Aldrich, USA) substrates was reacted with various concentrations of Mpro (0, 20, 74, 148, 296, 592, and 1184 nM) for 4 h. Then, the fluorescence intensities were measured by excitation at 444 nm and emission at 510 nm using the microplate reader.
Monitoring the efficacy of Mpro inhibitors using MCAP-AuNPs
Mpro (50 ng) was added to Mpro inhibitors at various concentrations (ebselen: 0.009 μM to 9.25 μM, hesperetin: 4.625 to 185 μM, lopinavir: 0.925 to 92.5 μM, leupeptin: 0.094 to 281.25 μM, and hesperidin: 11.563 to 370 μM) dissolved in PBS (1.5% DMSO), and the total volume of the solution was 800 μL. These solutions were incubated at room temperature for 20 min and then filtered using a 200-μm-pore PVDF syringe filter (Biopil, China) to remove the undissolved drugs. Then, 600 μL of the filtered solution was added to 200 μL of MCAP-AuNP solution and incubated at 37 °C for 1 h. After incubation, the UV-vis absorption spectra of the solutions were measured using a spectrophotometer. The degree of particle aggregation was represented by the relative UV-vis absorbance ratio A650/A525.
Monitoring the enzymatic activity of Mpro using MCAP-AuNP
Mpro (12 μg) was added to Mpro inhibitors (ebselen, hesperidin, hesperetin, lopinavir, and leupeptin) at various concentrations dissolved in PBS (10% DMSO), and the total volume of the solution was 500 μL. These solutions were then incubated at room temperature for 20 min and then filtered using a 200-μm-pore PVDF syringe filter to remove the undissolved drugs. Afterward, 70 μL of the filtered solutions were added into 1.88 μM, 3.7 μM, 5.66 μM, 7.55 μM, 9.43 μM, and 11.32 μM of MCAP solutions with a final volume of 220 μL. Using a microplate reader (Molecular Device, USA), inhibitor-controlled Mpro activity with respect to MCAP-AuNP concentration was calculated using the relative UV-vis absorbance (A650/A525). Enzymatic activities were calculated using the Michaelis-Menten equation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting this research are available within the article and its Supplementary Information files or from the corresponding author upon request. Source data are provided in this paper. Source data file includes raw data underlying the respective main text (Figs. 1, 2, 3, and 5) and Supplementary Information (Supplementary Figs. S3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, and 22). The data for molecular docking are from the Protein Data Bank (PDB accession code: 6LU7) and the PubChem compound database under accession codes: 3194, 72281, 72429, and 92727. To visualize the origins of the amyloid sequence and Mpro cleavage sequence in Fig. 1b, the crystal structures of the yeast prion protein Sup35 (PDB accession code: 1R5B) and 2019-nCoV nsp7-nsp8c complex (PDB accession code: 6M5I) were used. Input and output files of AutoDock Vina calculations are provided in Supplementary Data 1. Source data are provided in this paper.
References
Hu, B., Guo, H., Zhou, P. & Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 19, 141–154 (2021).
WHO Coronavirus (COVID-19) Dashboard | WHO Coronavirus (COVID-19) dashboard with vaccination data. [accessed 6 Nov 2023]. Available from: https://covid19.who.int/.
DeGrace, M. M. et al. Defining the risk of SARS-CoV-2 variants on immune protection. Nature 605, 640–652 (2022).
Keyel, A. C. et al. SARS-CoV-2 vaccine breakthrough by Omicron and Delta variants, New York, USA. Emerg. Infect. Dis. 28, 1990–1998 (2022).
Liu, X.-H. et al. Structures of the SARS-CoV-2 spike glycoprotein and applications for novel drug development. Front. Pharmacol. 13, 955648 (2022).
Gil, C. et al. COVID-19: drug targets and potential treatments. J. Med. Chem. 63, 12359–12386 (2020).
Kumari, M. et al. A critical overview of current progress for COVID-19: development of vaccines, antiviral drugs, and therapeutic antibodies. J. Biomed. Sci. 29, 1–36 (2022).
Barkas, F., Styla, C.-P., Bechlioulis, A. & Milionis, H. Liberopoulos EJJoCD, Disease. Sinus bradycardia associated with remdesivir treatment in COVID-19: a case report and literature review. J. Cardiovasc. Dev. Dis. 8, 18 (2021).
Ragland, D. A. et al. Drug resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J. Am. Chem. Soc. 136, 11956–11963 (2014).
Johnson, C. L. & Owen, D. M. Gale MJJoBC. Functional and therapeutic analysis of hepatitis C virus NS3· 4A protease control of antiviral immune defense. J. Biol. Chem. 282, 10792–10803 (2007).
Jin, Z. et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Bhardwaj, T. et al. Amyloidogenic proteins in the SARS-CoV and SARS-CoV-2. Proteomes 14, 945 (2023).
Owen, D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Boras, B. et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 12, 6055 (2021).
Zhu, T. et al. Safety, tolerability, and pharmacokinetics of intravenous doses of PF‐07304814, a phosphate prodrug protease inhibitor for the treatment of SARS‐CoV‐2, in healthy adult participants. Clin. Pharmacol. Drug Dev. 11, 1382–1393 (2022).
Wang, F. et al. Efficacy and safety of SIM0417 (SSD8432) plus ritonavir for COVID-19 treatment: a randomised, double-blind, placebo-controlled, phase 1b trial. Lancet Reg. Health West. Pac. 38, 100835 (2023).
Zhu, W. et al. Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol. Transl. Sci. 3, 1008–1016 (2020).
Liu, J. et al. A high-throughput drug screening strategy against coronaviruses. Int. J. Infect. Dis. 103, 300–304 (2021).
Zhou, Y., Wang, F., Tang, J., Nussinov, R. & Cheng, F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Health 2, e667–e676 (2020).
Yan, G. et al. Development of a simple and miniaturized sandwich-like fluorescence polarization assay for rapid screening of SARS-CoV-2 main protease inhibitors. Cell Biosci. 11, 1–14 (2021).
Llanos, M. A. et al. Strengths and weaknesses of docking simulations in the SARS-CoV-2 era: the main protease (Mpro) case study. J. Chem. Inf. Model. 61, 3758–3770 (2021).
Makin, O. S., Atkins, E., Sikorski, P., Johansson, J. & Serpell, L. C. Molecular basis for amyloid fibril formation and stability. Proc. Natl Acad. Sci. 102, 315–320 (2005).
Lee, D. et al. Biomimetically engineered amyloid-shelled gold nanocomplexes for discovering α-synuclein oligomer-degrading drugs. ACS Appl. Mater. Interfaces. 15, 2538–2551 (2022).
Majithia, R., Patterson, J., Bondos, S. E. & Meissner, K. E. On the design of composite protein–quantum dot biomaterials via self-assembly. Biomacromolecules 12, 3629–3637 (2011).
Lee, D. et al. Plasmonic nanoparticle amyloid corona for screening Aβ oligomeric aggregate-degrading drugs. Nat. Commun. 12, 1–11 (2021).
Li, C., Adamcik, J. & Mezzenga, R. Biodegradable nanocomposites of amyloid fibrils and graphene with shape-memory and enzyme-sensing properties. Nat. Nanotechnol. 7, 421–427 (2012).
Lee, D., Lee, G. & Yoon, D. S. J. A. Anti-Aβ drug candidates in clinical trials and plasmonic nanoparticle-based drug-screen for Alzheimer’s disease. Analyst 143, 2204–2212 (2018).
Gladytz, A., Abel, B. & Risselada, H. J. Gold‐induced fibril growth: the mechanism of surface‐facilitated amyloid aggregation. Angew. Chem. Int. Ed. 55, 11242–11246 (2016).
Zhao, Y. et al. Structural basis for replicase polyprotein cleavage and substrate specificity of main protease from SARS-CoV-2. Proc. Natl Acad. Sci. 119, e2117142119 (2022).
V’kovski, P., Kratzel, A., Steiner, S., Stalder, H. & Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 19, 155–170 (2021).
Jackson, C. B., Farzan, M., Chen, B. & Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 23, 3–20 (2022).
López de la Paz, M. & Serrano, L. Sequence determinants of amyloid fibril formation. Proc. Natl Acad. Sci. 101, 87–92 (2004).
Tzotzos, S. & Doig, A. J. J. P. S. Amyloidogenic sequences in native protein structures. Protein Sci. 19, 327–348 (2010).
Chen, P. et al. Amyloidosis inhibition, a new frontier of the protein corona. Nano Today 35, 100937 (2020).
Nel, A. E. et al. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 8, 543–557 (2009).
Brahmkhatri, V. P. et al. Multilayer protein corona on gold nanorod surface: first evidence of soft corona protein-protein interactions using solution NMR spectroscopy. Appl. Surf. Sci. Adv. 11, 100272 (2022).
García-Álvarez, R. & Vallet-Regí, M. Hard and soft protein corona of nanomaterials: analysis and relevance. Nanomaterials 11, 888 (2021).
Wu, W.-T., Chen, C.-H., Chiang, C.-Y. & Chau, L.-K. Effect of surface coverage of gold nanoparticles on the refractive index sensitivity in fiber-optic nanoplasmonic sensing. Sensors 18, 1759 (2018).
Gupta, A. et al. Ultrastable and biofunctionalizable gold nanoparticles. ACS Appl. Mater. Interfaces 8, 14096–14101 (2016).
Du, S. et al. Aggregation and adhesion of gold nanoparticles in phosphate-buffered saline. J. Nanopart. Res. 14, 1–14 (2012).
Ramos-Guzmán, C. A., Ruiz-Pernía, J. J., & Tuñón, I. Unraveling the SARS-CoV-2 main protease mechanism using multiscale methods. ACS Catal. 10, 12544–12554 (2020).
Amporndanai, K. et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 12, 1–7 (2021).
Menéndez, C. A., Byléhn, F., Perez-Lemus, G. R., Alvarado, W. & de Pablo, J. J. Molecular characterization of ebselen binding activity to SARS-CoV-2 main protease. Sci. Adv. 6, eabd0345 (2020).
Fu, L. et al. Mechanism of microbial metabolite leupeptin in the treatment of COVID-19 by traditional Chinese medicine herbs. Mbio 12, e02220–e02221 (2021).
Kneller, D. W. et al. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure 28, 1313–1320 (2020).
Ghosh, R., Chakraborty, A., Biswas, A. & Chowdhuri, S. Depicting the inhibitory potential of polyphenols from Isatis indigotica root against the main protease of SARS CoV-2 using computational approaches. J. Biomol. Struct. Dyn. 40, 4110–4121 (2022).
Fatriansyah, J. F., Rizqillah, R. K. & Yandi, M. Y. Molecular docking and molecular dynamics simulation of fisetin, galangin, hesperetin, hesperidin, myricetin, and naringenin against polymerase of dengue virus. J. Trop. Med. 2022, 12 (2022).
Nogara, P. A. et al. In silico studies on the interaction between Mpro and PLpro From SARS‐CoV‐2 and Ebselen, its metabolites and derivatives. Mol. Inform. 40, 2100028 (2021).
Verma, D. et al. Potential inhibitors of SARS-CoV-2 (COVID 19) proteases PLpro and Mpro/3CLpro: molecular docking and simulation studies of three pertinent medicinal plant natural components. Curr. Res. Pharmacol. Drug Discov. 2, 100038 (2021).
Elfiky, A. A. SARS-CoV-2 papain-like protease as a target for anti-HCV and anti-HIV proteases: in silico perspective. Int. J. Mol. Sci. 13, 242–250 (2022).
Choi, S.-S., Lee, S.-H. & Lee, K.-A. A comparative study of hesperetin, hesperidin and hesperidin glucoside: antioxidant, anti-inflammatory, and antibacterial activities in vitro. Antioxidants 11, 1618 (2022).
Copeland, R. A. Evaluation of Enzyme Inhibitors in Drug Discovery: a Guide for Medicinal Chemists and Pharmacologists. John Wiley & Sons (2013).
Harvey, W. T. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424 (2021).
Das, S., Sarmah, S., Lyndem, S., & Roy, A. S. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 39, 3347–3357 (2021).
Cheng, B. & Li, T. J. B. Discovery of alliin as a putative inhibitor of the main protease of SARS-CoV-2 by molecular docking. BioTechniques 69, 108–112 (2020).
Gupta, S. et al. Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. J. Biomol. Struct. Dyn. 39, 4334–4345 (2021).
Glaab E., Manoharan G. B. & Abankwa D. J. Pharmacophore model for SARS-CoV-2 3CLpro small-molecule inhibitors and in vitro experimental validation of computationally screened inhibitors. J. Chem. Inf. Model. 61, 4082–4096 (2021).
Souza, T. M. L., & Morel, C. M. The COVID-19 pandemic and the relevance of biosafety facilities for metagenomics surveillance, structured disease prevention and control. Biosaf. Health 3, 1–3 (2021).
Fan, S. et al. Research progress on repositioning drugs and specific therapeutic drugs for SARS-CoV-2. Fut. Med. Chem. 12, 1565–1578 (2020).
Mahdi, M. et al. Analysis of the efficacy of HIV protease inhibitors against SARS-CoV-2′ s main protease. Virol. J. 17, 1–8 (2020).
Nguyen, T. et al. The inhibitory effects of plant derivate polyphenols on the main protease of SARS coronavirus 2 and their structure–activity relationship. Molecules 26, 1924 (2021).
Dickson, M. & Gagnon, J. P. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 3, 417–429 (2004).
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIP) (NRF-2022R1A2C1091756, NRF-2020R1A2C2102262, NRF-2021R1A2C2004760 and NRF-2022R1A6A3A03066467), BK21 FOUR Institute of Precision Public Health, Health Fellowship Foundation, and NRF under project BK21 FOUR operated by the Center for Teaching and Learning of Korea University. This work was also supported by the Korea Health Industry Development Institute (KHIDI) grant HU21C0053 and the Bio & Medical Technology Development Program of the National Research Foundation funded by the Korean government (MSIT) (No. 2023M3E5E3080743).
Author information
Authors and Affiliations
Contributions
D. Lee, H. G. Jung, D. Park, J. H. Lee and D. S. Yoon conceived and designed the study. D. Lee, H. G. and J. Bang conducted the analysis of MCAP and overall experiments. D. Park performed molecular structure analysis. D. Y. Cheong, G. Lee and Y. H. Kim supported the ThT assay and AFM analysis. J. W. Jang, Y. Kim, S. Lee, S. W. Lee and J. H. Hong supported data analysis. D. Lee, H. G. Jung, D. Park, K. S. Hwang, J. H. Lee, D. S. Yoon drafted the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Thimmaiah Govindaraju, Byung Hee Hong and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, D., Jung, H.G., Park, D. et al. Bioengineered amyloid peptide for rapid screening of inhibitors against main protease of SARS-CoV-2. Nat Commun 15, 2108 (2024). https://doi.org/10.1038/s41467-024-46296-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-46296-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
