
Abstract
The construction of N–N axially chiral motifs is an important research topic, owing to their wide occurrence in natural products, pharmaceuticals and chiral ligands. One efficient method is the atroposelective dihydropyrimidin-4-one formation. We present herein a direct catalytic synthesis of N–N atropisomers with simultaneous creation of contiguous axial and central chirality by oxidative NHC (N-heterocyclic carbenes) catalyzed (3 + 3) cycloaddition. Using our method, we are able to synthesize structurally diverse N–N axially chiral pyrroles and indoles with vicinal central chirality or bearing a 2,3-dihydropyrimidin-4-one moiety in moderate to good yields and excellent enantioselectivities. Further synthetic transformations of the obtained axially chiral pyrroles and indoles derivative products are demonstrated. The reaction mechanism and the origin of enantioselectivity are understood through DFT calculations.
Similar content being viewed by others
Introduction
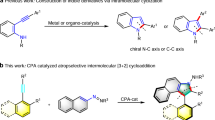
Recently, the wide applications of axially chiral biaryls in catalysis and drug delivery have excited much research interest in their synthesis1,2. Among them, atropisomeric indole derivatives are particularly important as synthetic precursors of many pharmaceuticals3,4. The last decade has witnessed many excellent research works in the construction of axially chiral indole-based frameworks5. Nitrogen-nitrogen atropisomers, an important subclass arising from sterically hindered rotation around a single N–N bond, are of high natural abundance6,7,8,9, as exemplified by Dixiamycin A and Schischkiniin. In addition, 2,20-bis(diphenylphosphino)-1,10-bibenzimidazole (BIMIP) was reported as a diphosphine ligand (Fig. 1a)10,11. Irrespective of their significant utility, effective enantioselective synthesis of N–N atropisomers was not explored until recently, Liu12,13,14,15, Sparr16, You17, Sun18, Li19, Zhao20, Shi21,22, Lu and Houk23, and Li24,25 reported several methods for the synthesis of N–N bispyrrole, indole pyrrole, bisindole, and nonbiaryl atropisomers via asymmetric copper-catalysis, palladium-catalysis, iridium-catalysis, rhodium-catalysis, chiral phosphoric acid catalysis, or Brønsted base (Fig. 1b). However, these studies mainly focused on the atroposelective construction of N–N axial chirality. Besides, the highly atroposelective creation of N–N axially chiral pyrrole/indole-based heterocyclic six-membered ring remains unknown19. Recently, simultaneously controlling multiple chiral elements (axial chirality and central chirality) has emerged as an important research area with several pioneering works reported in the past decade26,27,28. Despite of the excellent progress made by these groups, it remains very challenging to construct contiguous axial and central chirality in a high diastereo- and enantioselective manner, including (1) efficient cyclization between two sterically demanding partners, (2) the control of enantioselectivity, and (3) creation of vicinal axial and central chirality in a single operation with potential diastereoselectivity issues. Very recently, elegant catalytic enantioselective syntheses of atropisomeric hydrazides were reported, through a one-pot sequence of two organocatalytic cycles29. However, the direct catalytic synthesis of N–N atropisomers with simultaneous creation of contiguous axial and central chirality remains elusive in the literature, which represented a significant gap in synthetic method development.

a Important N-N atropisomers wtih bis-heterocyclic skeletons. b Recent examples of N-N atropisomers bearing pyrrole and/or indole scaffolds. c This work: synthesis of N-N atropisomers wtih bis-heterocyclic skeletons by oxidative NHC catalysis.
N-heterocyclic carbene (NHC) catalysis has emerged as a powerful tool in the preparation of both central and atropisomeric chiral molecules30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50 and in the construction of six-membered rings through asymmetric (3 + 3) cycloaddition using α,β-unsaturated acylazoliums as C3 synthon36. To the best of our knowledge, no example of the synthesis of N–N axially chiral pyrroles and indoles catalyzed by N-heterocyclic carbene has been reported51. We are interested in NHC-catalyzed asymmetric reactions52,53,54,55,56,57,58. Here, we show an efficient access to diastereo- and atroposelective N–N axially chiral pyrroles and indoles with vicinal central chirality through a highly atroposelective (3 + 3) cycloaddition between readily available isothioureas with enals via oxidative NHC catalysis. Besides, this strategy also enabled facile access to N–N axially chiral pyrroles and indoles with excellent atroposelectivity starting from readily available thioureas with ynals through oxidative NHC catalysis (Fig. 1c).
Results
Diastereo- and atroposelective synthesis of N–N axially chiral indoles/pyrroles with vicinal central chirality
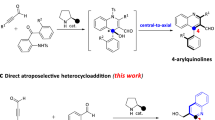
Initially, we designed the oxidative NHC catalytic synthesis of 5,6-dihydropyrimidin-4-ones from readily available isothiourea 1a and cinnamaldehyde 2a with central chirality followed by central-to-axial chirality conversion to produce N–N atropisomers with bis-heterocyclic skeletones (Fig. 2a). The Chi’s group reported an NHC-catalyzed addition of isothioureas to enals under oxidative conditions to form 5,6-dihydropyrimidin-4-ones bearing stereocenters with high optical purity59. When we carried out the reaction of 1a with cinnamaldehyde 2a in the presence of azolium catalyst, oxidant, NaOAc, and HOAc, to our great surprise, two diastereomeric compounds 3a and 3a’ (about 1.5:1 dr), could be easily separated by column chromatography; both isomers were obtained with high ee of 86% (Fig. 2b). The absolute configuration of 3a and 3a’ were unambiguously assigned by single crystal X-ray diffraction analysis, respectively. The configuration of the other products was assigned by analogy. Attracted by this unprecedented one-pot catalytic synthesis of N–N axially chiral indoles with vicinal central chirality, we turned our attention to the exploration of this intriguing transformation.

a Our design: construction of central chirality followed by central-to-axial chirality conversion. b Beyond design: synergistic construction of central and axial chirality via a single stereodetermining steps.
Our next goal is to optimize the reaction to improve both enantio- and diastereoselectivity. After a brief survey of NHC precatalysts, the aminoindanol-derived NHC generated from the chiral triazolium salt F bearing N-2,4,6-triisopropylphenyl group was found to the best catalyst. In the presence of NaOAc as the base, the major diastereomer 3a could be obtained in 68% NMR yield with 92% ee as well as the minor diastereomer 3a’ in 23% NMR yield at 30 °C in toluene (Table 1, entry 6 vs entries 1–5). The solvent screening revealed that DCM, ethyl acetate (EA), and THF resulted in reduced selectivity and yields (Table 1, entries 7 − 9). An extensive base screening revealed that bases such as DBU could not furnish the desired product (Table 1, entry 10), whereas DIPEA, KOtBu, and KOAc could afford the desired product in reduced selectivity and yield as compared to NaOAc (Table 1, entry 6 vs entries 11 − 13). Lastly, other additives were tested such as 3 Å MS, 5 Å MS, Na2SO4, and MgSO4. It was shown that the addition of MgSO4 could help to afford the 3a with the highest yield and excellent ee.
We then proceeded to examine the generality of this catalytic (3 + 3) cycloaddition reaction using the optimized reaction conditions (Fig. 3). A range of enals were examined as substrates using isothiourea 1a as the complementary reactant to furnish products 3b-3m. Various enals containing electron-donating and electron-withdrawing substituents at the para position of the β-aryl moiety were shown to be effectively converted under these developed conditions. In all cases examined, the indole-derived 5,6-dihydropyrimidin-4-one products were obtained in good yields and enantiomeric excess values (3b-3h). Furthermore, enals possessing meta-substituents were shown to undergo smooth (3 + 3) cycloaddition, furnishing the desired products in moderate to good yield with excellent stereoselectivity (3i-3j). Additionally, substitution at the β-position of the enal with a heteroaryl moiety did not impact the outcome of the reaction, and the corresponding furyl adduct 3k was synthesized successfully. Moreover, this methodology was found to be extensible to β-vinyl enals, generating the desired cycloadduct (3 l) in moderate yield and appreciable enantioselectivity. Additionally, alkyl enal such as hex-2-enal were amenable to the developed conditions, furnishing two diastereomeric products (3m) and (3m’) that could be readily isolated in 73-80% ee following flash column chromatography.

For details, please see Supplementary Information (SI).
Next, the variations on isothioureas were studied. The substrate with different substitutions on the phenyl moiety of isothioureas as well 2,3-dimethyl or cyclohexyl groups also gave the corresponding products (3n-3r) in moderate to high yields and excellent ee. To further expand the scope of the substrate, the pyrrole based isothiourea was also tested. Electron-withdrawing, and electron-donating groups at the para-position of cinnamaldehyde as well 3-(furan-2-yl)acrylaldehyde were well tolerated to afford the corresponding products 3s-3v in good yield and enantioselectivity.
The two rotamers 3a and 3a’ were oxidized by Iodine in DMSO at room temperature to furnish atropisomeric indole-based pyrimidin-4-one 4 and its enantiomer ent-4, respectively, without any loss of enantiomeric excess (Fig. 4a, b). Simultaneously, iodo substituent was introduced onto the 3-position of indole, which served as a potential transformation handle. The absolute configuration of 4 was unambiguously assigned by single crystal X-ray diffraction analysis.

a Oxidation of 3a and 3a’ into enantiomeric compounds 4 and ent-4. b Alternative strategy for synthesizing N–N axially chiral pyrroles. c Alternative strategy for synthesizing N–N axially chiral pyrroles.
Atroposelective synthesis of N–N axially chiral pyrroles and indoles
Besides, alkenyl acylazoliums, another important type of NHC-bound intermediates such as the alkynyl acylazoliums were also investigated as a C3 synthon. Our next goal is to create other type of N–N axially chiral pyrroles or indoles using (3 + 3) cycloaddition catalyzed through alkynyl acylazoliums (II)60,61,62,63. Initially, the isothiourea 1a was chosen as the model substrate, no desired product was obtained after a lot of conditions screening. Later, we switched the substrate isothiourea to thiourea. Fortunately, the N–N axially chiral pyrrole bearing a 2,3-dihydropyrimidin-4-one moiety 7a was obtained in 36% yield and −77% ee (Fig. 4c). Nevertheless, NHC optimization revealed that the yield and ee of product 7a was improved to 51% and 97%, respectively. It is noteworthy that no axially chiral product 8 was detected and this reaction proceeded in a chemoselective fashion. The representative optimization studies are included in the Supplementary Information (Supplementary Table 1).
Initially, the commonly used strong electron withdrawing group (CF3) on the N-phenyl group of the substrate 5 could be well-tolerated to produce (7b-7c) with moderate yields and moderate to excellent enantioselectivities of 90–94% ee (Fig. 5). Unfortunately, the substrate bearing a neutral or electron donating group on the N-phenyl group led to complex reaction mixtures in this NHC catalytic conditions. With the identified optimal conditions in hand, the variations of either electron-donating or withdrawing group on para-position of phenyl moiety of 5 were examined and they were well-tolerated to yield the corresponding products 7d–7j in moderate yields and high to excellent enantioselectivities (85–93% ee). The different substituents on the meta or ortho position of phenyl ring were also tested, and the corresponding products 7k-7o were afforded with moderate yields and excellent enantioselectivities (90–99% ee). The absolute configuration of 7 l was unambiguously assigned by single crystal X-ray diffraction analysis. The configuration of the other products was assigned by analogy.

For details, please see Supplementary Information (SI).
Then we investigated the different substituents on the phenyl ring of the ynals in the reaction. The electronic effect showed obvious influence on the reaction outcome and the corresponding products 7p-7ac were obtained with moderate yields and excellent enantioselectivities (95–98% ee). Lastly, substitution at the β-position of ynals with heteroaryl moiety did not affect the outcome of the reaction and the corresponding thienyl product 7ad was formed. To further expand the scope of the substrate, the indole based thioureas were also tested. The 2-substituted indole based thioureas (methyl or phenyl group) were well tolerated to afford the corresponding products 7ae-7af in moderate yields and high enantioselectivities. The substrate bearing the useful group such as -P(O)Ph2 successfully provided the desired product 7ag in 65% yield with 86% ee. When the ortho CF3 group on the N-phenyl group of the substrates 5 were employed to afford the thiazine derivatives with C–N axial chirality (9a-9b) in moderate yields with moderate enantioselectivities of 54–77% ee. This type of thiazine derivatives with C–N axial chirality was reported by the Jin’s group. The ortho isopropyl group on the N-phenyl group of the thioureas were involved in his work64. The configuration of 9b was unambiguously assigned by single crystal X-ray diffraction analysis. The configuration of the other products was assigned by analogy. The symmetrical pyrrole based thiourea was also employed to give the desired product 9c in 51% yield with 60% ee. The configurational stability of this C–N linked axially chiral compounds was briefly investigated. Experimentally, the ee value of 9c deteriorated significantly at 100 °C in toluene and became racemic after stirring for 12 h. The barrier to rotation for 9c was measured at 100 °C in toluene (ΔG≠ = 29.6 kcal/mol). The details are included in the Supplementary Information (Supplementary Fig. 5).
Next, we investigated experimentally the configurational stability of this type of N–N linked axially chiral compounds. Selected compounds (3a, 4, 7 l, etc.) were heated in toluene for 72 h at 150 °C. No erosion of enantiopurity was observed, showing that the chiral axes in these compounds are indeed configurationally stable. Our experimental result was further corroborated by the computed rotation energies of the N–N bond (leading to racemization) of 3a, 4, and 7 l (Fig. 6), which were determined to be 37.1, 48.2, and 45.9 kcal/mol at 150 °C (with t1/2 of 13 days, >19,000, and 1270 years), respectively65.

DFT calculation on the rotation energy of 3a, 4, and 7 l.
Transformations and application
A one-mmol-scale reaction was conducted under standard conditions, affording 3a and 3a´ in 72% and 18% yield, respectively (Fig. 7a). The pyrimidin-4-one product bearing axial with or without vicinal central chirality could be further functionalized via simple operations. The treatment of 3a with Grignard reagent in THF at −15 °C followed by elimination of H2O during work-up to generate product 10 with 91% yield and 92% ee (Fig. 7b). Additionally, the axially chiral 4 bearing iodo, late-stage coupling with TMS acetylene followed by deprotection can afford the desired product 11 in 95% yield and 91% ee (Fig. 7c). Bromination of axially chiral 7a with CuBr2 at 40 °C delivered fully-substituted pyrrole 12 in 67% yield and 94% ee (Fig. 7d). The axially chiral phosphine oxide 7ag was transformed into the phosphine 13 by reduction with HSiCl3 reagent in 84% yield with 85% ee (Fig. 7e)66. Notably, compound 13 was used as a chiral ligand in the palladium-catalyzed enantioselective allyl substitution reaction of (E)-1,3-diphenylallyl acetate 14 and dimethyl malonate 15, affording the target product 16 in 81% yield with 40% ee (Fig. 7f). Although the enantioselectivity needs to be improved, this attempt demonstrated the potential application of the constructed axially chiral indole bearing a 2,3-dihydropyrimidin-4-one moiety scaffold for the development of chiral ligand.

a one-mmol-scale reaction. b–e Transformations of axial products. f Application of chiral phosphine ligand.
Reaction mechanism and origin of stereoselectivity
Interestingly, when the ee value of 3a was studied with respect to catalyst enantiopurity, a negative nonlinear effect (NLE) was observed (Fig. 8a), which suggests a second catalyst molecule is involved in the enantio-differentiating transition states, possibly activating the other substrate isothiourea 1a. Our result is consistent with earlier studies by Huang, and Jin, which also reported a NLE effect in NHC catalysis67,68. In comparison, a linear effect was observed in NHC pre-catalyst H (bearing an OH group) catalyzed synthesis of 7a, suggesting that in this case, only one catalyst was involved in the enantio-differentiating step (see Supplementary Information for details). One possible explanation for the different behaviors could be that the OH group of pre-catalyst H forms a hydrogen bond in the enantio-differentiating step, alleviating the need to involve a second catalyst molecule37.

a Nonlinear effect with respect to product ee and catalyst ee. b Chemical shift of the acidic proton of the NHC pre-catalyst F under various conditions.
To substantiate the proposed interaction between F and isothiourea 1a, we carried out 1H NMR analysis (Fig. 8b). Upon addition of isothiourea 1a to a mixture of pre-catalyst F and base NaOAc, the chemical shift of the acidic proton of F showed a downward shift of 0.15 ppm (Fig. 8b, 1 vs 2). This result is in support of the existence of interaction between F and isothiourea 1a. Taken together with the NLE experiment, it is possible that the second NHC molecule helps to activate isothiourea 1a through N-H···C hydrogen bond55,69,70,71,72,73,74,75. (for more details, see Supplementary Information).
With the above experimental mechanistic insights established, we next proceeded to study the mechanism computationally. Chemistry of NHC catalysis has been well established, and much mechanistic detail prior to the key stereoselective steps, e.g. formation of F-2 from F and 2a, can be understood without carrying out further experiments or calculations. Based on previous studies and our mechanistic investigations, we initially proposed a catalytic cycle for NHC catalyst F as illustrated in Supplementary Figure 350.
It was hypothesized that the rotational barrier around the N–N axis in intermediates F-3 and after is prohibitively high. Therefore, the level of diastereoselectivity is determined by the relative energies of diastereomeric nucleophilic addition transition states from F-2 to F-3. To substantiate this hypothesis, we calculated a model compound truncated from F-3, the result of which showed the rotational barrier is very high at 45.3 kcal/mol. At room temperature where the experiments were conducted, such a high barrier will render the axis configurationally stable. Furthermore, N–N axes in compounds similar to F-3 have also been reported to be configurationally stable by other groups23.
We investigated computationally the steps starting from the formation of F-3 from F-2 of the proposed catalytic cycle. A second carbene molecule was proposed to act as a Brønsted base to activate 1a through N-H···C hydrogen bond. Our calculated result in Fig. 9 showed the C–C bond cleavage transition state F-TS6-RRa has the highest energy of 59.4 kJ/mol. The first N–C bond-formation transition state F-TS1-RRa, the second proton-transfer transition state F-TS2-RRa and the third ring-closure transition state F-TS3-RRa have lower energies of 40.6, 53.9, and 6.5 kJ/mol respectively. Barring computational errors, this will imply that the reaction falls within the Curtin-Hammett paradigm and the reaction stereoselectivity will be governed by the C–C bond-cleavage transition states. We noticed that F-TS6-RRa yields a protonated 3a as the immediate product, and a proton transfer to F is required to regenerate it. We next investigated if the high barrier of this step could be lowered by added base or acid. Indeed, a new reaction pathway with lowered energies was located. Its first step is an acetic acid-catalyzed tautomerization of F-5 (2.1 kJ/mol) to a lower-energy tautomer F-8 (−30.9 kJ/mol), which appears to be quite facile. In the presence of the conjugate base sodium acetate, the energy of the C–C bond-cleavage transition state, now as F-TS5-RRa, could be lowered to 41.7 kJ/mol. Based on the calculated reaction profile and Curtin-Hammett principle, we concluded that the stereochemistry of the reaction is governed by diastereomeric F-TS2 transition states. However, the presence of many low-lying molecular vibrations (frequencies <30 cm−1), due to the association of a second carbene molecule through a weak hydrogen bond, makes a quantitative prediction of reaction stereoselectivity very challenging. It should be emphasized that our calculations are not in full agreement with the experimental stereoselectivity and there still lacks a clear mechanistic explanation of the negative NLE (for more details, see Supplementary Information).

Calculated reaction profile at M06-2X/6-311 + G(2d,p)-SMD(toluene)//M06-2X/6-31 G(d)-SMD(toluene) level of NHC-catalyzed atroposelective construction of N–N axially chiral indoles.
Discussion
In conclusion, we have developed an oxidative NHC-catalyzed atroposelective synthesis of N–N axially chiral compounds from readily available starting materials. The contiguous N–N axial and central chirality could be simultaneously constructed in a single operation. The reaction took place smoothly under mild conditions and showed good functional group tolerance, which can be allowed for the synthesis of a variety of N–N axially chiral pyrroles and indoles (more than 50 examples) in moderate to good yields, moderate to good diastereoselectivities, and excellent enantioselectivities. Mechanistic studies indicate that non-covalent interactions between the isothiourea and an NHC•HX catalyst play an important role in activating isothiourea towards nucleophilic addition, which was corroborated by DFT calculations. The success of this work not only provides a useful method for the simultaneous construction of contiguous N–N axial and central chirality within a single step but also represents the rare example of catalytic enantioselective synthesis of N–N atropisomers catalyzed by NHC. The development of catalytic procedures to access other challenging structures bearing different types of stereogenic elements is currently under investigation.
Methods
General procedure for the Diastereo- and atroposelective synthesis of 3
A dry 4 mL vial with a stir bar was charged with isothioureas 1 (0.1 mmol,1.0 equiv.), PreNHC F (20 mol%), NaOAc (0.15 mmol, 1.5 equiv.), 3,3’,5,5’-Tetra-tert-butyl-4,4’-dibenzoquinone (0.125 mmol, 1.25 equiv.) and MgSO4 (100 mg). The mixture was taken into the glovebox, where AcOH (0.03 mmol, 0.3 equiv.), enals 2 (0.18 mmol, 1.8 equiv.) and toluene (2.0 mL) were added. The reaction mixture was taken outside the glovebox. The vial was then sealed and the mixture was allowed to stir in the fume hood at 30 °C (oil bath) for 48 h. When the substrate was consumed completely, the mixture was concentrated under vacuum and purified by column chromatography on silica gel (hexane/ethyl acetate = 2:1) to afford the pure product 3.
Synthesis of 4
A mixture of 3a (0.2 mmol, 1.0 equiv.) and iodine (0.4 mmol, 2.0 equiv.) in DMSO (2.0 mL) was warmed at 60 °C in an oil bath for 8 h. On completion of the reaction, the reaction mixture was poured onto a saturated solution of sodium thiosulfate. The precipitated solid was collected and the desired product was purified by column chromatography using silica gel with increasing percentage of ethyl acetate in hexane as eluting solvent. The desired product 4 was obtained in 95% yield with 90% ee.
General procedure for the atroposelective synthesis of 7
To a 4 mL vial was added the thioureas 5 (0.2 mmol, 1.0 equiv.), PreNHC H (0.04 mmol, 20 mol%), 3,3’,5,5’-Tetra-tert-butyl-4,4’-dibenzoquinone (0.2 mmol, 1.0 equiv.) and NaOAc (0.2 mmol, 1.0 equiv.). The mixture was taken into the glovebox, where ynals 6 (0.24 mmol, 1.2 equiv.) and EtOAc (2.0 mL) were added. The reaction mixture was taken outside the glovebox. The vial was then sealed and the reaction mixture was allowed to stir in the fume hood at 30 °C (oil bath) for overnight. The crude reaction mixture was directly purified by silica gel column chromatography with hexanes/ethyl acetate (5:1 v/v) as eluent to afford the pure products 7.
Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers 2206276 (3a), 2218428 (3a’), 2221389 (4), 2202253 (7 l) and 2262465 (9b). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. Data related to materials and methods, optimization of conditions, experimental procedures, mechanistic experiments, and spectra are provided in the Supplementary Information. Coordinates of the optimized structures are provided in the supplementary data file. All data are available from the corresponding authors upon request. Source data are provided with this paper.
References
LaPlante, S. R. et al. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 54, 7005–7022 (2011).
Nguyen, T. Giving atropisomers another chance. Chem. Eng. N. 96, i33 (2018).
Ito, C., Thoyama, Y., Omura, M., Kajiura, I. & Furukawa, H. Alkaloidal Constituents of Murraya koenigii. Isolation and structural elucidation of novel binary carbazolequinones and carbazole alkaloids. Chem. Pharm. Bull. 41, 2096–2100 (1993).
Shoeb, M. et al. Isolation, structure elucidation and bioactivity of schischkiniin, a unique indole alkaloid from the seeds of Centaurea schischkinii. Tetrahedron 61, 9001–9006 (2005).
Zhang, H.-H. & Shi, F. Organocatalytic atroposelective synthesis of indole derivatives bearing axial chirality: Strategies and applications. Acc. Chem. Res. 55, 2562–2580 (2022).
Blair, L. M. & Sperry, J. Natural products containing a nitrogen–nitrogen bond. J. Nat. Prod. 76, 794–812 (2013).
Zhang, Q. et al. N–N-coupled indolo-sesquiterpene atropo-diastereomers from a marine-derived actinomycete. Eur. J. Org. Chem. 2012, 5256–5262 (2012).
Xu, Z., Baunach, M., Ding, L. & Hertweck, C. Bacterial synthesis of diverse indole terpene alkaloids by an unparalleled cyclization sequence. Angew. Chem. Int. Ed. 51, 10293–10297 (2012).
Rosen, B. R., Werner, E. W., O’Brien, A. G. & Baran, P. S. Total synthesis of dixiamycin B by electrochemical oxidation. J. Am. Chem. Soc. 136, 5571–5574 (2014).
Benincori, T. et al. Chiral atropisomeric five-membered biheteroaromatic diphosphines: New ligands of the bibenzimidazole and biindole series. J. Organomet. Chem. 529, 445–453 (1997).
Antognazza, P., Benincori, T., Mazzoli, S., Sannicolo, F. & Pilati, T. Resolution and characterization of 2,2’-Bis(Diphenylphosphino)-1,1’-bibenzimidazole (BIMIP): the first chiral atropisomeric diphosphine ligand with hindered rotation around a N–N Bond. Phosphorus Sulfur Silicon Relat. Elem. 144, 405–408 (1999).
Wang, X.-M. et al. Enantioselective synthesis of nitrogen−nitrogen biaryl atropisomers via copper-catalyzed friedel−crafts alkylation reaction. J. Am. Chem. Soc. 143, 15005–15010 (2021).
Xu, Q. et al. Cu(I)-catalyzed asymmetric arylation of pyrroles with diaryliodonium salts toward the synthesis of N−N atropisomers. Org. Lett. 24, 31385–3143 (2022).
Zhang, P. et al. Enantioselective synthesis of N−N bisindole atropisomers. Angew. Chem. Int. Ed. 61, e202212101 (2022).
Yao, W. et al. Enantioselective Synthesis of N–N Atropisomers by Palladium-Catalyzed C–H Functionalization of Pyrroles. Angew. Chem. Int. Ed. 62, e202218871 (2023). During the revision of our work, the preparation of indole-pyrrole bearing a N–N chiral axis was reported using an atroposelective C–H functionalization of pyrroles, see:.
Hutskalova, V. & Sparr, C. Control over Stereogenic N–N Axes by Pd-Catalyzed 5-endo-Hydroaminocyclizations. Synthesis 55, 1770–1782 (2023).
Yin, S.-Y., Zhou, Q., Liu, C.-X., Gu, Q. & You, S.-L. Enantioselective Synthesis of N–N Biaryl Atropisomers through Iridium(I)-Catalyzed C–H Alkylation with Acrylates. Angew. Chem. Int. Ed. 62, e202305067 (2023). During the revision of our work, the preparation of indole-pyrrole and bis-pyrroles bearing a N–N chiral axis was reported using an enantioselective C–H alkylation approach, see:.
Wang, C. & Sun, J. Atroposelective synthesis of N–N axially chiral bipyrroles via rhodium-catalyzed C–H insertion reaction. Org. Lett. 25, 4808–4812 (2023). During the revision of our work, the preparation of bis-pyrroles bearing a N–N chiral axis was reported using an enantioselective C−H bond insertion reaction, see:.
Zhu, X. et al. Rhodium-catalyzed annulative approach to N–N axially chiral biaryls via C-H activation and dynamic kinetic transformation. Chem. Sci. 14, 8564–8569 (2023). During the revision of our work, the preparation of indole derivatives bearing a N–N chiral axis was reported using an enantioselective (4 + 2) oxidative annulation of internal alkynes with benzamides. see:.
Gao, Y., Wang, L.-Y., Zhang, T., Yang, B.-M. & Zhao, Y. Atroposelective synthesis of 1,1’-bipyrroles bearing a chiral N–N axis: Chiral phosphoric acid catalysis with lewis acid induced enantiodivergence. Angew. Chem. Int. Ed. 61, e202200371 (2022).
Chen, K.-W. et al. Organocatalytic atroposelective synthesis of N–N axially chiral indoles and pyrroles by de novo ring formation. Angew. Chem. Int. Ed. 61, e202116829 (2022).
Chen, Z.-H. et al. Organocatalytic enantioselective synthesis of axially chiral N,N’-bisindoles. Angew. Chem. Int. Ed. 62, e202300419 (2023).
Mei, G.-J. et al. Rational design and atroposelective synthesis of N–N axially chiral compounds. Chem 7, 27435–2757 (2021).
Lin, W. et al. Asymmetric synthesis of N-N axially chiral compounds via organocatalytic atroposelective N-acylation. Chem. Sci. 13, 141–148 (2022).
Pan, M., Shao, Y.-B., Zhao, Q. & Li, X. Asymmetric synthesis of N–N axially chiral compounds by phase-transfer-catalyzed alkylations. Org. Lett. 24, 374–378 (2022).
Bai, X.-F., Cui, Y.-M., Cao, J. & Xu, L.-W. Atropisomers with axial and point chirality: Synthesis and applications. Acc. Chem. Res. 55, 2545–2561 (2022).
Wu, P. et al. Design and synthesis of axially chiral aryl-pyrroloindoles via the strategy of organocatalytic asymmetric (2 + 3) cyclization. Fundamental Res. 3, 237–248 (2023).
Wang, H.-Q., Wu, S.-F., Yang, J.-R., Zhang, Y.-C. & Shi, F. Design and organocatalytic asymmetric synthesis of indolyl-pyrroloindoles bearing both axial and central chirality. J. Org. Chem. 88, 7684–7702 (2023).
Portolani, C. et al. Synthesis of atropisomeric hydrazides by one-pot sequential enantioselective and diastereoselective catalysis. Angew. Chem. Int. Ed. 61, e202209895 (2022).
Grossmann, A. & Enders, D. N-heterocyclic carbene catalyzed domino reactions. Angew. Chem. Int. Ed. 51, 314–325 (2012).
Bode, J. W. Carbene catalysis: An internal affair. Nat. Chem. 5, 813–815 (2013).
Hopkinson, M. N., Richter, C., Schedler, M. & Glorius, F. An overview of N-heterocyclic carbenes. Nature 510, 485–496 (2014).
Flanigan, D. M., Romanov-Michailidis, F., White, N. A. & Rovis, T. Organocatalytic reactions enabled by N-heterocyclic carbenes. Chem. Rev. 115, 9307–9387 (2015).
Wang, M. H. & Scheidt, K. A. Cooperative catalysis and activation with N-heterocyclic carbenes. Angew. Chem. Int. Ed. 55, 14912–14922 (2016).
Zhang, C., Hooper, J. F. & Lupton, D. W. N-heterocyclic carbene catalysis via the α,β-unsaturated Acyl Azolium. ACS Catal. 7, 2583–2596 (2017).
Mondal, S., Yetra, S. R., Mukherjee, S. & Biju, A. T. NHC-catalyzed generation of α,β-unsaturated acylazoliums for the enantioselective synthesis of heterocycles and carbocycles. Acc. Chem. Res. 52, 425–436 (2019).
Chen, X.-Y., Gao, Z.-H. & Ye, S. Bifunctional N-heterocyclic carbenes derived from l-pyroglutamic acid and their applications in enantioselective organocatalysis. Acc. Chem. Res. 53, 690–702 (2020).
Sumoda, Y. & Ohmiya, H. Direct excitation strategy for radical generation in organic synthesis. Chem. Soc. Rev. 50, 6320–6332 (2021).
Gao, J., Feng, J. & Du, D. Generation of azolium dienolates as versatile nucleophilic synthons via N-heterocyclic carbene catalysis. Org. Chem. Front. 8, 6138–6166 (2021).
Chen, K.-Q., Sheng, H., Liu, Q., Shao, P.-L. & Chen, X.-Y. N-heterocyclic carbene-catalyzed radical reactions. Sci. China Chem. 64, 7–16 (2021).
Biju, A. T. N-Heterocyclic carbenes in organocatalysis. Wiley-VCH Verlag GmbH & Co. KGaA, (2019).
Song, R., Xie, Y., Jin, Z. & Chi, Y. R. Carbene-catalyzed asymmetric construction of atropisomers. Angew. Chem. Int. Ed. 60, 26026–26037 (2021).
Wang, J., Zhao, C. & Wang, J. Recent progress toward the construction of axially chiral molecules catalyzed by an N-heterocyclic carbene. ACS Catal. 11, 12520–12531 (2021).
Feng, J. & Du, D. Asymmetric synthesis of atropisomers enabled by N-heterocyclic carbene catalysis. Tetrahedron 100, 132456–132470 (2021).
Zhang, C.-L., Gao, Y.-Y., Wang, H.-Y., Zhou, B.-A. & Ye, S. Enantioselective synthesis of axially chiral benzothiophene/benzofuran-fused biaryls by N-heterocyclic carbene catalyzed arene formation. Angew. Chem. Int. Ed. 60, 13918–13922 (2021).
Wu, Y., Li, M., Sun, J., Zheng, G. & Zhang, Q. Synthesis of axially chiral aldehydes by N-heterocyclic-carbene-catalyzed desymmetrization followed by kinetic resolution. Angew. Chem. Int. Ed. 61, e202117340 (2022).
Lv, Y. et al. Catalytic atroposelective synthesis of axially chiral benzonitriles via chirality control during bond dissociation and CN group formation. Nat. Commun. 13, 36 (2022).
Yang, X. et al. Atroposelective access to 1,3-oxazepine-containing bridged biaryls via carbene-catalyzed desymmetrization of imines. Angew. Chem. Int. Ed. 61, e202211977 (2022).
Chu, Y. et al. N-heterocyclic carbene-catalyzed atroposelective synthesis of pyrrolo[3,4-b]pyridines with configurationally stable C–N axial chirality. Org. Lett. 24, 3884–3889 (2022).
Wang, G. et al. N-heterocyclic carbene-catalyzed atroposelective synthesis of axially chiral 5-aryl 2-pyrones from enals. Sci. China Chem. 65, 1953–1961 (2022).
Balanna, K. et al. N-heterocyclic carbene-catalyzed atroposelective synthesis of N–N axially chiral 3-amino quinazolinones. ACS Catal. 13, 8752–8759 (2023). During the revision of our work, the preparation of 3-amino Quinazolinones bearing a N–N chiral axis was reported using an atroposelective N-Acylation, see:.
Lu, S., Poh, S. B. & Zhao, Y. Kinetic resolution of 1,1’-Biaryl-2,2’-diols and amino alcohols through NHC-catalyzed atroposelective acylation. Angew. Chem. Int. Ed. 53, 11041–11045 (2014).
Lu, S., Ong, J.-Y., Poh, S. B., Tsang, T. & Zhao, Y. Transition-metal-free decarboxylative propargylic substitution/cyclization with either azolium enolates or Acyl anions. Angew. Chem. Int. Ed. 57, 5714–5719 (2018).
Poh, S. B., Ong, J.-Y., Lu, S. & Zhao, Y. Highly regio- and stereodivergent access to 1,2-amino alcohols or 1,4-fluoro alcohols by NHC-catalyzed ring opening of epoxy enals. Angew. Chem. Int. Ed. 57, 1645–1649 (2018).
Lu, S. et al. Diastereo- and atroposelective synthesis of bridged biaryls bearing an eight-membered lactone through an organocatalytic cascade. J. Am. Chem. Soc. 141, 17062–17067 (2019).
Lu, S., Poh, S. B., Rong, Z.-Q. & Zhao, Y. NHC-catalyzed atroposelective acylation of phenols: Access to enantiopure NOBIN analogs by desymmetrization. Org. Lett. 21, 6169–6172 (2019).
Jiang, J. et al. Enantioselective cascade annulation of α-amino-ynones and enals enabled by gold and oxidative NHC relay catalysis. Angew. Chem. Int. Ed. 61, e202115464 (2022).
Zhang, S.-C. et al. Enantioselective access to triaryl-2-pyrones with monoaxial or contiguous C–C diaxes via oxidative NHC catalysis. ACS Catal. 13, 2565–2575 (2023).
Maiti, R. et al. Carbene-catalyzed selective addition of isothioureas to enals for access to sulphur-containing 5,6-dihyropyrimidin-4-ones. Org. Chem. Front. 8, 743–747 (2021).
Mou, C. et al. Carbene-catalyzed LUMO activation of alkyne esters for access to functional pyridines. Chem. Commun. 53, 13359–13362 (2017).
Zhao, C. et al. Enantioselective [3+3] atroposelective annulation catalyzed by N-heterocyclic carbenes. Nat. Commun. 9, 611 (2018).
Yan, J. et al. Carbene-catalyzed atroposelective synthesis of axially chiral styrenes. Nat. Commun. 13, 84 (2022).
Zhang, S. et al. Atroposelective synthesis of triaryl α-pyranones with 1,2-Diaxes by N-heterocyclic carbene organocatalysis. Angew. Chem. Int. Ed. 61, e202212005 (2022).
Li, T. et al. N-heterocyclic carbene-catalyzed atroposelective annulation for access to thiazine derivatives with C−N axial chirality. Angew. Chem. Int. Ed. 60, 9362–9367 (2021).
Kobayashi, T. et al. Analysis of interconversion between atropisomers of chiral substituted 9,9’-Bicarbazole. Eur. J. Org. Chem. 2021, 449–451 (2021).
Zhang, H.-H. et al. Design and enantioselective construction of axially chiral naphthyl-indole skeletons. Angew. Chem. Int. Ed. 56, 116–121 (2017).
Chen, J. & Huang, Y. Asymmetric catalysis with N-heterocyclic carbenes as non-covalent chiral templates. Nat. Commun. 5, 3437 (2014).
Peng, X., Xu, J., Li, T., Chi, Y. R. & Jin, Z. Chemo-selective cross reaction of two enals via carbene-catalyzed dual activations. Chem. Sci. 11, 12533–12539 (2020).
Phillips, E. M., Riedrich, M. & Scheidt, K. A. N-heterocyclic carbene-catalyzed conjugate additions of alcohols. J. Am. Chem. Soc. 132, 13179–13181 (2010).
Chen, J., Meng, S., Wang, L., Tang, H. & Huang, Y. Highly enantioselective sulfa-Michael addition reactions using N-heterocyclic carbene as a non-covalent organocatalyst. Chem. Sci. 6, 4184–4189 (2015).
Santra, S., Porey, A., Jana, B. & Guin, J. N-Heterocyclic carbenes as chiral Brønsted base catalysts: A highly diastereo- and enantioselective 1,6-addition reaction. Chem. Sci. 9, 6446–6450 (2018).
Santra, S., Maji, U. & Guin, J. Enantioselective α‑amination of acyclic 1,3-dicarbonyls catalyzed by N‑heterocyclic carbene. Org. Lett. 22, 468–473 (2020).
Guo, F., Chen, J. & Huang, Y. A bifunctional N-heterocyclic carbene as a noncovalent organocatalyst for enantioselective Aza-Michael addition reactions. ACS Catal. 11, 6316–6324 (2021).
Li, E., Chen, J. & Huang, Y. Enantioselective seleno-michael addition reactions catalyzed by a chiral bifunctional N-heterocyclic carbene with noncovalent activation. Angew. Chem. Int. Ed. 61, e202202040 (2022).
Maji, U., Mondal, B. D. & Guin, J. Asymmetric aminative dearomatization of 2‑naphthols via non-covalent N‑heterocyclic carbene catalysis. Org. Lett. 25, 2323–2327 (2023).
Acknowledgements
S.L. is grateful for the generous financial support from the Natural Science Basic Research Plan in Shaanxi Province of China (2020JM-107), the Program for Young Talents of Shaanxi Province (5113200043), the Joint Research Funds of Department of Science & Technology of Shannxi Province and Northwestern Polytechnical University (2020GXLH-Z-023), and the Fundamental Research Funds for the Central Universities. We acknowledge Prof. Yu Zhao (National University of Singapore) for his insightful discussions and generous help with the manuscript preparation.
Author information
Authors and Affiliations
Contributions
S.L. and M.W.W. conceived and designed the study. S.-J.W., X.W., X.X. and S.Z. performed the experiments and prepared the Supplementary Information. H.Y. performed the DFT studies and prepared the Supplementary Information. S.L., H.Y. and M.W.W. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Feng Shi, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, SJ., Wang, X., Xin, X. et al. Organocatalytic diastereo- and atroposelective construction of N–N axially chiral pyrroles and indoles. Nat Commun 15, 518 (2024). https://doi.org/10.1038/s41467-024-44743-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-44743-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
