
Abstract
Tissue structure identification is a crucial task in spatial omics data analysis, for which increasingly complex models, such as Graph Neural Networks and Bayesian networks, are employed. However, whether increased model complexity can effectively lead to improved performance is a notable question in the field. Inspired by the consistent observation of cellular neighborhood structures across various spatial technologies, we propose Multi-range cEll coNtext DEciphereR (MENDER), for tissue structure identification. Applied on datasets of 3 brain regions and a whole-brain atlas, MENDER, with biology-driven design, offers substantial improvements over modern complex models while automatically aligning labels across slices, despite using much less running time than the second-fastest. MENDER’s identification power allows the uncovering of previously overlooked spatial domains that exhibit strong associations with brain aging. MENDER’s scalability makes it freely appliable on a million-level brain spatial atlas. MENDER’s discriminative power enables the differentiation of breast cancer patient subtypes obscured by single-cell analysis.
Similar content being viewed by others

Introduction
Recent advances in spatially resolved single-cell (SRSC) technologies allow the profiling of cellular gene expression in the tissue context, allowing comprehensive spatial characterization of various systems1,2,3,4,5,6,7. Coordinated by different cell states with varying gene expression patterns, spatial domains are higher-order functional units that recurrently distribute across tissue space, and have close relationships with tissue physiology8,9. In complex diseases such as cancer, mounting evidence has suggested the pivotal roles of specified spatial domains in disease diagnosis and monitoring10,11,12. Given the ever-increasing SRSC data13,14, many computational methods have been developed to identify spatial domains15,16,17.
In a typical SRSC dataset, the spatial coordinates and gene expression profiles of each cell are measured. Such data representation naturally forms a spatial graph with cells as nodes and gene expression as node attributes, which motivated the two major modeling paradigms in this field, i.e., Graph Neural Network (GNN)18,19,20, and Bayesian Network (BN)21,22,23. Along the developmental paths of both paradigms, the vast majority of methods were designed to improve performance by increasing model complexity. GNN-based methods introduced dedicated neural modules, loss functions, and network architectures. BN-based methods extend additional hidden variables, variable dependencies, and specified priors. Although increasingly complex models often lead to better performance, the improvements are, in some recent studies, seeing a diminishing marginal return24. Besides, additional model complexity may subject the algorithms to non-trivial parameter-tunning, low time efficiency, and/or reduced generalizability. As such, all these issues call for a new paradigm to break through the developmental bottlenecks in this field.
In this work, we evaluate both advantages and disadvantages of the existing state-of-the-art methods to elicit the bottleneck problems that a new method must solve. We next analyze and observe the consistency of cellular neighborhood structure across 24 data from 8 different spatial technologies and tissue systems. Based on this, we design a biology-driven cellular context representation, which obtains consistent prediction improvements over current state-of-the-art GNN models in 3 different supervised learning settings on 13 spatial data across different technologies. Inspired by the above analyses, we present Multi-range cEll coNtext DEciphereR (MENDER) for unsupervised spatial domain identification. MENDER has 3 highlighted points, which are considered major bottlenecks of existing methods: (1) multi-slice spatial domain identification that challenges many advanced methods; (2) scalability to million-level datasets; and (3) improved running time efficiency without the need of GPU. Comprehensive benchmark analyses show MENDER’s substantial improvements in terms of accuracy, continuity, and running time over complex GNN and BN models on various datasets with increasing challenges. On the million-level brain spatial atlas, MENDER is the only method that successfully delineates major brain domains largely consistent with established Allen brain reference, without any human intervention. On a model of mouse brain aging, MENDER identifies subdomains consistent across 3 aging stages and also domains that specifically occurred in young mice. On a 40-patient triple-negative breast cancer (TNBC) dataset, MENDER can differentiate three subtypes of TNBC by explaining the cellular spatial organization differences. We also extended MENDER’s application on a wider range of spatial data types, showing its generalizability.
Results
Motivation and overview
Limitations of existing methods
We first explain why a new method for spatial domain identification is still needed given the existence of many methods in the field. We select 8 existing methods published in the last two years and evaluate from 6 criteria, including support for multi-slice analysis, stability, interpretability, scalability, speed, and availability of cell context representation (Supplementary Fig. 1A). The definition of each criterion is briefly explained in Supplementary Fig. 1A and detailed explained in “Six aspects to view existing methods” section of “Methods”. These methods include 4 GNN-based (SpaGCN18, STAGATE19, CCST25, and SpaceFlow20) and 4 BN-based (BayesSpace21, BASS23, SpatialPCA26, and SOTIP27).
One can observe that most evaluation criteria are strongly associated with the method principle (Supplementary Fig. 1A). All GNN-based methods have better scalability and speed (conditional on GPU) than BN-based methods and they can also output the context representations for cells. The common limitations of GNN-based methods are the lack of stability and interpretability inherited from general deep-learning models. BN-based methods, on the contrary, have better output stability and interpretability than GNN-based methods since they are generally built on well-defined probabilistic variable dependencies. But they cannot guarantee good scalability to large datasets with short running time, and generally don’t output the cell context representations (SpatialPCA26 as an exception). These evaluations were also verified in recent studies20,23 and in the subsequent benchmark analysis of this manuscript.
In particular, some criteria in the above analysis are critical in the advent of the big data era of space omics. Many large consortia efforts have generated spatial datasets containing millions of cells collected from a bunch of slices28,29,30. In such scenarios, the scalability of methods to large datasets, running time, and the support of multi-slice analysis are especially needed. Although there are many methods for spatial domain identification, new innovations still are needed to meet the above criteria as possible.
Consistent neighborhood structures across different data
We analyze the distance of neighboring cells from different spatial technologies and different tissue systems (Supplementary Fig. 1B). To do so, we collect 24 datasets generated by 6 different spatial technologies from SODB31 [https://gene.ai.tencent.com/SpatialOmics/]. These spatial data cover the major part of currently mainstream technologies14, including MERFISH29,30,32, DARTFISH14, BaristaSeq33, STARmap34, osmFISH4, and seqFISH35 (see “Methods”). For each dataset, we first construct a 1-NN graph of all cells based on each cell’s spatial coordinates, then the distance between each cell and its directly connected cell is recorded to form a distribution. One can observe that the major mass (between first and third quartiles) of the distribution is between 10 and 20 μm, concentrating around 15 μm (Supplementary Fig. 1B), even though these datasets are from distinct spatial technologies and tissue systems.
Multi-range cEll coNtext DEciphereR (MENDER)
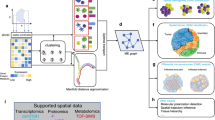
Previous studies have used the cell type composition within the cellular neighborhood around each index cell as its context representation, followed by clustering on the representation (termed cellular neighborhood clustering, CNC)12. However, this approach only considers the context information in one spatial range, limiting the consideration of the cellular relationships across multiple ranges. Motivated by CNC, and given the consistent neighborhood structures, we present Multi-range cEll coNtext DEciphereR (MENDER) by building the cell state composition of multiple ranges into the cellular context representation (Fig. 1, see “Methods”). MENDER takes spatial omics data as input (Fig. 1A), then constructs the spatial graph based on the cell distance matrix and defines cell state based on the gene expression profiles (Fig. 1B). Then the cell state frequencies across multiple ranges are recorded to form the cell context representation (Fig. 1C). The applications of MENDER include identifying spatial domains from multiple slices, i.e., multi-slice analysis (Fig. 1D), detecting condition-specific spatial signatures (Fig. 1E), decoding the MENDER representation into cell spatial organization (Fig. 1F), and extending to large-scale datasets (Fig. 1G). Note that MENDER doesn’t rely on accurately annotated cell clustering, instead, cell clustering is obtained by simply adopting standard Leiden algorithm. We have tested MENDER’s robustness to different cell clustering methods and parameters, as well as noisy cell cluster labels (Supplementary Figs. 2–5, see “Cell state” in “Methods”).

A The input of MENDER is spatial omics data, containing a gene expression matrix and a spatial coordinate matrix. B, C The main body of MENDER. The cell distance matrix is computed using the spatial coordinate matrix and the cell state is determined by the gene expression matrix (B). The cell state frequencies are recorded across multiple ranges away from each cell (C). Applications of MENDER. MENDER can perform multi-slice spatial domain identification (D), identify condition-specific spatial signatures (E), interpret the context representation to biological entities (F), and scale to large datasets (G).
Better prediction power than graph neural network models
To evaluate MENDER’s representation power towards spatial domain prediction and compare with alternative methods, we employed a supervised learning strategy to compare the prediction accuracy of different methods (Fig. 2). The compared methods included SpaGCN18, SpaceFlow20, and STAGATE19, which are considered as the state-of-the-art spatial domain identification methods that can output cellular context representation. We also included SingleRange (the single-range version of MENDER), and CNC (cellular neighborhood clustering)12. We collected 13 SRSC datasets (see “Methods”) with spatial domain annotations regarded as ground truth. We reported the classification accuracy across tenfold across-validation using 3 different classifiers, i.e., Linear Support Vector Machines (SVM) (Fig. 2A), Radial Basis Function (RBF) SVM (Fig. 2B), and Random Forest (Fig. 2C), respectively. A consistent improvement was observed in the cellular context representation obtained by MENDER vis-a-vis modern GNN models independent of the classifier chosen (Fig. 2A–C). We also analyzed the influence of parameters of MENDER, the prediction accuracy of MENDER saturated after the number of ranges reaches 6, and the cell state clustering parameter didn’t affect the classification accuracy (Supplementary Fig. 6).

Linear SVM (A), RBF SVM (B), and Random Forests (C) are performed on the top of each spatial representation method across 13 data, respectively. Each method is run for 5 times. The accuracy is measured using tenfold cross-validation. Error bars are based on mean and 95% confidence interval. Source data are provided as a Source Data file.
Evaluation
The strength of MENDER over complex models in supervised learning situations was evident from earlier analyses. As spatial domain identification is typically an unsupervised learning process that doesn’t use training data, we compared MENDER with other methods in unsupervised contexts (Fig. 3).

A–F Benchmarking on STARmap dataset. The dataset is from the mouse prelimbic area (A) from 3 slices (B). NMI and PAS are used to evaluate different methods for each slice separately (C, E), and jointly (D, F). Error bars are based on mean and 95% confidence interval. G–L Benchmarking on BaristaSeq dataset. The dataset is from the mouse primary visual area (G) from 3 slices (H). NMI and PAS are used to evaluate different methods for each slice separately (I, K), and jointly (J, L). Error bars are based on mean and 95% confidence interval. M–R Benchmarking on MERFISH dataset. The dataset is from the mouse frontal cortex area from 31 slices (M–P). NMI and PAS are used to evaluate different methods for each slice (Supplementary Fig. 7), and jointly (Q, R). Error bars are based on mean and 95% confidence interval. Source data are provided as a Source Data file.
The significance of multi-slice spatial domain identification has been highlighted by the ever-increasing large-scale studies, which collect samples from various tissue sections or individuals. Performing multi-slice analysis can ensures consistent cell labeling and uniform clustering granularities across different slices. We benchmarked MENDER against 4 spatial methods (i.e., STAGATE19, BASS23, CNC12, and SOTIP27) that supported multi-slice analysis, 2 non-spatial algorithms (i.e., Louvain and Leiden36), and 1 method that is single-range version of MENDER (i.e., SingleRange). The 3 benchmark datasets contain 3, 3, and 31 slices measured by STARmap, BaristaSeq, and MERFISH, respectively, each containing the expert-annotated spatial domain labels regarded as ground truth (see “Methods”). Consistent with previous studies20,25,26, the evaluation metrics (see “Methods”) included Normalized Mutual Information (NMI, metrics for accuracy, higher better) and Percentage of Abnormal Spots (PAS, metrics for continuity).
Evaluation on STARmap dataset
We applied the 8 different methods (i.e., Louvain, Leiden, STAGATE, BASS, SOTIP, SingleRange, CNC, and MENDER) on a spatial transcriptomics dataset of the mouse prelimbic area (Fig. 3A) measured by STARmap, containing 3190 cells from 3 slices (Fig. 3B), with expression measurements for 166 genes34. Resulted from 5 replicated runs for each method, the multi-slice spatial domain identification performance of MENDER was consistently the best among alternative methods of all slices, both in accuracy (Figs. 3C, 5 runs for each bar) and continuity (Figs. 3E, 5 runs for each bar). The aggregated performance across all slices also indicated MENDER’s best performance (Fig. 3D, F, 15 runs for each bar).
Evaluation on BaristaSeq dataset
We evaluated on a larger dataset with more spatial domains, which was collected from the mouse primary visual area (Fig. 3G) measured by BaristaSeq, containing 11,426 cells from 3 slices (Fig. 4H), with expression measurements for 79 genes33. Quantitative results of 5 replicated runs showed that MENDER got consistently the best accuracy (Fig. 3I) across different slices, and comparable continuity with BASS substantially better than others (Fig. 3K). The aggregated performance across all slices also indicated MENDER’s best accuracy (Fig. 3J, 15 runs for each bar) in terms of NMI, and second-highest continuity comparable with BASS in terms of PAS (Fig. 3L, 15 runs for each bar).

A–H Application of MENDER on a mouse brain atlas using MERSCOPE. A The dataset contains 9 slices from 3 different brain positions. For each slice, we show the MENDER result (top). We also highlight one slice for each position to compare with the Allen brain reference (bottom). Specific brain domains are shown, including Pontine Gray (B), Isocortex Layers (C), Corpus Callosum (D), Hippocampal region (E), Thalamus (F), and Caudoputamen (G). H The UMAP embedding on the top of MENDER cellular context representation. I Summary of benchmark datasets for unsupervised spatial domain identification. J Running time comparison of different spatial methods across the 4 datasets. Error bars are based on mean and 95% confidence interval. Source data are provided as a Source Data file.
Evaluation on MERFISH dataset
We challenged these methods on an even more complex dataset, collected from the mouse frontal cortex area measured by MERFISH, containing 378,918 cells from 31 slices of 3 ages, with expression measurements for 374 genes (Fig. 3M–P). Compared with the former datasets, this MERFISH dataset contained 33-fold more cells organized as more complex tissue structures and measured more genes. More importantly, the 31 slices were collected from multiple individuals from 3 different aging stages, potentially leading to non-shared spatial domains across slices. Due to the large data size, SOTIP and BASS raised running time issues, so we only compared Louvain, Leiden, STAGATE, SingleRange, CNC, and MENDER. Quantitative results of 5 replicated runs showed that MENDER got substantial improvements in both accuracy and continuity across 31 slices (Supplementary Fig. 7). The aggregated performance of all slices also showed the best accuracy (Fig. 3Q, 155 runs for each bar) and highest continuity (Fig. 3R, 155 runs for each bar) of MENDER.
Evaluating single-slice versus multi-slice analysis
The benefit of multi-slice analysis over single-slice analysis, as emphasized in recent studies23,27, is its ability to perform spatial domain identifications across multiple slices simultaneously. This facilitates the comparison of identified results across slices. Utilizing single-slice analysis for multiple slices separately introduces challenges, such as the need for additional domain matching, especially when the number of slices increases, and the risk of inconsistent clustering granularity across slices. We further assessed whether multi-slice analysis offers per-slice improvement. We conducted single-slice analysis for each slice across three datasets, resulting in a total of 37 single-slice analyses. We then compared the accuracy (in terms of NMI) of single-slice and multi-slice analyses for each slice (see “Methods”). Although statistically significant, the per-slice improvement of multi-slice over single-slice analysis is relatively small (Supplementary Fig. 8).
Extended evaluation
We conducted supplementary studies to evaluate MENDER’s performance outside its primary scope. Specifically, we tested MENDER’s capacity for cell type identification, a task distinct from spatial domain identification, using both supervised and unsupervised settings. Please see Supplementary Notes “Extended analysis on cell type identification task”. Our supplementary analyses demonstrated that MENDER, as well as other state-of-the-art spatial domain identification methods such as STAGATE and SpaceFlow, align more closely with Domain annotations than Cell Type annotations. Quantitative measures, such as Normalized Mutual Information (NMI), showed that MENDER’s performance reduced when compared against Cell Type annotation instead of Domain annotation. Furthermore, when performing supervised cell type identification, the accuracy of MENDER’s representation was found to be approximately 50%, indicating a substantial number of mislabeled cells. This pattern was also observed in other leading spatial domain identification methods, suggesting that these methods are not ideally suited for cell type identification tasks.
Scalability on million-level brain atlas
Another distinct feature of MENDER is the scalability to large datasets, which stems from its deterministic recording of a few spatial neighbors for each cell, in contrast with other complex models that require repeatedly accessing the large spatial graph during the iterations of stochastic optimization. We tested MENDER on a single-cell spatial transcriptomics dataset of the whole mouse brain sections measured by MERSCOPE [https://info.vizgen.com/mouse-brain-data], containing 734,696 cells from 9 slices of 3 different brain positions (Fig. 4A), with expression measurements for 483 genes. The challenges of this dataset include a large cell count, the complex spatial structure of the whole brain, and inconsistent domains between different positions. The advantage of this dataset is that each position has three replicates, which can help verify the consistency of the identified spatial domains of multi-slice analysis. The Allen brain map37 supported additional reference to be compared with [https://mouse.brain-map.org/static/atlas]. Due to these challenges, MENDER was the only spatial method that can handle this dataset.
We closely examined the 9-slice joint analysis results provided by MENDER. A comprehensive assessment of all 9 slices revealed visual consistency within the same position, thus verifying the method’s reliability (Fig. 4A, top). Upon closer inspection of the detailed results for the 3 positions, respectively, a clear correspondence between MENDER’s predicted results and the Allen brain map was identified (Fig. 4A, bottom). For instance, MENDER accurately outlined the laminar patterns in the Isocortex in all three positions (Fig. 4A, C), and also identified domains shared by the 3 positions while being morphologically distinct, such as the Corpus Callosum (Fig. 4A, D) and the Hippocampal region (Fig. 4A, E). Additionally, domains not shared across positions were also detected, examples include the Pontine Gray (Position 1, Fig. 4A, B), the Thalamus (Position 1 and 2, Fig. 4A,F), and the Caudoputamen (Position 2 and 3, Fig. 4A, G). The UMAP embedding of MENDER-induced cell context representation (MENDER-UMAP, Fig. 4H) exhibited both continuous tissue patterns and clear boundaries of distinct tissue compartments. These analyses demonstrate MENDER’s scalability on million-level spatial datasets and the potential for condition-specific domain discovery with multi-slice analysis.
Running time
Another important feature of MENDER is its speed. We summarized the running time of different spatial domain methods in previous datasets (Fig. 4I) and identified a substantial improvement of MENDER over other methods across the 4 datasets (Fig. 4J). Specifically, MENDER was 9.4-fold, 9.3-fold, and 139.1-fold faster than the second-fastest method in STARmap dataset (Dataset1), BaristaSeq dataset (Dataset2), and MERFISH dataset (Dataset3), respectively (Fig. 4J). In the last MERSCOPE dataset (Dataset4) containing 734,696 cells, MENDER is the only appliable method.
Generalizability
We’ve made strides towards testing MENDER’s generalizability on more spatial data modalities, particularly in light of the rapid emergence of new spatial technologies. We evaluated MENDER and other methods on additional datasets (Supplementary Fig. 9). These datasets include single-cell resolution (including Stereo-seq38, osmFISH4, and STARmapPLUS39) and non-single-cell resolution (including Spatial Transcriptomics40, 10x Visium, and Slide-seq6). With the non-single-cell resolution data, we aimed to assess whether MENDER could identify expected tissue structures, even though its initial design is not specifically for such data.
Firstly, we used two Slide-seq datasets (Supplementary Fig. 10). The first dataset originates from the mouse cerebellum and contains 23,096 genes measured on 39,496 beads. We compared the results from different methods (Supplementary Fig. 10B) with the tissue structure reference (Supplementary Fig. 10A). It was evident that all methods consistently identified major cerebellum structures like Molecular Layer (ML) and Granule Layer (GL). However, only MENDER pinpointed the Purkinje Layer (PL), which other methods overlooked. These identified structures were then matched using known structural markers (Supplementary Fig. 10C). Marker genes Gpm6b (ML marker, Supplementary Fig. 10C 3rd column), Calb1 (PL marker, Supplementary Fig. 10C 2nd column), and Cblb3 (GL marker, Supplementary Fig. 10C 1st column) were specifically upregulated in the expected structures. The overlay of these genes also displayed patterns consistent with the expected structures and MENDER’s results (Supplementary Fig. 10C 4th column).
The second dataset, derived from the mouse hippocampus, covered 23,264 genes measured on 53,208 beads. By comparing the results of various methods (Supplementary Fig. 10E) with H&E images and structure annotations from the Allen reference atlas (Supplementary Fig. 10D), it became clear that all methods identified the overall structure of the hippocampal region. However, BASS and SOTIP had difficulties to differentiate finer structures of Cornu Ammonis (CA) and Dentate Gyrus (DG), whereas STAGATE, CNC, and MENDER succeeded in differentiating sub-structures of CA (CA1 and CA3) and DG (Supplementary Fig. 10E). Validation of these identified structures using known structural markers (Supplementary Fig. 10F) revealed Wfs1, a marker gene of CA1, was notably enriched in the region annotated by MENDER (Supplementary Fig. 10F 1st column). The same was true for Chgb in CA3 (Supplementary Fig. 10F 2nd column) and C1ql2 in DG (Supplementary Fig. 10F 3rd column). The overlay of these marker genes (Supplementary Fig. 10F 4th column) matched well with the predicted structures.
We also collected mouse olfactory bulb (MOB) datasets from four distinct spatial technologies: Spatial Transcriptomics (ST), 10x Visium, Slide-seq, and Stereo-seq. These datasets’ resolutions span from single-cell (Stereo-seq) to nearly single-cell (Slide-seq) to tissue level (10x Visium and ST), enabling a thorough assessment of method generalizability across different spatial resolutions. The MOB tissue exhibited well-structured layer patterns with known layer markers, shown for each dataset (Supplementary Fig. 11C, F, I, L). For ST and 10x Visium data, we provided the histological images from the original publications40,41 (Supplementary Fig. 11A, D). From the ST and 10x Visium results, MENDER consistently identified major MOB layers, including Granule Cell Layer (GCL), Mitral Cell Layer (MCL), Glomerular Layer (GL), and Olfactory Nerve Layer (ONL) (Supplementary Fig. 11B, E). Other methods, such as BASS, SOTIP, and STAGATE also detected some of these structures (Supplementary Fig. 11B, E). From Slide-seq and Stereo-seq results, MENDER highlighted finer tissue structures, including the Rostral Migratory Stream (RMS) and Internal Plexiform Layer (IPL), leveraging the enhanced spatial resolution (Supplementary Fig. 11H, K). Other methods, particularly SOTIP and STAGATE, also detected some expected structures (Supplementary Fig. 11H, K).
Subsequently, we collected four datasets of brain cortex tissue from two distinct spatial technologies: 10x Visium and osmFISH. While osmFISH offers single-cell resolution, 10x Visium data is at the spot level. The cortical structures facilitated an evaluation of method performance. Similar to our prior analysis, we provided layer markers for reference (Supplementary Fig. 12C, F, I). Paired histological images were available for 10x Visium data (Supplementary Fig. 12A, D), and structure annotations from the original publication were available for osmFISH data (Supplementary Fig. 12G). For the two 10x Visium datasets, BASS and MENDER results yielded the best laminar structures compared to other methods (Supplementary Fig. 12B, E). For the osmFISH data, referencing the tissue anatomy (Supplementary Fig. 12G), all methods identified expected layers such as Pia, Layer1-6, white matter, and hippocampus. Yet, MENDER achieved sharper layer boundaries (Supplementary Fig. 12H).
Next, we collected eight data obtained by a newer spatial technology, STARmapPLUS39 (Supplementary Fig. 13). The assayed tissue includes both the cortex and hippocampus regions (Supplementary Fig. 13). Since these datasets are of high quality, sourced from standard mouse brain coronal section, a comparison between method results and the Allen reference atlas readily reveals which methods can better identify expected tissue structures. Across these eight samples, all methods differentiated between the cortex and Hippocampal formation (HPF). Focusing on HPF sub-structures, almost all methods detected CA1, CA3, and DG, but only MENDER consistently identified CA2 (Supplementary Fig. 13). Regarding cortex sub-structures, MENDER delineated the clearest layer boundaries compared to other methods (Supplementary Fig. 13).
We have developed an online webpage [https://mender-tutorial.readthedocs.io/], which provide essential guidance on the applications of MENDER for various data types.
Identify age-consistent and age-specific spatial domains
We again analyzed the MERFISH dataset (Fig. 3M–P) since it contains spatial single-cell data at different aging stages (i.e., 4 weeks, 24 weeks, and 20 months)42, which might lead to age-associated biological insights. MENDER-UMAP showed agreement between the low-dimensional embedding and ground truth labels, and also implied the existence of subdomains ignored by original annotation (Fig. 5A, annotated by red, green, pink, and orange dashed circles). We herein sub-clustered the data using MENDER to 9 domains (Fig. 5A, right). Compared with MENDER results before sub-clustering (i.e., the analysis in Fig. 3M–R, where the number of clusters was set to 8), the new clustering result got significantly improved accuracy, consistently across 31 slices (Fig. 5B).

A MENDER-UMAP of the MERFISH aging dataset, colored by the expert annotation from the original publication (left) and MENDER results (right). B The ARI and NMI before and after MENDER sub-clustering. Each point is a slice (the number of slices in total is 31). The green line indicates improved performance after sub-clustering and the orange line indicates decreased performance after sub-clustering. The original annotation and MENDER’s annotation were plotted on the three stages, 4 weeks (C), 24 weeks (D), and 20 months (E), respectively. The aging stage labels are annotated on the MENDER-UMAP plot both on a single plot (F) and separately (G). H The stage distribution across different domains from the original annotation (top) and MENDER (bottom), respectively. I The domain distribution across different slices from 4-week-old mice. J Spatial distribution of MENDER-annotated domains across different aging stages. Different colors indicate different spatial domains. K The spatial signature analysis of MENDER-annotated domains across slices from all aging stages. Each row is a feature of MENDER-computed context representation. The cell states are indicated by different colors, and the ranges are indicated by the number beside the cell state label. Each column shows the spatial signature of an identified domain. The values of the context representation matrix are reflected by the size and colors of the dots. Source data are provided as a Source Data file.
We sought to examine the discrepancy between the original annotation and MENDER prediction (Fig. 5A). We first focused on the Olfactory region (OLF) (Fig. 5A, left), which was sub-clustered as two domains, D6 (pink dashed circle) and D7 (orange dashed circle), by MENDER (Fig. 5A). The spatial single-cell plot displayed not only consistent existence of the two subdomains across different aging stages but also similar localization relationships between them. Spatial signature analysis (see “Methods”) showed dominant distribution of InN-Olf (Inhibitory Neurons) and ExN-Olf (Exhibitory Neurons) in the two sub-domain of OLF (i.e., OLF1 and OLF2), respectively, across ranges 0~4 (Fig. 5K, purple dashed boxes), indicating distinct neuron activities between the two subdomains of OLF conserved across ages. This analysis demonstrated MENDER’s potential to identify biologically meaningful subdomains overlooked by previous analyses.
We next explored an apparent discrepancy between the original annotation and MENDER’s prediction, specifically concerning D4 and D8 (Fig. 5A, right, red and green dashed circle). Both of these were initially annotated as Corpus Callosum (CC) (Fig. 5A, left). The UMAP embedding of MENDER (MENDER-UMAP) revealed an uneven distribution of the three stage labels (i.e., 4 weeks, 24 weeks, and 20 months) across the CC in feature space (Fig. 5F, G). Quantitatively, D8 was almost entirely concentrated in the 4-week stage, while D4 was mostly populated by the 24-week and 20-month stages (Fig. 5H bottom), contrasting the original annotation (Fig. 5H, top). The consistent distribution of D8 across 10 replicates of 4-week mice suggested that this result was not an artifact (Fig. 5I). This paragraph primarily focused on discovering these subdomains within the CC and examining their distribution across different time points.
Advancing from the discovery of these sub-CC domains, we further investigated their spatial distribution. One of the advantages of MENDER’s multi-slice analysis is that it allows for a direct comparison of domain labels across different slices. We selected representative tissue slices from the three aging stages and presented the spatial distribution of MENDER-identified spatial domains for comparison (Fig. 5J). The resulting spatial map exhibited highly similar laminar structures (from outermost to innermost layers: Pia mater, Layer II/III, Layer V, Layer VI, CC, and Striatum) and sharp boundaries, as expected. Notably, the CC domain exhibited different colors (corresponding to different domain labels) between stages, i.e., green at 4 weeks and red at 24 weeks and 20 months. The spatial signature analysis revealed the primary difference between CC (4w) and CC (24w & 20 m) was the distinct distributions of oligodendrocyte subtypes (Fig. 5K, red boxes). These observations underscore MENDER’s potential to identify condition-specific domains and illustrate the spatial relationships between these domains across different stages.
Differentiate spatial-related subtypes of breast cancer
Having already benchmarked the performance and scalability of MENDER and demonstrated the biological insights that MENDER-identified spatial domains provide, our next objective was to investigate whether MENDER could identify spatial domains with biomedical significance. For this purpose, we employed the multi-slice analysis of MENDER on a large-scale MIBI-TOF spatial proteomics dataset11 of 40 Triple-Negative Breast Cancer (TNBC) patients, comprising approximately 200,000 cells in total (see Fig. 6A). This large volume of data poses a challenge to other related methods (as demonstrated with former sections), but can be easily resolved by the high computational efficiency of MENDER.

A Information of the datasets. The datasets comprise 3 patient groups, namely cold, mixed and compartmentalized, consisting of 6, 19 and 15 patients, respectively. B The cell type annotations and MENDER-predicted domains on representative patients from different groups are shown. PCA plots of the patients characterized by the proportions of cell type (fine), termed CT-fine repr (C), cell type (coarse), termed CT-coarse repr (D), and MENDER-identified domains termed MENDER repr (E), respectively. N = 6 independent samples for cold, N = 19 for mixed, and N = 15 for compartmentalized. Boxplot setting: the lower and upper hinges show the first and third quartiles (the 25th and 75th percentiles); the center lines correspond to the median; the upper whisker extends from the upper hinge to the largest value, which should be less than 1.5× the interquartile range and the lower whisker extends from the lower hinge to the smallest value, which is at most the 1.5× interquartile range. F The separability of patients by 3 different proportions is quantified by the classification accuracy of 2 supervised classifiers, i.e., KNN and SVM. The y-axis shows the classification accuracy (ACC) reported fivefold cross-validation, using either KNN or SVM as classifiers. The patient-level labels are cold, mixed and compartmentalized. The p-values (one-sided t-test) indicate the significance of the difference between CT-fine repr and MENDER repr, and between CT-coarse repr and MENDER repr, respectively. Error bars are based on mean and 95% confidence interval. G Similar to F, except that the features of these proportions are PCA-reduced to the same dimensions before classification. The number of PCs ranges from 2 to 17. The y-axis also shows the classification accuracy (ACC) reported by fivefold cross-validation. N = 5 independent experiments. Error bars are based on mean and 95% confidence interval. Source data are provided as a Source Data file.
The patients were categorized into three TNBC subtypes based on the accompanying metadata, namely the cold group, mixed group, and compartmentalized group (see Fig. 6A). The three groups were reported to have significantly different survival outcomes11. Notably, the original study had previously shown that differences between the three subtypes could not be explained by cell type abundance alone. Given that MENDER-identified spatial domains integrate the spatial relationships of diverse cell types, we posited that the abundance of spatial domains across patients may be better suited to distinguish between the three TNBC subtypes.
In this dataset, we observed distinct topological variations across the TNBC subtypes via the spatial domains identified by MENDER (Fig. 6B). We proceeded to represent each patient using the proportion of cell type and domain. The original publication supplied both fine and coarse cell classifications. In subsequent discussions, we define patient representation using MENDER domain proportion as “MENDER repr” and patient representation deploying cell type (fine/coarse) as “CT-fine repr” and “CT-coarse repr”. Aligning with prior findings, neither CT-fine repr nor CT-coarse repr could effectively distinguish among the three subtypes (Fig. 6C, D).
Contrastingly, MENDER repr successfully differentiated the subtypes, and captured the progressive prognosis from the cold group, to the mixed group, and ultimately to the compartmentalized group, as shown in the PCA plot (Fig. 6E). In quantitative terms, for both CT-fine repr and CT-coarse repr, while PC1 could significantly differentiate compartmentalized from cold/mixed for the cell type proportion, PC2 was unable to distinguish between the three groups (Fig. 6C, D). Conversely, for MENDER repr, PC1 could significantly differentiate cold from mixed/compartmentalized, and PC2 could significantly differentiate compartmentalized from cold/mixed (Fig. 6E). By combining the two main PCs, MENDER repr could easily tell apart the three TNBC subtypes (Fig. 6E).
The PCA analysis underscored the visual separability of the patient groups when MENDER was deployed. In order to evaluate the differentiating ability of different patient representations, we adopted the procedure used in representation learning literature43,44,45 and constructed classification tasks using different representations, reporting the classification accuracy as a measure of the differentiating power of each representation (see “Methods”). For the three representations—MENDER repr, CT-fine repr, and CT-coarse repr—we applied two supervised classifiers: K-nearest neighbors (KNN) and Support Vector Machines (SVM). The results underscored that MENDER repr clearly outperformed CT-fine repr and CT-coarse repr in classifier accuracy, whether KNN (Fig. 6F left) or SVM (Fig. 6F right) was employed as the classifier, suggesting a significantly higher ease of classification for the patient groups using MENDER-derived representations. To control for the effects of varying feature numbers, we also evaluated the classification accuracy in three feature spaces by projecting the same number of principal components (see “Methods”). The findings affirmed that the superior predictive capacity of the MENDER-identified domains remained unaffected by feature dimensionality (Fig. 6G).
Discussions
Spatial domain identification is a crucial task in spatial biology and is an important intersection of the machine learning and spatial omics fields. For this task, new methods often followed the established paradigms and conducted incremental developments by increasing model complexity. But whether complex models could deliver consistent gains has not been discussed. To this end, our analysis hinted that a simple model might bring better performance over modern complex models, thus inspiring a new paradigm to break through current bottlenecks.
There are primarily two factors that can influence the determination of spatial domain labels. The first factor is cellular context because MENDER relies on the representation of cellular context to determine spatial domain labels. However, it’s important to note that the presence of the same spatial domains doesn’t necessarily imply the absence of cellular context variations. For instance, consider the original spatial domain region in Fig. 6B, which can still contain cellular context variations, as demonstrated by the color variations in the \({R}_{h}\) region in Supplementary Fig. 35A. Here, we used UMAP-reduced cellular context representation and mapped it to the CIELAB color space for each cell to illustrate these variations. The second factor is the Leiden clustering resolution. When we increased the clustering resolution, we observed that the \({R}_{l}\) region generated different spatial domain labels (Supplementary Fig. 35C). Conversely, when we decreased the Leiden resolution, we noticed that the domain labels within \({R}_{l}\) became more homogeneous (Supplementary Fig. 35B).
There were two folds of analytical contributions. First, we identified consistent neighborhood statistics across different spatial technologies in different tissue systems. Second, we found that simple cellular context analysis might have improved performance compared to state-of-the-art complex models (e.g., Graph Neural Networks and Bayesian Networks) in both supervised and unsupervised settings. There were also two folds of practical contributions. First, we solved the multi-slice analysis in the spatial domain identification task which was little considered by previous methods. Second, we solved the scalability and running time problems, which were the main issue of previous methods in the applications on million-level datasets. We conducted a memory usage comparison between MENDER and other competing methods (Supplementary Table 1). We recorded the peak memory usage for each method on every dataset. The results indicated that SingleRange, CNC, and MENDER exhibit the best memory efficiency, as they only require the maintenance of one fixed spatial graph and context representation in memory. It’s worth noting that even on the MERSCOPE dataset with over 700,000 cells, MENDER only requires 25 min and 80GB+ of memory, showcasing its potential capability to handle datasets of million-level scale.
MENDER’s innovation is best understood within the computational community of spatial transcriptomics, where the mainstream methodological paradigm of spatial domain identifications (also known as spatial clustering) follows a two-step approach8,17. The first step involves encoding the cellular context information into a context-aware representation, and the second step involves clustering the context-aware representation to obtain the spatial domain labels.
Regarding the first step, some methods use graph convolutional networks (GCN) to obtain the context-aware representation18,19,46, while others use probabilistic graphical models (PGM)47,48. MENDER utilizes a new concept which presents a descriptor on how cell state is spatially organized within the local context, as an alternative to GCN and PGM methods. We avoided GCN and PGM methods due to their limitations: GCN methods were reported to have unstable outputs across different runs on different machines25, and PGM methods were reported to have longer running time23. Additionally, both existing GCN and PGM methods lack scalability to large datasets, causing the failure of applying existing methods on datasets larger than \({10}^{6}\) cells.
For the second step, Leiden is used for clustering the context-aware representation, which was commonly used by other spatial clustering methods’ 2nd step. Although some methods use other clustering algorithms for the second step (e.g., STAGATE uses mclust when the number of classes is known19), they are fundamentally similar to MENDER, as they all use existing clustering algorithms to cluster the context-aware representation to obtain the spatial domain labels.
Due to the relatively early developmental stage of the spatial omics field, most available datasets are from the brain and other healthy tissues, and there is still a lack of complex disease or tumor data. In such disease cases, the cell spatial organization and effect range (radius) might be more complex and various across different tissue positions, progression stages, or patients. How to design an adaptive effect range is a future challenge. Due to the complex tissue structures to be studied, especially in diseases, identifying hierarchical tissue structures is another important direction. A straightforward solution is to embed hierarchical clustering methods into current spatial clustering methods. A recent innovation solved this problem in a different way, extracting spatial structure through co-expression hotspots, enabling the identification of multi-scale, multi-layer, interpretable organizational structures49.
Methods
MENDER
MENDER takes multiple slices of spatial omics data as input (see “Input” in the following and Fig. 1A), which contains two matrices, i.e., the gene expression matrix and the spatial coordinate matrix. Next, MENDER uses the gene expression matrix to determine the cell states (see “Cell Group Computation” in the following), and uses the spatial coordinate matrix to obtain the multi-range neighborhood of each cell (see “Multi-range neighborhood Representation Computation” in the following) (Fig. 1B). Then for each index cell, MENDER records the number of each cell state in each range and concatenates them to get the context representation of the index cell (Fig. 1C), which is finally clustered to get the spatial domains (see “MENDER-UMAP Visualization and MENDER Spatial Domains Computation” in the following) (Fig. 1D-G). For technical details, please refer to Supplementary Fig. 14 while reading the following descriptions. Data elements (including input, output, and intermediate data), boxed in purple in Supplementary Fig. 14, are emphasized using “Double quotes” in the following descriptions.
Input
The input of MENDER is multiple slices of spatially resolved single-cell data. “Multiple slices” means these tissue slices are collected from multiple tissue sections and don’t share a common spatial coordinate system. MENDER processes a spatial dataset containing \(G\) genes measured on \(N\) cells from \(S\) slices, with three pieces of input information: (1) The “Gene expression matrix” \(({Nrows}\times {Gcolumns})\); (2) The “Spatial matrix” (\({N\; rows}\times 2{columns},\) for 2D data, or \({Nrows}\times 3{columns},\) for 3D data); (3) The slice ID identifier (a vector of length N), specifying the origin slice of each cell. In order to be compatible with common single-cell and spatial omics analysis packages50,51, the data accepted by MENDER is prepared in Anndata format. The multi-slice data should be merged into the same Anndata object, using the keyword “slice_id” in Anndata.obs to identify different slices.
Cell group computation
Before the construction of cell context representation, MENDER relies on the determination of each cell’s cell state (i.e., “Cell group” in Supplementary Fig. 14) based on the gene expression matrix. In our practice, if batch effects exist across slices, Harmony52 (scanpy.externel implementation, default setting) is used for data integration, followed by neighborhood searching and Leiden clustering (scanpy.tool implementation, with resolution = 2) on the “Harmonized embedding”. If no batch effect is present, the PCA-reduced gene expression profiles are directly subjected to neighborhood searching and Leiden clustering.
Some resource papers provided expert-annotations of cell types, if such reliable annotation is provided as prior knowledge, the above procedure can be bypassed to directly acquire the “Cell group”. This approach has the potential to enhance accuracy and shorten the running time. In real-world applications, particularly when confronted with complex disease cases and possible batch effects, we highly recommend using reliable cell type annotation across multiple slices to evade potential inaccuracies.
We evaluated the robustness of MENDER with respect to various cell clustering methods and parameters across multiple datasets (Supplementary Figs. 2–4). Specifically, we assessed four single-cell clustering methods: UMAP + KMeans, Louvain, Leiden, and SC3s53. We then reported MENDER’s performance (quantified in terms of NMI) in relation to diverse clustering parameters associated with each method. As clustering granularity increased, MENDER’s performance rapidly approached its peak and maintained without significant degradation. This behavior demonstrates MENDER’s robustness to variations in cell clustering methods and their corresponding parameters across datasets. Broadening our exploration, we examined MENDER’s robustness against low-quality cell clusters (Supplementary Fig. 5). To achieve this, we introduced varying noise levels to cell cluster labels. Our results indicate that MENDER’s performance suffers only a slight drop when the noise level remains under 0.5, emphasizing MENDER’s robustness even in the presence of noisy cell group labels.
In more general cases, the disengagement of the cell state clustering step could provide a divide-and-conquer solution when inputting low-quality spatial data. Besides the above approach to determine the “Cell Group”, one can also apply reference-based54 cell type annotations using methods such as scArches55, Tangram56, and Spatial-ID57, to determine the cell state labels. Such feature of MENDER makes it a highly flexible framework that can effectively integrate rich resources of bioinformatics tools.
Multi-range neighborhood Representation Computation
The “Spatial Matrix”, “Slice ID”, and the “Cell Group” (obtained previously) are utilized to calculate the “Multi-range Neighborhood Representation”. The “Slice ID” separates the “Spatial Matrix” into multiple matrices, each representing the spatial matrix of a slice, to prevent cells from different slices from becoming spatial neighbors. For each spatial matrix corresponding to a slice, spatial neighborhood searching (Squidpy51 implementation, default parameters) is performed independently by constructing a multi-process parallelization pool (Python multiprocessing implementation).
For each slice, around every cell, \(S\) ranges (the setting of \(S\) depends on the spatial resolution, as noted in Supplementary Fig. 14: 2 for 10X Visium/ST, 4 for Slide-seq, and 6 for single-cell resolution technologies) of spatial neighborhoods are created, forming \(S\) ring areas around the central cell (the radius is set to 15um by default). The cell index located within each ring area is recorded for each central cell. Note that although 15um was consistently set across this manuscript, it may not necessarily hold if broader range of spatial data is obtained from a larger variety of tissues in the future. Therefore, we have developed a function module called estimate_radius that enables the evaluation of distance distributions for new datasets.
Combining the “Cell Group” computed earlier, for each cell, the frequencies of cell types located within the central cell’s associated multi-range neighborhood are recorded. These frequencies are then concatenated to form the “Multi-range Neighborhood Representation”.
Formally, suppose the total number of cells across all input slices is \(N\), the first step partitioned all cells into \(C\) distinct cell states, i.e., “Cell group” in Supplementary Fig. 14, noted as \(G=\left\{{g}_{c}\right\},{c}\in [1,C]\). The cell state of the \(i\)-th cell is noted as \({cel}{l}_{i},{i}\in [1,N]\). The origin slice of the \(i\)-th cell is noted as \({slic}{e}_{i},{i}\in [1,N].\) The spatial coordinate of the \(i\)-th cell is noted as \(\left({x}_{i},{y}_{i}\right),{i}\in [1,N]\). The number of ranges is set to \(S\). The radius is set to \(R\). Then the multi-range neighborhood representation matrix, \(M\in {{Z}^{+}}^{N\times (S\times C)}\), in which the \(i\)-th row is the context-aware representation of the \(i\)-th cell.
MENDER-UMAP visualization and MENDER spatial domains computation
Utilizing the “Multi-range Neighborhood Representation” of each cell, dimension reduction and clustering are executed to generate the “MENDER-UMAP Visualization” and the “MENDER Spatial Domains”. To create the “MENDER-UMAP Visualization”, neighborhood graph (scanpy.pp.neighbors) is constructed on the normalized and PCA-reduced “Multi-range Neighborhood Representation” (implemented by scanpy.pp.normalize_total then scanpy.pp.log1p and scanpy.pp.pca). Then UMAP (implemented by scanpy.tool.umap) is applied on the neighborhood graph. To generate the “MENDER Spatial Domains”, the Leiden clustering is employed to cluster on the neighborhood graph (same as before). The clustering resolution of Leiden is set in the following manner: if the expected number of domains is known, a function is implemented to automatically estimate the suitable Leiden resolution. This can be accomplished by executing run_clustering_normal (with a positive value as the parameter, for the expected number of domains). Conversely, if the expected number of domains is not available (as in exploratory studies), the Leiden resolution defaults to 0.5. This is achieved by running run_clustering_normal (with a negative value for clustering resolution).
Evaluation and biomedical applications
Finally, once the spatial domains are obtained, one might want to evaluate the accuracy of the identified domains and carry out biomedical applications. For evaluation purposes, MENDER includes Compute_NMI, a tool for comparing the similarity between predicted domains and ground truth domains using Normalized Mutual Information (NMI). Compute_PAS is also provided to assess the spatial coherence of the predicted domains.
For biomedical applications, it is suggested to use the proportions of domains of each patient as their representation. In the case of unsupervised analysis, Principal Component Analysis (PCA) is applied to the patient representation to embed each patient in a low-dimensional space. This process can automatically partition patients into different groups with significantly different outcomes. More details can be found in “Evaluation of patient representations” in “Methods”.
Determine the optimal clustering resolution
The challenge of determining the appropriate resolution or number of regions in spatial clustering is a common hurdle in the field. To address this challenge, we introduced the “res_search” method in MENDER. This approach enables users to iteratively search for the optimal Leiden resolution, given the expected number of regions (Supplementary Fig. 36). To demonstrate, Supplementary Fig. 37 highlights the effectiveness of the “res_search” method in resolution selection. Using a MERSCOPE brain dataset, we showed that MENDER, with default resolution settings, identifies fine-grained structures. However, when applying “res_search” with an expected number of regions set to 5, MENDER accurately discerns broader brain regions, aligning with the Allen Brain Atlas.
Six aspects to view existing methods
Support for multi-slice analysis
The multi-slice analysis is a concept proposed relative to single-slice analysis23. It aims to perform spatial domain identifications on multiple slices at the same time so that the labels of the identified results can be compared across slices. If the single-slice analysis is used to analyze multiple slices separately, it will lead to two problems. First, the labeling result of each slice is independent, which means that the domain A in one slice and the domain A in another slice do not necessarily refer to the same domain, resulting in the need for additional domain matching, which is more challenging in scenarios where the number of slices increases. Second, single-slice analysis of multiple slices may result in inconsistent clustering granularity across slices. For example, domain A in one slice may be split into domain C and domain D in another slice.
The importance of multi-slice analysis was highlighted in recent studies23,27, and only 3 existing methods provided the interface for users to perform multi-slice analysis, including STAGATE19, BASS23, and SOTIP27. STAGATE provides a tutorial for multi-slice analysis at [https://stagate.readthedocs.io/en/latest/AT1.html], SOTIP provides at [https://github.com/TencentAILabHealthcare/SOTIP/tree/master/SOTIP_analysis/multi_sample], and BASS provides at [https://zhengli09.github.io/BASS-Analysis/].
We can categorize the methods that support multi-slice analysis into three paradigms: “early-support”, “late-support”, and “data-end-support”, based on the point in the workflow where the integration occurs.
The “early-support” paradigm typifies methods such as SOTIP and BASS, which perform data integration early in the procedure. In other words, they initially harmonize gene expressions across different slices before proceeding to construct spatial graphs independently for each slice. The final modeling is then performed on all slices jointly, yielding spatial domain results comparable between slices.
On the other hand, STAGATE exemplifies the “late-support” paradigm, where single-slice analysis of STAGATE is performed independently for each slice. This process generates a context-aware representation for each slice. The data integration operation is then performed on these representations, with the final clustering carried out in the integrated embedding space.
The third paradigm, “data-end-support”, is exemplified by BayesSpace. This paradigm modifies the spatial coordinates of different slices to lay on the same spatial coordinates, and maintains a substantial gap between slices, so that spots from different slices are not neighbors. The algorithm then proceeds with single-slice analysis, yielding spatial clustering results.
At present, SpaGCN, CCST, SpaceFlow, and SpatialPCA currently lack the necessary functionalities in their code and documentation for multi-slice analysis. However, with modifications to their code, they could be adapted to facilitate multi-slice analysis, using either the “early-support” or “late-support” paradigms previously discussed. On the other hand, BayesSpace’s methodology does not generate context-aware cell representations, making it unsuitable for extension via the “late-support” paradigm.
Stability
Stability is the ability of a computational method to produce similar output as possible in different runs when given identical input58. Reproducibility in the scientific community has received widespread attention in recent years, and method stability is an important part of it. The randomness and non-convexity of modern deep-learning models make them difficult to produce stable results across different runs on different machines. Relatively better stability was reported by probabilistic graphical models although they also cannot guarantee complete stability.
Interpretability
Interpretability judges whether the parameters and variables involved in the model can be mapped to biological entities or relationships. Good interpretability can lead to more meaningful model outputs and better means of model diagnosis. Deep-learning models have inherent disadvantages in interpretability compared to Bayesian models, which were generally built upon biological items as variables and dependencies as conditional probabilities.
Scalability and speed
Since computational methods in spatial omics need to additionally consider spatial relationships of cells, compared with single-cell (non-spatial) methods (such as single-cell clustering), they are more challenging to scale to large-scale data. At present, most existing spatial methods are only applied and benchmarked on relatively smaller data (<100,000 cells). GNN-based methods tend to be more scalable than BN-based methods since they can rely on mature deep-learning communities and tools. The scalability bottleneck of existing methods is considered as the ability to generate output within a reasonable running time. But for even larger datasets, memory may be the next bottleneck (GNN may be at a disadvantage since larger VRAM is less accessible than RAM).
Availability of cell context representation
This feature enables the output of both cell context representation (the cellular context information is encoded) and the spatial domain label for each cell. The context representation is a fixed-length vector for each cell, which may be useful for additional downstream analysis like those in single-cell analysis. The additional analysis might include pseudo-space analysis20,59 (similar to the pseudo-time analysis in single-cell analysis60,61,62), data visualization (for example using t-SNE63, UMAP64, and PHATE65), differential expression analysis66,67 (this requires the representation to be biologically meaningful, i.e., interpretable), and other analysis implemented in single-cell packages like SCANPY50 or Seurat68. GNN-based methods generally output both the context representation and spatial domain labels, and the latter is generally obtained by clustering on the former. Among BN-based methods, those based on Markov random fields (e.g., BayesSpace and BASS) generally do not output the context representation.
Analyzing the distance of neighboring cells
Datasets
MERFISH primary motor cortex dataset is from Ref. 29, MERFISH hypothalamic preoptic region dataset is from Ref. 32, MERFISH nucleus accumbens dataset is from Ref. 30, DARTFISH occipital cortex dataset is from Ref. 14, BaristaSeq primary visual area dataset is from Ref. 33, STARmap primary visual cortex dataset is from Ref. 34, osmFISH somatosensory cortex dataset is from Ref. 4, seqFISH embryo dataset is from Ref. 35. These datasets contain the major single-cell-level resolution spatial technologies. We didn’t analyze datasets from other single-cell-level technologies such as slide-seq5 or slide-seqV26,69,70 because the sequencing units (i.e., beads) were array-like distributed in the space and thus didn’t reflect real cell distance.
Analysis
For each slice in each dataset, we recorded the distance of each cell with its nearest cell, and all cells in the slice were collected as a distribution (i.e., boxplot in Fig. 1B). Specifically, for each slice, we first computed the pairwise distance of all cells to get a distance matrix. This was done using the “pdist” and “squareform” functions of scipy71. To avoid the zero values along the diagonal, we set the diagonal of the distance matrix to infinite value (“fill_diagonal” function of numpy72), then for each row, we record its minimum value as the shortest distance. Some datasets contain many slices, to save space, we only randomly selected 5 slices to show the distribution.
Boxplot
The lower and upper hinges show the first and third quartiles (the 25th and 75th percentiles); the center lines correspond to the median. Distance ranges from 10 μm to 20 μm were highlighted with orange, and distance of 15 μm was indicated with the red dashed line (Supplementary Fig. 1B). Boxplots were generated using Seaborn [https://seaborn.pydata.org/].
Supervised learning settings
General settings
To compare the spatial domain prediction performance between Graph Neural Network models and our simple cellular context representation, we used 3 state-of-the-art methods, i.e., SpaGCN18, SpaceFlow20, and STAGATE19, that can output cell context representation as the input for prediction. For a fair comparison, we set the number of neurons of the hidden layer (i.e., the number of dimensions of context representation) of each method to 50, and the number of epochs is set to 500. Each method on each dataset was performed for 5 times.
SpaGCN
For SpaGCN, we followed the tutorial in [https://github.com/jianhuupenn/SpaGCN/blob/master/tutorial/tutorial.ipynb], the difference is that we set the “histology” parameter to false since there are not H&E images available. Since SpaGCN involved a PCA step (to reduce the gene expression vector to 50) prior to the GNN network, which would raise an error if the number of genes is smaller than 50. We thus made a modification to the original code so that the “n_comp” of the PCA is set to the smaller value between (50 and the number of measured genes). The resulted cell context representation was obtained by the “embed” attribute of SpaGCN object.
SpaceFlow
For SpaceFlow, the authors provided tutorials for single-cell spatial transcriptomics data [https://github.com/hongleir/SpaceFlow], we followed their recommended parameters and obtained the context representation by the “embedding” attribute of SpaceFlow object.
STAGATE
For STAGATE, the authors nicely provided multiple tutorials for different spatial data types, including Slide-seqV2, 10X Visium, stereo-seq, and STARmap datasets. Since our test datasets’ characteristics were most similar to STARmap dataset, we used the recommended steps in [https://stagate.readthedocs.io/en/latest/T9_STARmap.html]. We also tuned the “rad_cutoff” parameter of STAGATE for best performance (in practice, this parameter is best tuned so that the number of neighbors per cell on average is around 10).
Evaluation
We used three classifiers to evaluate the representation powers of SpaGCN, SpaceFlow, STAGATE, and MENDER. The three classifiers were Linear SVM, RBF SVM, and Random Forest, which were standard classification algorithms for linear classifier, non-linear classifier, and tree-based classifier. We recorded the median classification accuracy across tenfold cross-validations. The classifiers and cross-validation implementations were used from Scikit-learn73.
Unsupervised learning settings
The unsupervised task in this study is multi-slice analysis for spatial domain identification. We benchmarked MENDER against other methods, including 4 spatial methods that were available for multi-slice analysis (STAGATE, BASS, CNC, and SOTIP), and 2 non-spatial methods (Louvain and Leiden). We used two evaluation metrics as done by previous benchmarks26.
Normalized Mutual Information (NMI) can be used to evaluate the accuracy of predicted spatial domain labels compared with ground truth, and high NMI means good performance. NMI quantifies the similarity between two label assignments, supposed as \(P\) and \(T\), to the same set of objects. \(H(P)\) and H\((T)\) are denoted as their entropies. Then NMI is computed as:
Percentage of Abnormal Spots (PAS) can be used to evaluate the spatial continuity of predicted domain labels given the spatial coordinates, and low PAS means good performance. PAS was calculated as the percentage of cells whose spatial domain label differed from at least 6 of its 10 neighbors.
Given the complexity and heterogeneity inherent in spatial biological data, a single evaluation metric may not sufficiently capture the performance of spatial domain identification methods. While metrics like PAS, Local Inverse Simpson’s Index (LISI), and spatial chaos score (CHAOS) offer insights into the spatial continuity of the predicted domains25,26, higher spatial continuity doesn’t not necessary mean better spatial domain prediction, and they must be interpreted carefully and in the context of other performance measures such as NMI or ARI, which directly compare the predicted labels against the ground truth. The simultaneous consideration of these metrics can provide a more nuanced understanding of the method’s performance. More importantly, it can help to avoid potential misinterpretations that may arise when these metrics are considered in isolation.
Multi-slice analysis of STAGATE
We used the tutorial provided in [https://stagate.readthedocs.io/en/latest/AT1.html]. The difference is that that tutorial used 10X Visium datasets that were different from our single-cell resolution datasets. So we pre-process our datasets as done in [https://stagate.readthedocs.io/en/latest/T9_STARmap.html], and used the multi-slice code to construct the spatial graph separately. As to the “rad_cutoff” parameter, like before, we tuned this parameter so that the number of neighbors per cell on average is around 10, to get the best performance.
Multi-slice analysis of BASS
The authors of BASS directly provided the code for multi-slice analysis on single-cell resolution spatial data [https://zhengli09.github.io/BASS-Analysis/STARmap.html]. So we directly employed the steps.
Multi-slice analysis of SOTIP
The authors provided the code for multi-slice analysis in [https://github.com/TencentAILabHealthcare/SOTIP/tree/master/SOTIP_analysis/multi_sample]. We directly adopted the code for benchmark analysis.
Parameter setting
Please refer to Supplementary Fig. 9. We have also provided the corresponding reproducibility code.
Elaborating on the parameters, the ‘scale’ refers to the number of ranges employed during the construction of multi-range neighborhoods. For technologies of single-cell resolution, such as STARmap, BaristaSeq, MERFISH, MERSCOPE, Stereo-seq, osmFISH, ExSeq, and STARmapPlus, we have consistently set the ‘scale’ to 6.
The parameters ‘nn’ or ‘radius’ dictate the size of each range. ‘nn’ is utilized for spatial technologies with an array-like spot distribution, such as 10X Visium and ST. Specifically, ‘nn’ is set to 6 for 10X Visium, reflecting its six nearest neighborhoods per spot, and 4 for ST, corresponding to its four nearest neighborhoods per spot. On the other hand, for spatial technologies with non-array-like distributions, we set the ‘radius’ parameter to a consistent 15 µm. We also provided a function in MENDER package called estimate_radius to recommend suitable ‘radius’ for potential users.
Leiden clustering is employed twice, initially to define the ‘Cell group’ (Supplementary Fig. 14) and finally to obtain ‘MENDER spatial domains’ (Supplementary Fig. 14). For the initial clustering, we consistently apply Leiden with a resolution of 2. The final Leiden clustering requires one parameter, ‘k’. If ‘k’ is positive, MENDER’s ‘res_search’ function is executed to automatically ascertain the optimal Leiden resolution that will yield ‘k’ domains. This is particularly useful when the expected number of domains is known a priori, such as in our benchmark studies with ground truth domain annotations. If ‘k’ is negative, Leiden clustering is executed with a resolution equal to the absolute value of ‘k’. This is preferred when the number of domains is not provided as prior knowledge to the method user. In such scenarios, multiple different resolutions should be tested.
Per-slice performance comparison between multi-slice and single-slice analysis
Single-slice analysis is conducted using the MENDER.MENDER_single module, while multi-slice analysis is performed using the MENDER.MENDER module in the MENDER package. The parameter settings for both analyses are (scale = 6 | radius = 15 µm | k = #domains). The NMI is compared as follows (using the STARmap dataset (Fig. 3B) as an example). For single-slice analysis, the three slices of the dataset are analyzed independently, each with 10 replicates, resulting in 10 NMI values per slice. For multi-slice analysis, the dataset (three slices jointly) is used for joint spatial clustering, also for 10 replicates. Therefore, each slice has 10 versions of predicted domains and 10 NMI values. Hence, both multi-slice and single-slice analyses of the STARmap dataset yield 30 NMI values, as compared in Supplementary Fig. 8. The p-value is obtained using a one-sided Wilcoxon rank-sum test. The same approach was applied to the BaristaSeq and MERFISH datasets.
Evaluation of patient representations
In our final application, we assessed the performance of three distinct methods for patient-level representation in separating patient groups. Patient-level annotations are available in the original manuscript11, with three distinct labels: cold, compartmentalized, and mixed. The three patient representations evaluated are all proportions of cell-level labels within each patient. The first two representations, termed CT-fine repr and CT-coarse repr, derive from cell type labels within each patient, as annotated by the original paper, but with different cell type granularities (i.e., fine and coarse). The third representation, termed MENDER repr, is derived from the spatial domain labels identified by MENDER (default parameters: radius = 15 µm, scale = 6, k = −0.5). Each representation matrix has rows equivalent to the total number of cells and columns equivalent to the number of unique labels across the dataset.
Unsupervised analysis (Fig. 6C–E) was performed by initially applying PCA to the three patient representations, followed by mapping all patients into the resultant three PCA spaces (the top two principal components). We used a two-sided Student’s t-test to assess differences between patient groups for each top principal component. PCA was conducted using Scanpy’s default settings, and the Student’s t-test was conducted using Scipy’s implementation.
The supervised analysis (Fig. 6F, G) consisted of two parts. The first part (Fig. 6F) involved supervised classification on the raw feature space of the three patient representations, namely CT-fine repr, CT-coarse repr, and MENDER repr. The second part (Fig. 6G) involved supervised classification on the PCA-reduced (top 2~17 principal components) feature space of the three patient representations. In the first part, we employed a K-nearest-neighbor (KNN) classifier to classify the three patient representations. We reported classification accuracy (using sklearn.metrics.accuracy_score implementation) with fivefold cross-validation. The results are displayed in Fig. 6F (left), where the y-axis represents the KNN classification accuracy. The p-value was calculated using Scipy’s implementation of the Student’s t-test. The procedure for Fig. 6F (right) was similar to that for Fig. 6F (left), except we substituted the KNN classifier with an SVM classifier. In the second part (Fig. 6G), as in the first part, KNN and SVM classifiers were used to evaluate the different patient representations. The difference was that Fig. 6G tested the PCA-reduced versions of the three representations. Specifically, for Fig. 6G (top), the three representations were reduced to k (2~17) top principal components, and for each k, we reported the KNN classification accuracy using fivefold cross-validation. A similar approach was taken for the SVM analysis (Fig. 6G, bottom).
Annotations
All domain annotations have been manually annotated in previously published papers. The STARmap data was obtained from Ref. 34 and manually annotated by Xiang Zhou’s group23, based on the Allen reference map and the gene expression patterns of the prelimbic area. The data and annotation can be downloaded from https://github.com/zhengli09/BASS-Analysis/tree/master/data. The BaristaSeq data was manually annotated by the SpaceTx Consortium14, which comprises 13 labs from 11 universities and institutes. The primary aim of the Consortium is to provide data resources for benchmarking computational methods in the field of spatial transcriptomics. The data and annotations can be downloaded from https://spacetx.github.io. The MERFISH data was annotated in their original study42. Every single slice was first clustered into small patches based on gene expression and spatial locations. These patches were then manually merged with reference to brain anatomical structures and known gene expressions. The data and annotations can be downloaded from https://cellxgene.cziscience.com/collections/31937775-0602-4e52-a799-b6acdd2bac2e.
Running time
For all computational experiments, we used the Python library “time” to record the running time. Each method’s data preprocessing step was included in the duration, along with the main body of the method. For deep-learning methods (STAGATE), since the running time was strongly related to the number of epochs, we set the epochs to 500 as indicated in the tutorials. An increased number of epochs might bring improved accuracy but increased running time.
Spatial signature analysis
Given the interpretability of MENDER, i.e., the representation vector obtained by MENDER could be mapped to biological entities, such as cell state and distances to the index cell. One can identify the spatial differences of one domain (that is, a group of cells clustered by the context representation) compared to another domain. To do this, we employed the Wilcoxon rank-sum test between two groups, found the top 5 features for each domain with the highest scores (the score is computed for each gene using both the p-value and expression levels, implemented by SCANPY), and plotted as dot-plots in Fig. 5K. In the dot-plot, each row represented a feature of the cell context representation by MENDER, and the feature encodes two levels of information, one is the cell state, and another is the distance range away from the index cell (each range is a 15um radius ring centered at the index cell). The dot plot then displayed the spatial signatures of every spatial domain, so that one can observe what specific cell spatial organization is associated with each domain.
Computational resource
CPU: Intel(R) Xeon(R) Gold 5218R CPU @ 2.10 GHz 80 cores. Memory: 263724496 kB in total.
Statistics & reproducibility
No statistical method was used to predetermine sample size. No data were excluded from the analyses. Randomization was achieved by setting random seeds. The algorithm developer and the data analyzer were the same person, so totally blinding was impossible.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All relevant data supporting the key findings of this study are available within the article and its Supplementary Information files. STARmap Prelimbic area data: [https://github.com/zhengli09/BASS-Analysis/tree/master/data]; BaristaSeq Visual cortex data: [https://spacetx.github.io/data.html]; MERFISH Frontal cortex data: [https://cellxgene.cziscience.com/collections/ 31937775-0602-4e52-a799-b6acdd2bac2e]; MERSCOPE (Vizgen) Brain data: [https://info.vizgen.com/mouse-brain-data]; ST Olfactory bulb data: [https://www.spatialresearch.org/resources-published-datasets/]; Visium data: [https://www.10xgenomics.com/resources/datasets]; Slide-seq data: [https://singlecell.broadinstitute.org/single_cell/study/SCP815/sensitive-spatial-genome-wide-expression-profiling-at-cellular-resolution#study-summary]; Stereo-Seq data: [https://db.cngb.org/stomics/datasets/STDS0000058]; osmFISH data: [http://linnarssonlab.org/osmFISH/]; ExSeq data: [10.5281/zenodo.4075515]; STARmapPLUS data: [https://singlecell.broadinstitute.org/single_cell/study/SCP1375]; Allen Reference Atlas: [https://mouse.brain-map.org/experiment/thumbnails/100048576?image_type=atlas]; We also provide the benchmark datasets as h5ad format via SODB31, please find how to load them in the tutorial [https://mender-tutorial.readthedocs.io/en/latest/]. Source data are provided with this paper.
Code availability
The Python implementation of MENDER is available at Github [https://github.com/yuanzhiyuan/MENDER] and Zenodo74. A tutorial on MENDER package is also available at [https://mender-tutorial.readthedocs.io/en/latest/].
References
Shah, S., Lubeck, E., Zhou, W. & Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016).
Eng, C. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. Y. & Zhuang, X. W. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Codeluppi, S. et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 15, 932 (2018). +.
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463 (2019).
Stickels, R. R. et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. https://doi.org/10.1038/s41587-020-0739-1 (2020).
Moses, L. & Pachter, L. Museum of spatial transcriptomics. Nat. Methods 19, 534–546 (2022).
Palla, G., Fischer, D. S., Regev, A. & Theis, F. J. Spatial components of molecular tissue biology. Nat. Biotechnol. 40, 308–318 (2022).
Rao, A., Barkley, D., Franca, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220 (2021).
Lewis, S. M. et al. Spatial omics and multiplexed imaging to explore cancer biology. Nat. Methods 18, 997–1012 (2021).
Keren, L. et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174, 1373 (2018).
Schürch, C. M. et al. coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182, 1341–1359.e19 (2020).
Fan, Z., Chen, R. & Chen, X. SpatialDB: a database for spatially resolved transcriptomes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz934 (2019).
Long, B., Miller, J. & Consortium, T. S. SpaceTx: a roadmap for benchmarking spatial transcriptomics exploration of the brain. Preprint at https://arxiv.org/abs/2301.08436 (2023).
Zeng, Z., Li, Y., Li, Y. & Luo, Y. Statistical and machine learning methods for spatially resolved transcriptomics data analysis. Genome Biol. 23, 83 (2022).
Walker, B. L., Cang, Z., Ren, H., Bourgain-Chang, E. & Nie, Q. Deciphering tissue structure and function using spatial transcriptomics. Commun. Biol. 5, 220 (2022).
Dries, R. et al. Advances in spatial transcriptomic data analysis. Genome Res. 31, 1706–1718 (2021).
Hu, J. et al. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021).
Dong, K. & Zhang, S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat. Commun. 13, 1739 (2022).
Ren, H., Walker, B. L., Cang, Z. & Nie, Q. Identifying multicellular spatiotemporal organization of cells with SpaceFlow. Nat. Commun. 13, 4076 (2022).
Zhao, E. et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 39, 1375–1384 (2021).
Yang, Y. et al. SC-MEB: spatial clustering with hidden Markov random field using empirical Bayes. Brief. Bioinform. 23, bbab466 (2022).
Li, Z. & Zhou, X. BASS: multi-scale and multi-sample analysis enables accurate cell type clustering and spatial domain detection in spatial transcriptomic studies. Genome Biol. 23, 168 (2022).
Cheng, A., Hu, G. & Li, W. V. Benchmarking cell-type clustering methods for spatially resolved transcriptomics data. Brief. Bioinform. 24, bbac475 (2023).
Li, J., Chen, S., Pan, X., Yuan, Y. & Shen, H.-B. Cell clustering for spatial transcriptomics data with graph neural networks. Nat. Comput. Sci. 2, 399–408 (2022).
Shang, L. & Zhou, X. Spatially aware dimension reduction for spatial transcriptomics. Nat. Commun. 13, 7203 (2022).
Yuan, Z. et al. SOTIP is a versatile method for microenvironment modeling with spatial omics data. Nat. Commun. 13, 7330 (2022).
Fang, R. et al. Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH. Science 377, 56–62 (2022).
Zhang, M. et al. Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature 598, 137–143 (2021).
Chen, R. et al. Decoding molecular and cellular heterogeneity of mouse nucleus accumbens. Nat. Neurosci. 24, 1757–1771 (2021).
Yuan, Z. et al. SODB facilitates comprehensive exploration of spatial omics data. Nat. Methods 20, 387–399 (2023).
Moffitt, J. R. et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, https://doi.org/10.1126/science.aau5324 (2018).
Chen, X., Sun, Y.-C., Church, G. M., Lee, J. H. & Zador, A. M. Efficient in situ barcode sequencing using padlock probe-based BaristaSeq. Nucleic Acids Res. 46, e22–e22 (2018).
Wang, X. et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, aat5691 (2018).
Lohoff, T. et al. Integration of spatial and single-cell transcriptomic data elucidates mouse organogenesis. Nat. Biotechnol. 40, 74–85 (2021).
Traag, V. A., Waltman, L. & Van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 1–12 (2019).
Wang, Q. et al. The Allen mouse brain common coordinate framework: a 3D reference atlas. Cell 181, 936–953 e920 (2020).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. https://doi.org/10.1016/j.cell.2022.04.003 (2022).
Zeng, H. et al. Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer’s disease. Nat. Neurosci. 26, 430–446 (2023).
Stahl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Lebrigand, K. et al. The spatial landscape of gene expression isoforms in tissue sections. Nucleic Acids Res. 51, e47 (2023).
Allen, W. E., Blosser, T. R., Sullivan, Z. A., Dulac, C. & Zhuang, X. Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell 186, 194–208.e18 (2022).
Li, X. et al. Network embedding-based representation learning for single cell RNA-seq data. Nucleic Acids Res. 45, e166 (2017).
Gidaris, S., Singh, P. & Komodakis, N. Unsupervised representation learning by predicting image rotations. Preprint at https://arxiv.org/abs/1803.07728 (2018).
Doersch, C., Gupta, A. & Efros, A. A. Unsupervised visual representation learning by context prediction. In Proc 2015 IEEE International Conference on Computer Vision (ICCV), 1422–1430 (2015).
Fu, H. et al. Unsupervised spatial embedded deep representation of spatial transcriptomics. Preprint at bioRxiv, https://doi.org/10.1101/2021.06.15.448542 (2021).
Shang, L. & Zhou, X. Spatially aware dimension reduction for spatial transcriptomics. https://doi.org/10.1101/2022.01.19.476966 (2022).
Chidester, B., Zhou, T., Alam, S. & Ma, J. SPICEMIX enables integrative single-cell spatial modeling of cell identity. Nat. Genet. https://doi.org/10.1038/s41588-022-01256-z (2023).
Walker, B. L. & Nie, Q. NeST: nested hierarchical structure identification in spatial transcriptomic data. Nat. Commun. 14, 6554 (2023).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Palla, G. et al. Squidpy: a scalable framework for spatial omics analysis. Nat. Methods. https://doi.org/10.1038/s41592-021-01358-2 (2022).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Quah, F. X. & Hemberg, M. SC3s: efficient scaling of single cell consensus clustering to millions of cells. BMC Bioinform. 23, 536 (2022).
Li, B. et al. Benchmarking spatial and single-cell transcriptomics integration methods for transcript distribution prediction and cell type deconvolution. Nat. Methods. https://doi.org/10.1038/s41592-022-01480-9 (2022).
Lotfollahi, M. et al. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-01001-7 (2021).
Biancalani, T. et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 18, 1352–1362 (2021).
Shen, R. et al. Spatial-ID: a cell typing method for spatially resolved transcriptomics via transfer learning and spatial embedding. Nat. Commun. 13, 7640 (2022).
Saelens, W., Cannoodt, R., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547–554 (2019).
Zuo, C. et al. Elucidating tumor heterogeneity from spatially resolved transcriptomics data by multi-view graph collaborative learning. Nat. Commun. 13, 5962 (2022).
Haghverdi, L., Buttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Haghverdi, L., Buettner, F. & Theis, F. J. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31, 2989–2998 (2015).
Wolf, F. A. et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019).
Maaten, L. V. D. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
McInnes, L., Healy, J., Saul, N. & Grossberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 3, 861 (2018).
Moon, K. R. et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492 (2019).
Vandenbon, A. & Diez, D. A clustering-independent method for finding differentially expressed genes in single-cell transcriptome data. Nat. Commun. 11, 4318 (2020).
Ntranos, V., Yi, L., Melsted, P. & Pachter, L. A discriminative learning approach to differential expression analysis for single-cell RNA-seq. Nat. Methods 16, 163 (2019).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Chen, H. et al. Dissecting mammalian spermatogenesis using spatial transcriptomics. Cell Rep. 37, 109915 (2021).
Marshall, J. L. et al. High-resolution Slide-seqV2 spatial transcriptomics enables discovery of disease-specific cell neighborhoods and pathways. Iscience 25, 104097 (2022).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Harris, C. R. et al. Array programming with NumPy. Nature 585, 357–362 (2020).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Yuan, Z. MENDER: fast and scalable tissue structure identification in spatial omics data. Zenodo https://doi.org/10.5281/zenodo.10216989 (2023).
Acknowledgements
Z.Y. acknowledges the support by National Nature Science Foundation of China (62303119), Chenguang Program of Shanghai Education Development Foundation and Shanghai Municipal Education Commission (22CGA02), Shanghai Science and Technology Development Funds (23YF1403000), Tencent AI Lab Rhino-Bird Focused Research Program (RBFR2023008), Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01), ZJ Lab, and Shanghai Center for Brain Science and Brain-Inspired Technology, and 111 Project (No. B18015).
Author information
Authors and Affiliations
Contributions
Z.Y. conceived and designed the study, developed the computational methods, performed the analysis, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Peer review
Peer review information
Nature Communications thanks Qing Nie and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yuan, Z. MENDER: fast and scalable tissue structure identification in spatial omics data. Nat Commun 15, 207 (2024). https://doi.org/10.1038/s41467-023-44367-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-44367-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
