
Abstract
The surge in anthropogenic CO2 emissions from fossil fuel dependence demands innovative solutions, such as artificial photosynthesis, to convert CO2 into value-added products. Unraveling the CO2 photoreduction mechanism at the molecular level is vital for developing high-performance photocatalysts. Here we show kinetic isotope effect evidence for the contested protonation pathway for CO2 photoreduction on TiO2 nanoparticles, which challenges the long-held assumption of electron-initiated activation. Employing isotopically labeled H2O/D2O and in-situ diffuse reflectance infrared Fourier transform spectroscopy, we observe H+/D+-protonated intermediates on TiO2 nanoparticles and capture their inverse decay kinetic isotope effect. Our findings significantly broaden our understanding of the CO2 uptake mechanism in semiconductor photocatalysts.
Similar content being viewed by others

Introduction
The continued dependence on fossil fuels has led to a substantial increase in anthropogenic carbon dioxide (CO2) emissions, culminating in deleterious environmental impacts and energy crises1,2. An optimal strategy for addressing these challenges involves the conversion of CO2 into value-added products, such as CO and CH4, through artificial photosynthesis, which directly exploits incident sunlight and water3,4. However, a comprehensive understanding of the complex CO2 photoreduction reaction at the molecular level, particularly at the CO2/H2O/catalyst gas-liquid-solid interface, remains elusive owing to the involvement of numerous proton-coupled electron transfer processes and potential reaction pathways with various intermediates5,6,7. Elucidating the CO2 reduction pathway on the semiconductor catalyst surface is crucial for designing high-performance photocatalysts8.
Upon light exposure, a comprehensive CO2 photoreduction process typically encompasses water oxidation (or organic sacrificial agents, if utilized) and CO2 reduction half-reactions. The water oxidation half-reaction is often regarded as analogous to the oxygen-evolving reaction (OER) in water-splitting9,10. The CO2 reduction reaction encompasses multiple step-wise proton/electron transfer processes. Identifying the rate-determining step in such multi-step chemical reactions is an arduous task, yet essential for optimizing reaction systems. For example, the classic CO2 + 2e− + 2H+ → CO + H2O (−0.53 V vs. NHE) reaction on a semiconductor photocatalyst necessitates the enrichment and activation of CO2 molecules at the gas-vapor-catalyst or gas-liquid-catalyst interface, followed by a reduction reaction through a series of fundamental steps involving consecutive proton and electron transfers11. As a linear non-polar molecule, CO2 is among the most stable carbon compounds. Nevertheless, the oxygen atoms in CO2 can donate their lone pair of electrons to surface Lewis acid centers or be protonated by Brønsted acids12. The carbon atom can also accept electrons from Lewis base centers, forming carbonate-like species13. Moreover, the π electrons of the C=O bond can interact with electron centers, leading to bond cleavage and hybridization changes from O-sp2 to O-sp3. On the surface of the semiconductor catalyst, the adsorption configuration of CO2 is also notably altered and influenced by the presence of water or other molecular proton donors14,15,16. All these potential reaction configurations constitute the initial steps of CO2 activation.
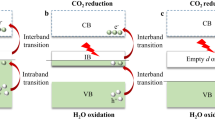
Figure 1 illustrates two feasible reaction pathways for CO2 photoreduction to CO in an aqueous solution: the electron-initiated pathway (path I) and the protonation pathway (path II). For a prolonged time, the initial step of CO2 activation was presumed to occur through path I, with a negatively charged CO2δ•- species as the sole intermediate product17,18. However, the single-electron transfer to CO2 is highly endergonic due to the molecule’s negative adiabatic electron affinity19. Additionally, the initial CO2 uptake on hydrophilic surfaces of MOx/MSx semiconductor photocatalysts is challenging, which impedes direct single-electron transfer20,21. Instead, a protonation pathway (path II) that first polarizes CO2 molecules, akin to the photocatalytic dehalogenation of non-polar halogenated aromatics22, appears more plausible. However, both pathways lack definitive, direct evidence for confirmation.

Two feasible reaction pathways for photoreduction of CO2 in aqueous solution.
The kinetic isotope effect (KIE) is a crucial and sensitive tool for investigating reaction mechanisms by tracking the transition state of the rate-determining step using isotopically-labeled reagents23,24. In this study, we employed isotopically labeled H2O/D2O to determine an inverse kinetic solvent isotope effect (KSIE) of 0.2~0.9 on the photoreduction of CO2 to CO on TiO2 nanoparticles. Our findings confirm the protonation pathway with O sp2−hybridized O=C=O-H+/D+ intermediates (Fig. 2), providing the elucidation of the protonation pathway for CO2 photoreduction and shedding light on the nature of CO2 uptake on semiconductor photocatalysts.

The water-splitting reaction (left) and the CO2 photoreduction to CO (right) with isotopically labeled H2O/D2O. CPET represents concerted proton-coupled electron transfer, PT represents proton transfer, ET represents electron transfer.
Results
Inverse KIE of CO2 photoreduction
We commenced our investigation by examining the KSIE of CO2 photoreduction to CO in a TiO2/water system, employing isotopically labeled H2O/D2O, and compared it to the water-splitting reaction in analogous systems (with or without CO2). We first used the commercially available anatase TiO2 (with ~20 nm-sized nanoparticles), a prevalent photocatalyst for water-splitting and CO2 reduction, as a representative example of conventional metal oxide (MOx) semiconductor catalysts with hydrophilic surfaces. We quantitatively detected the reduction products (i.e., H2, D2, CO) of the water-splitting and CO2 photoreduction reactions through gas chromatography (Supplementary Fig. 1). Control experiments conducted without CO2 yielded negligible amounts of CO, suggesting that CO2 reduction primarily contributes to the product formation (Supplementary Fig. 2).
Figure 3a illustrates that H2 production from the overall water-splitting in the Pt-TiO2/H2O system (with Pt as the hydrogen evolution reaction (HER) cocatalyst) proceeds more swiftly than with D2O, exhibiting a normal KSIEH2O/D2O(H2) of 2.8 at 15 °C. Diminishing the reaction system’s temperature augments the KSIE value to 5.8. The same experimental phenomena could be observed regardless of whether the cocatalyst was preloaded or loaded during the reaction (Supplementary Fig. 3). This temperature-dependent KSIE is consistent with the primary KIE’s characteristics for O-H/O-D cleavage during the oxygen evolution reaction (OER), indicating direct O-H cleavage as the rate-determining step of water-splitting25,26,27. However, we observed an inverse KSIEH2O/D2O(CO) using the same catalyst in the presence of CO2 (Fig. 3b). As the temperature declined from 15 to 3 °C, the KSIEH2O/D2O(CO) decreased from 0.9 to 0.2. Except for adding CO2, all experimental conditions were congruent with the water-splitting reaction. Furthermore, the KSIEH2O/D2O(H2) under identical experimental conditions displayed >1 normal values (Supplementary Fig. 4), suggesting different rate-determining steps between CO2 photoreduction and water-splitting. Without Pt loading, the CO2 photoreduction on pristine TiO2 exhibited analogous inverse KSIEH2O/D2O(CO) values (Fig. 3c). This outcome implies that the rate-determining step encompasses hybridization changes from sp2 to sp3 in the secondary inverse KIE phenomenon, consistent with the double-bond break of O=C=O molecules instead of direct O-H cleavage in OER. By employing H2O/D2O as labeled isotopes, the observed inverse KIE denotes a configuration transition between protonated intermediates O=C=O-H+/D+ (O sp2) and O=C•−O-H/D (O sp3) during electron transfer, offering robust evidence for a protonation pathway involving the formation of the protonated intermediate O=C=O-H+ as the initial step of CO2 photoreduction (path II, Fig. 1). This mechanism challenges the widely accepted electron-initiated pathway (path I, Fig. 1). Note that such a protonation pathway does not rely on the presence of a water solvent. We introduced water in the form of vapor into the reaction instead of as a solvent, and the same inverse KIE could be observed (Supplementary Fig. 5). This suggests that the protonation of CO2 can be achieved through water vapor.

a KSIE (H2) values obtained by comparing the H2 production kinetics of the water-splitting reaction on anatase TiO2 in H2O/D2O systems at different temperatures (Pt was loaded as cocatalysts, 3% chloroplatinic acid); b KSIE (CO) values obtained by comparing the kinetics of the CO2 reduction reaction on anatase TiO2 in H2O/D2O systems at different temperatures (Pt was loaded as cocatalysts); c KSIE (CO) values are given by comparing the CO production kinetics of the CO2 reduction reaction in the H2O/D2O systems at different temperatures without Pt cocatalysts. Error bar represents three independent experiments obtaining the standard deviation.
It is well-acknowledged that the characteristics of employed TiO2 catalysts can significantly influence their interaction with target molecules and thus lead to the change in reaction kinetics. To better ascertain whether the observed KIE changes originated from the reaction pathway itself or were influenced by the catalyst material, we conducted supplementary experiments across various TiO2 systems to bolster our findings. We first examined the influence of the TiO2 crystal structure by comparing the KIE for the CO2 reduction on anatase and rutile TiO2 (characterized by XRD and TEM/HR-TEM, see Supplementary Fig. 6 and Supplementary Fig. 7). We found that the KSIEH2O/D2O (CO) on both anatase and rutile catalysts exhibited inverse KIE (<1), suggesting that the observed inverse KIE and the protonation pathway in CO2 reduction are common to both crystal structures. Furthermore, we examined the effect of exposed facets of the TiO2 catalyst. As a comparison, we synthesized anatase TiO2 nanosheet with high exposure of the {001} facet according to a reported method28, which was characterized using XRD, TEM, HR-TEM, and SAED (Supplementary Fig. 8). The KSIEH2O/D2O (CO) for CO2 reduction on these {001}-exposed TiO2 nanosheets still exhibited inverse KIE < 1. These results further confirmed that the exposed facet of the TiO2 nanoparticles does not influence the CO2 reduction pathway under our experimental conditions. Finally, we evaluated the effect of oxygen defects. Oxygen vacancies on the TiO2 surface are often considered active sites for the oxygen evolution reaction (OER)29. However, their direct influence on CO2 reduction is less clear. We prepared oxygen-deficient TiO2 nanoparticles according to a reported method of NaBH4 calcination30, and characterized them using XRD, TEM, and ESR, which confirmed the presence of oxygen vacancies (Supplementary Fig. 9). The KSIEH2O/D2O (CO) for CO2 reduction on these oxygen-deficient nanoparticles remained <1, exhibiting the secondary inverse KIE and aligning with the protonation pathway. These additional characterizations and experiments confirmed that the inverse KIE observed in the CO2 reduction reaction is intrinsic to the TiO2 material generalized to a broader range of TiO2-based photocatalytic systems, regardless of the crystal structure, exposed facet, or oxygen vacancy concentration.
As a conventional metal oxide semiconductor with a hydrophilic surface and moderate reduction ability, TiO2 demonstrates inadequate CO2 uptake capacity31. As a result, the endergonic single-electron transfer of CO2 → CO2δ•- on TiO2 signifies a high-energy reaction. Nevertheless, in the photocatalytic dehalogenation of non-polar halogenated aromatics (e.g., polybrominated diphenyl ethers, PBDEs), a protonation pathway involving initial proton adhesion on the aromatic ring of PBDE molecules before subsequent electron transfer has been substantiated22. Additionally, in our recent work, we uncovered a step-wise proton transfer/electron transfer (PT/ET) pathway on TiO2 for the single-electron/single-proton reduction of tBu3ArO• and TEMPO• to tBu3ArOH and TEMPOH32. These investigations support the feasibility of the protonation pathway for CO2 photoreduction on TiO2 catalysts.
In-situ DRIFTS measurements
To investigate the protonation pathway and monitor O=C=O-H+/D+ intermediates during photocatalytic CO2 reduction, we employed in-situ diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) at the TiO2/H2O/CO2 (TiO2/D2O/CO2) interface. The experiment was carried out under 365 nm irradiation (3 W, LED) for 15 min, with H2O and CO2 (5 mL/min) introduced into the chamber by N2 flow (5 mL/min) until equilibrium was reached. We used the pre-reaction equilibrium system as a blank background and observed negative or positive IR signals during the reaction, indicating the loss or gain of species at the TiO2/H2O/CO2 (TiO2/D2O/CO2) interface. Control experiments demonstrated that in the absence of incident light, the reaction did not occur (Supplementary Fig. 10).
Figure 4a reveals negative peaks at 3700–2800 cm−1 and 1665 cm−1 at the TiO2/H2O interface upon constant irradiation, corresponding to the O-H stretching and H-O-H bending vibrations of H2O molecules33, respectively. The weak signal at 3705 cm−1 represented the terminal O-H group on the TiO2 surface34. When H2O was replaced with D2O, noticeable redshifts of both O-D stretching and D-O-D bending vibrations to 2700–2100 cm−1 and 1218 cm−1 were observed (Fig. 4b), in line with the theoretical H/D replacement effect35,36. The decay kinetics of O-H/O-D stretching vibrations showed that the O-H signal decays much faster than the O-D signal, resulting in a direct KIE of 2.11 (Fig. 4e), consistent with the measured normal KSIEH2O/D2O(H2) values and representing features of the direct O-H/-D cleavage during overall water-splitting.

DRIFTS spectra collected at the TiO2/H2O (a), TiO2/D2O (b), TiO2/H2O/CO2 (c), TiO2/D2O/CO2 (d) and TiO2/H2O/13CO2 (e) interfaces under constant 365 nm (3 W, LED) irradiation in 15 min; f Time profiles of IR signals at 3447 cm−1 in (a) and 2619 cm−1 in (b) from light-on to light-off, representing the decay kinetics of O-H and O-D in water-splitting; g Time profiles of IR signals at 3447 cm−1 in (c) and 2619 cm−1 in (d) from light-on to light-off, representing the decay kinetics of O-H and O-D in CO2 photoreduction; h Time profiles of IR signals at 2335 cm−1 and 2306 cm−1 (after baseline corrections to maintain positive values) from light-on to light-off, representing the formation and decay kinetics of O=C=O-H+ and O=C=O-D+. An inverse KIE was obtained during the decay process after light-off. Pink shading represents the peak position of the COOH+ intermediate, and green shading represents the peak position of the COOD+ intermediate.
In the TiO2/H2O/CO2 system (Fig. 4c), negative peaks at 2377 cm−1 and 2298 cm−1 corresponding to the C=O stretching vibrations of CO2 were observed, along with an emerging positive signal peak at 2335 cm−1 adjacent to the decayed stretching vibration signals of CO2, likely due to the formation of the protonated CO2 intermediate (O=C=O-H+). The adhesion of a proton to the oxygen atom would alter the C=O bond and alter the effective mass of oxygen, thereby changing the vibration frequency. In addition, according to Hooke’s Law, the adhesion of protons to the oxygen nucleus in C=O bonds increases the effective mass of the oxygen atom, which subsequently results in a change in the frequency of the stretching vibrations of the C=O bond37. Moreover, the increasing positive signal at 1089 cm−1 is likely from the C=O-H+ bending vibration. Negative peaks at 3727 cm−1, 3701 cm−1, 3624 cm−1, and 3597 cm−1 corresponded to the weak overtone region of CO2 molecules38, and signals at 1591 cm−1, 1522 cm−1, 1442 cm−1, and 1359 cm−1 were assigned to -COOH* species, monodentate carbonate (m-CO2-3) groups, as well as the antisymmetric and symmetric stretching bands of bidentate carbonate (b-CO2-3) groups39,40, respectively.
To verify that the observed changes in CO2 FT-IR signals resulted from a surface reaction rather than a modification in the surface adsorption configuration of CO2 under incident light, we carried out a control experiment. This involved first running the reaction for a specified time under light, followed by the removal of the gas phase using a N2 flow. By subtracting the equilibrium background in N2 prior to the experiment, we were able to observe changes in surface-adsorbed species over time. Given that the removal of the CO2 gas phase would cut off the replenishment of surface CO2, a fading CO2 signal would suggest that the observed signals stemmed from the reaction rather than adsorption. Otherwise, we would observe unchanged, stable adsorbate signals. As illustrated in Supplementary Fig. 11c, d, after the abrupt removal of CO2, both the negative and positive signals of C=O vibration from CO2 species around 2330 cm−1 to 2340 cm−1 continued to decrease over time and vanished within tens of seconds. This suggests that the observed CO2 signals are not from a stable adsorbate but from a surface reaction. Furthermore, to validate the assignment of the protonated O=C=O-H+ intermediate, we replaced H2O with deuterated-labeled D2O under identical conditions. The diagnostic signal peak of the protonated intermediate shifted towards a lower wavenumber from 2335 cm−1 to 2306 cm−1 upon replacing O=C=O-H+ with O=C=O-D+ (Fig. 4d). The negative signal peaks (both stretching bands and overtone region) of CO2 molecules remained unchanged. This H/D replacement effect on the C=O stretching vibration of O=C=O-H+/D+ intermediates is consistent with the results of Hooke’s Law (detailed calculation formula see supplementary methods). However, the C=O-D+ bending vibration was not observed in O=C=O-D+, which likely shifts a lower frequency, beyond our in-situ DRIFTS detection range (Fig. 4d). Together with the H/D replacement experiments without CO2, the shift of the diagnostic peak of O=C=O-D+ compared to that of the unlabeled O=C=O-H+ provides direct evidence for the formation of protonated O=C=O-H+ intermediates during the CO2 photoreduction process at the TiO2/H2O/CO2 interface. Furthermore, we have an additional DRIFTS experiment using 13C-labeled 13CO2. As depicted in Fig. 4e, a distinct redshift from 2335 cm−1 to 2293 cm−1 of the C=O stretching vibration was observed when employing 13CO2, corresponding to the shift of the 13C=O stretching vibration signal in O=13C=O-H+ compared to the unlabeled 12C=O in O=C=O-H+/D+ (2335 cm−1/2306 cm−1) due to the 12C/13C isotope replacement effect. Moreover, the bending vibration of C=O-H+ at 1089 cm−1 was also shifted to 1054 cm−1 in the 13CO2 system corresponding to 13C=O-H+. These findings are highly consistent with our KIE experimental results and further validates our assignment.
Quantum chemical calculations
We further conducted quantum chemical calculations to simulate the infrared signals of the H+/D+ protons adhered to the oxygen atom in CO2. The results are consistent with our assumption that the C=O stretching vibration in CO2 does not form a C-O-H sp3 structure after adhering to a H+/D+ proton, thereby a C-O signal does not appear (Supplementary Fig. 12). It remains at 2300–2400 cm−1 (the discrepancy between the calculation and actual data should come from different adsorption interfaces; the calculation only simulates the situation in a vacuum). The vibration frequency changes from protonated species and pristine CO2 due to the influence of bond energy and the effective mass of oxygen. Moreover, replacing H+ with D+ indeed causes the simulated C=O stretching vibration to shift to a lower frequency (2403 cm−1 → 2394 cm−1). Interestingly, quantum calculations also reveal possible O-H/O-D stretching vibrations (3406 cm−1/2490 cm−1), which are not clearly observed in the actual experiment due to the significant influence of water signals. More importantly, we found that the 960 cm−1 in O=C=O-H+ corresponds to the bending vibration of C=O-H+, which correspond to the positive signal at 1089 cm−1 observed in in-situ DRIFTS. In O=C=O-D+, the bending vibration of C=O-D+ shifts to a lower frequency, beyond our in-situ DRIFTS detection range, fully consistent with our observation. However, when 13C is used for simulation, the bending vibration of 13C=O-H+ can be seen to shift from 960 cm−1 to 952 cm−1. In our actual in-situ DRIFTS, when using 13CO2, we indeed observed a shift towards a lower wavenumber of the 13C=O-H+ bending vibration (1054 cm−1) from C=O-H+ (1089 cm−1) using unlabeled CO2 (Fig. 4e). This result fully support our assignment of the O=C=O-H+ signal.
Discussion
In prior research, the generation of CO2δ•- anion radicals have been detected during the photocatalytic degradation of formate on TiO2 nanoparticles using infrared (IR) and electron spin resonance (ESR) spectroscopy41,42. Although the CO2δ•- anion radical is often cited as the exclusive intermediate of the initial step in CO2 photoreduction, no definitive evidence has been provided for its presence in CO2 photoreduction systems. On a polar TiO2 surface surrounded by H2O molecules, chemisorbed species, mainly OH−, produce distinct π or δ resonances, while physisorbed species have weak signals43. This limits the opportunities for single-electron transfer by neutral physisorbed CO2 molecules, which are scarce at the polar H2O/TiO2 interface. Instead, an ionized CO2 moiety promotes interfacial CO2 uptake44, facilitating subsequent electron/proton transfer. Under our experimental conditions, the only visible positive signal peak after light illumination corresponds to the protonated O=C=O-H+/D+ signal. This finding contradicts previous understandings of the CO2 photoreduction mechanism and suggests a protonation pathway45.
We also compared the decay kinetics of O=C=O-H+/D+ and O-H/O-D during the reaction. The inverse kinetic isotope effect (KIE) of the O sp2 → O sp3 hybrid transition process is a classic phenomenon in reaction kinetics associated with the disparity in the vibration frequency of chemical bonds22,46. The observed inverse KIE of O=C=O-H+/D+ decay (KIE = 0.55) provides strong evidence for the protonation pathway (Fig. 4h; Supplementary Fig. 13), which involves the O=C=O-H+/D+ → O=C•−O-H/D double-bond break with a hybridization change from O sp2 → O sp3 via additional electron transfer (Fig. 5). In most reactions, the overall rate is determined by the slowest step, known as the rate-determining step47. In our system, without CO2, the direct breakage of the O-H/O-D bond undeniably constitutes the rate-determining step, hence its KIE is greater than 1 (Fig. 4f). However, the decay kinetics of O-H/O-D stretching vibration also exhibited an inverse KIE = 0.827 in the presence of CO2 (Fig. 4g), indicating that the slower reduction reaction of CO2 (in this case, the reduction of the protonated intermediate) becomes the rate-determining step.

Schematic illustrations and energetic profiles of the O=C=O-H+/D+ → O=C•−O-H/D electron transfer process. TS represents transition state, ZPE represents zero-point energy.
In this study, we unveil a mechanism governing the photoreduction of CO2 on semiconductor catalysts, which transpires via a protonation pathway. We report the formation of an O=C=O-H+ intermediate, which exhibits an inverse KIE during the subsequent electron transfer process. This electron transfer process prompts the conversion of the sp2−hybridized O=C=O-H+/D+ species into the sp3−hybridized O=C•−O-H/D species. Utilizing isotopically labeled in-situ DRIFTS, we successfully discern the formation of H+/D+-protonated O=C=O-H+/D+ intermediates on TiO2 nanoparticles and capture their inverse decay KIE. This research substantially broadens our comprehension of the CO2 uptake mechanism in semiconductor photocatalysts, necessitating a re-examination of long-held assumptions within the field. Our findings hold significant potential for advancing the development of more efficient and sustainable photocatalytic CO2 reduction technologies in the future.
Methods
Materials
Commercial titanium dioxide (TiO2, anatase, 20 nm), sodium borohydride (NaBH4), tetrabutyl titanate, hydrofluoric acid (HF, 40 wt%), chloroplatinic acid (H2PtCl6·6H2O), ethanol and deuterium oxide (D2O, 99.9 atom % D) were purchased from Shanghai McLean Biochemical Technology Co., Ltd. All reagents used in the synthesis were analytically pure and had not been further purified. Deionized water was obtained from a purified distillation unit in the laboratory. Before any photocatalytic reaction experiments, TiO2 samples were first calcinated and then illuminated by an ultraviolet lamp (365 nm, 160 mW∙cm−2) in water.
Synthesis of (001) exposed TiO2 nanosheet
In a typical synthesis, 12.5 mL of tetrabutyl titanate was mixed with 2 mL of HF solution, under stirring for 30 min. The solution was then transferred into a 50-mL Teflon-lined autoclave, and kept at 180 °C for 24 h. After the solvothermal reaction, the resulting white precipitates were collected and washed with ethanol and distilled water for three times. The samples were dried in a vacuum oven at 60 °C for 12 h.
Synthesis of oxygen-deficient TiO2
1 g TiO2 nanoparticle powder was mixed with 2 g NaBH4 and the mixture was ground for 30 min thoroughly. Then the mixture was transferred into a porcelain boat, and placed in a tubular furnace, heated from room temperature to 350 °C/1 h under an Ar atmosphere at a heating rate of 10 °C min−1. After naturally cooling down to room temperature, the colored TiO2 was obtained, simply washed with deionized water and ethanol several times to remove unreacted NaBH4, and dried at 70 °C.
Water-splitting experiments
In a typical procedure, 50 mg TiO2 powder was dispersed in 10 mL deionized water (H2O) and 10 mL deuterium water (D2O), respectively. Next, 3 wt% Pt as cocatalysts was loaded via in-situ photo deposition using H2PtCl6·6H2O without any sacrificial agents. After irradiation with an ultraviolet lamp (365 nm, 160 mW∙cm−2), Gas products were determined by using a gas chromatography (GC-7900) equipped with the TCD thermal conductivity detector and the carrier gas was chosen Ar.
CO2 photoreduction experiments
CO2 photoreduction was carried out in a sealed self-made 150 mL stainless-steel reactor with an ultraviolet lamp (365 nm, 160 mW∙cm−2) as the light source. In a typical procedure, 50 mg catalyst was dispersed in 10 mL deionized water (H2O) and 10 mL deuterium water (D2O), respectively. CO2 was then introduced into the reactor and bubbled for 25 min to completely remove air. Gas products were detected by the gas chromatography (GC-7920, China) equipped with hydrogen flame ionization detector (FID) and thermal conductivity detector (TCD). In addition, the control experiment had the same experimental conditions as described above except for the addition of 3 wt% Pt as cocatalysts; In the gas-solid reaction system, 50 mg catalyst was dispersed in quartz grooves, add 2 ml of water or deuterated water to the bottom of the 150 ml reactor with no direct contact with the catalyst, assuring that water participates in the reaction in vapor state. CO2 flow was then introduced into the reactor for 25 min before light-on.
In-situ DRIFTS experiments
In-situ diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) experiments were conducted on a Nicolet iS10 (Thermo) machine according to our previous work47. In a typical procedure, catalyst sample was sealed in the reaction chamber with a quartz window. CO2 and H2O (or D2O) were carried into the reaction chamber by N2 flow until equilibrium. After taking the equilibrium system before reaction as the blank background, IR signals were collected in-situ during the incident irradiation of a 365 nm LED lamb (3 W) through the quartz glass window.
Hooke’s law
Taking diatomic as an example, when the diatomic is telescopic and vibrating, they can be approximated as a simple harmonic oscillator. Given two bodies, one with mass m1 and the other with mass m2, the equivalent one-body problem, with the position of one body with respect to the other as the unknown, is that of a single body of mass; where the equivalent mass of O=C=O-H+ is m1 = 12 (C), m2 = 17 (O-H, v1 = 2335 cm−1); The equivalent mass of O=C=O-D+ is m1 = 12 (C), m2 = 18 (O-D, v2 = 2306 cm−1).
When v1 = 2335 cm−1:
The force constants of chemical bonds:
When the equivalent mass of O=C=O-D+ is m1 = 12, m2 = 18:
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information files. All other relevant source data are available from the corresponding author upon request. Source data are provided with this paper.
References
He, M., Sun, Y. & Han, B. Green carbon science: efficient carbon resource processing, utilization, and recycling towards carbon neutrality. Angew. Chem. Int. Ed. 61, e202112835 (2022).
Li, X., Yu, J., Jaroniec, M. & Chen, X. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 119, 3962–4179 (2019).
Bian, J. et al. Dimension‐matched zinc phthalocyanine/BiVO4 ultrathin nanocomposites for CO2 reduction as efficient wide‐visible‐light‐driven photocatalysts via a cascade charge transfer. Angew. Chem. Int. Ed. 131, 10989–10994 (2019).
Li, X. et al. Selective visible-light-driven photocatalytic CO2 reduction to CH4 mediated by atomically thin CuIn5S8 layers. Nat. Energy 4, 690–699 (2019).
Habisreutinger, S., Schmidt-Mende, L. & Stolarczyk, J. Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 52, 2–39 (2013).
Yin, G. et al. Hydrogenated blue titania for efficient solar to chemical conversions: preparation, characterization, and reaction mechanism of CO2 reduction. ACS Catal. 8, 1009–1017 (2018).
Schrauben, J. et al. Titanium and zinc oxide nanoparticles are proton-coupled electron transfer agents. Science 336, 1298–1301 (2012).
Pi, J. et al. Surface and defect engineering coupling of halide double perovskite Cs2NaBiCl6 for efficient CO2 photoreduction. Adv. Energy Mater. 12, 2202074 (2022).
Chen, Q. et al. Accelerated water oxidation kinetics triggered by supramolecular porphyrin nanosheet for robust visible‐light‐driven CO2 reduction. Small 18, 2204924 (2022).
Xie, S. et al. Facilitated photocatalytic CO2 reduction in aerobic environment on a copper‐porphyrin metal‐organic framework. Angew. Chem. Int. Ed. 62, e202216717 (2023).
Maeda, K. Metal‐complex/semiconductor hybrid photocatalysts and photoelectrodes for CO2 reduction driven by visible light. Adv. Mater. 25, 1808205 (2019).
Chang, X., Wang, T. & Gong, J. CO2 photo-reduction: insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci. 9, 2177–2196 (2016).
Indrakanti, V., Kubicki, J. & Schobert, H. Photoinduced activation of CO2 on Ti-based heterogeneous catalysts: current state, chemical physics-based insights and outlook. Energy Environ. Sci. 2, 745–758 (2009).
Roberts, M. et al. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 25, 225–273 (1996).
Shkrob, I. et al. Heteroatom-transfer coupled photoreduction and carbon dioxide fixation on metal oxides. J. Phys. Chem. C 116, 9461–9471 (2012).
Pougin, A. et al. Identification and exclusion of intermediates of photocatalytic CO2 reduction on TiO2 under conditions of highest purity. Phys. Chem. Chem. Phys. 18, 10809–10817 (2016).
Liu, T. et al. Ag@imidazolium functionalized polymeric yolk–shell hybrid nanoparticles for economical CO2 photoreduction. Green Chem. 25, 301–309 (2023).
Porosoff, M., Yan, B. & Chen, J. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities. Energy Environ. Sci. 9, 62–73 (2016).
Takahashi, K., Sawamura, S., Dimitrijevic, N., Bartels, D. & Jonah, C. Transient negative species in supercritical carbon dioxide: electronic spectra and reactions of CO2 anion clusters. J. Phys. Chem. A 106, 108–114 (2002).
Tang, Q. & Luo, Q. Adsorption of CO2 at ZnO: a surface structure effect from DFT+ U calculations. J. Phys. Chem. C 117, 22954–22966 (2013).
Yang, K. et al. Recent advances in CdS-based photocatalysts for CO2 photocatalytic conversion. Chem. Eng. J. 418, 129344 (2021).
Chang, W. et al. Inverse kinetic solvent isotope effect in TiO2 photocatalytic dehalogenation of non‐adsorbable aromatic halides: a proton‐induced pathway. Angew. Chem. Int. Ed. 54, 2052–2056 (2015).
Haschke, S. et al. Direct oxygen isotope effect identifies the rate-determining step of electrocatalytic OER at an oxidic surface. Nat. Commun. 9, 4565 (2018).
Gunnemann, C. et al. Isotope effects in photocatalysis: an underexplored issue. ACS Omega 6, 11113–11121 (2021).
Tse, E. et al. Observation of an inverse kinetic isotope effect in oxygen evolution electrochemistry. ACS Catal. 6, 5706–5714 (2016).
Yang, X. et al. Mechanism of water splitting and oxygen−oxygen bond formation by a mononuclear ruthenium complex. J. Am. Chem. Soc. 132, 120–130 (2010).
Zhang, Y. et al. Pivotal role and regulation of proton transfer in water oxidation on hematite photoanodes. J. Am. Chem. Soc. 138, 2705–2711 (2016).
Long, R. et al. Isolation of Cu atoms in Pd lattice: forming highly selective sites for photocatalytic conversion of CO2 to CH4. J. Am. Chem. Soc. 139, 4486–4492 (2017).
Li, J. et al. Oxygen vacancies on TiO2 promoted the activity and stability of supported Pd nanoparticles for the oxygen reduction reaction. J. Mater. Chem. A 6, 2264–2272 (2018).
Tan, H. et al. A facile and versatile method for preparation of colored TiO2 with enhanced solar-driven photocatalytic activity. Nanoscale 6, 10216–10223 (2014).
Wang, S. et al. Porous hypercrosslinked polymer-TiO2-graphene composite photocatalysts for visible-light-driven CO2 conversion. Nat. Commun. 10, 676 (2019).
Liu, Z. et al. Water molecule switching heterogeneous proton-coupled electron transfer pathway. Chem. Sci. 14, 4564–4570 (2023).
Imoto, S., Xantheas, S. & Saito, S. Ultrafast dynamics of liquid water: energy relaxation and transfer processes of the OH stretch and the HOH bend. J. Phys. Chem. B 119, 11068–11078 (2015).
Sheng, H. et al. Activation of water in titanium dioxide photocatalysis by formation of surface hydrogen bonds: an in situ IR spectroscopy study. Angew. Chem. Int. Ed. 54, 5905–5909 (2015).
Vinyard, D. et al. Photosystem II oxygen-evolving complex photoassembly displays an inverse H/D solvent isotope effect under chloride-limiting conditions. Proc. Natl. Acad. Sci. USA 116, 18917–18922 (2019).
Chatterjee, S. et al. Concerted proton–electron transfer in electrocatalytic O2 reduction by iron porphyrin complexes: axial ligands tuning H/D isotope effect. Inorg. Chem. 54, 2383–2392 (2015).
Burke, J. IR spectroscopy or Hooke’s law at the molecular level-a joint freshman physics-chemistry experience. J. Chem. Educ. 74, 1213 (1997).
Sheng, J. et al. Identification of halogen-associated active sites on bismuth-based perovskite quantum dots for efficient and selective CO2-to-CO photoreduction. ACS Nano 14, 13103–13114 (2020).
Di, J. et al. Surface local polarization induced by bismuth‐oxygen vacancy pairs tuning non‐covalent interaction for CO2 photoreduction. Adv. Energy Mater. 11, 2102389 (2021).
Wang, Y. et al. CO2 photoreduction with H2O vapor on highly dispersed CeO2/TiO2 catalysts: surface species and their reactivity. J. Catal. 337, 293–302 (2016).
Perissinotti, L. et al. Yield of carboxyl anion radicals in the photocatalytic degradation of formate over TiO2 particles. Langmuir 17, 8422–8427 (2001).
AlSalka, Y. & Al-Madanat, O. et al. Photocatalytic H2 evolution from oxalic acid: effect of cocatalysts and carbon dioxide radical anion on the surface charge transfer mechanisms. ACS Appl. Energy Mater. 3, 6678–6691 (2020).
Umezawa, N. et al. Reduction of CO2 with water on Pt-loaded rutile TiO2 (110) modeled with density functional theory. J. Phys. Chem. C 120, 9160–9164 (2016).
Cota, I. et al. Recent advances in the synthesis and applications of metal organic frameworks doped with ionic liquids for CO2 adsorption. Coordin. Chem. Rev. 351, 189–204 (2017).
Neatu, S. et al. Gold–copper nanoalloys supported on TiO2 as photocatalysts for CO2 reduction by water. J. Am. Chem. Soc. 136, 15969–15976 (2014).
Yang, Y. et al. Inverse kinetic isotope effects in the oxygen reduction reaction at platinum single crystals. Nat. Chem. 15, 271–277 (2023).
Yin, S. et al. Boosting water decomposition by sulfur vacancies for efficient CO2 photoreduction. Energ. Environ.Sci. 15, 1556–1562 (2022).
Acknowledgements
We gratefully acknowledge the financial support of the National Natural Science Foundation of China (Grant No. 22078131 (P. Huo) and 22208127 (Z. Zhu)); The Science and Technology Planning Social Development Project of Zhenjiang City (SH2021013 (P. Huo)); Graduate Research and Innovation Projects of Jiangsu Province (Grant No. KYCX22_3696 (S. Yin)).
Author information
Authors and Affiliations
Contributions
S.Y. and Y.Y. designed the whole experiment. S.Y., Y.Z., Z.L., H.W., and X.Z. conducted most experiments. S.Y. and Y.Y. wrote the paper. Z.Z. and P.H. contributed to the data analysis of the paper quality through discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jenny Schneider, Jennifer Strunk and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yin, S., Zhou, Y., Liu, Z. et al. Elucidating protonation pathways in CO2 photoreduction using the kinetic isotope effect. Nat Commun 15, 437 (2024). https://doi.org/10.1038/s41467-024-44753-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-44753-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
