
Abstract
Trapped ion mobility spectrometry (TIMS) adds an additional separation dimension to mass spectrometry (MS) imaging, however, the lack of fragmentation spectra (MS2) impedes confident compound annotation in spatial metabolomics. Here, we describe spatial ion mobility-scheduled exhaustive fragmentation (SIMSEF), a dataset-dependent acquisition strategy that augments TIMS-MS imaging datasets with MS2 spectra. The fragmentation experiments are systematically distributed across the sample and scheduled for multiple collision energies per precursor ion. Extendable data processing and evaluation workflows are implemented into the open source software MZmine. The workflow and annotation capabilities are demonstrated on rat brain tissue thin sections, measured by matrix-assisted laser desorption/ionisation (MALDI)-TIMS-MS, where SIMSEF enables on-tissue compound annotation through spectral library matching and rule-based lipid annotation within MZmine and maps the (un)known chemical space by molecular networking. The SIMSEF algorithm and data analysis pipelines are open source and modular to provide a community resource.
Similar content being viewed by others


Introduction
Mass spectrometry (MS) imaging gains traction in metabolic, lipidomic, and proteomic studies1 since the analyte distribution in biological tissues grants valuable insights into how a disease affects an organism. In recent years, advances in instrumentation led to higher spatial resolution2, increased specificity3, boosted sensitivity4, and increased sample throughput5,6. These characteristics are linked and influence each other, for example, higher spatial resolutions require efficient ionisation and increased sensitivity. A major bottleneck in many MS imaging studies remains the compound annotation, which often relies on accurate mass only, due to missing fragmentation data (MS2)2,7,8. Therefore, spectral library matching, manual annotation of fragmentation spectra, molecular networking9,10, and other fragment ion-based approaches, such as molecular structure prediction in the SIRIUS software, remain unavailable for these MS1-only workflows11,12,13. Generally, most public MS imaging studies lack MS2 data because of the limited availability of spatially resolved data-dependent acquisition (DDA) modes, where MS2 scans are usually scheduled based on the most abundant signals detected in MS1. While some studies have investigated MS imaging experiments with DDA, they rely on DDA methods initially designed for liquid chromatography (LC)-MS. Thus, they cannot leverage the spatial distribution for ideal MS2 coverage14,15,16,17. Nevertheless, this approach allowed more confident metabolite annotation based on low-resolution MS2 spectra acquired together with high-resolution MS1 images18. Other studies use a targeted approach with predefined isolation windows to acquire MS2-only images19, or manually set up inclusion lists and cycle MS1 and targeted MS2 experiments15,16,20. These strategies generate fragment ion information but are limited to a few targets leaving the chemical composition underexplored.
Recently, ion mobility spectrometry (IMS)-MS is gaining popularity in proteome and metabolome research21,22,23,24. The additional separation dimension based on the collision cross section (CCS)-to-charge ratio improves compound annotation and enables additional data acquisition strategies. A promising IMS technology is trapped IMS (TIMS), which traps ions in an electrostatic field and a counter gas flow25,26. The TIMS analyser consists of two separated IMS regions, one to accumulate ions and the other to separate and release an accumulated ion package. This technique has been incorporated into quadrupole time-of-flight (qTOF)-MS instruments with fast precursor isolation switching times. Synchronising the quadrupole isolation with the mobility separation enabled the development of additional DDA modes, such as parallel accumulation serial fragmentation (PASEF)27,28. Several additional PASEF modes have been described recently, such as data-independent acquisition (dia)PASEF29, parallel reaction monitoring (prm)PASEF30, midiaPASEF31, slicePASEF32, and synchroPASEF33. While these techniques provide improvements in the field of proteomics, they are designed and exclusively available in LC-IMS-MS analysis. Recently, use cases of a prototypic prm-MALDI workflow have been described to acquire multiple fragmentation spectra in a single TIMS ramp in a targeted analysis34. Thereby, fragment ion images of multiple precursor ions were acquired simultaneously. However, this approach relied on prior, manual data evaluation, precursor selection, hand-written precursor schedules, and manual data acquisition.
Comprehensive spatial metabolomics studies acquire complementary datasets by LC-MS2 and MS1 imaging, without fragmentation analysis. Here, laser capture microdissection enables sampling of regions from tissue thin sections for subsequent extraction and LC-MS analysis35,36,37. Integrative data analysis then aligns LC-MS2 features with ion images by their m/z. Furthermore, ion mobility or CCS can act as additional identifiers to align LC-IMS-MS and MALDI-IMS-MS imaging experiments by more than just m/z38. MZmine 3 is, to the best of our knowledge, the first tool to provide such an integrative data analysis workflow that results in a combined feature list with MS2 from LC-IMS-MS and spatial distributions from MS imaging39. However, this approach requires extensive sample preparation, which must be adequate to extract the metabolites of interest. Furthermore, the observed ion species (adduct) in MALDI-MS often differs from the adduct observed after electrospray ionisation, altering m/z and CCS values, effectively hampering a direct alignment.
Addressing the need for dataset-dependent MS2 acquisition in TIMS-MS imaging, we introduce workflows for spatial ion mobility-scheduled exhaustive fragmentation (SIMSEF) analysis. SIMSEF is wrapped into open source MZmine 3 modules to plan, acquire, and analyse spatially-resolved MS2 data. First, MALDI-TIMS-qTOF-MS analysis produces TIMS-MS1 images, which are analysed for subsequent, automated MS2 experiments to achieve an exhaustive precursor coverage. By tracking spatial coordinates, the module was designed to optimise the scheduling of MS2 events across a sample, considering the expected precursor purity of an isolation window, mobility separation, and spatial distribution. The final MS2-enriched datasets can be visualised and evaluated in MZmine 3, offering compound annotation and data integration with other platforms such as SIRIUS and Global Natural Products Social Molecular Networking (GNPS). The GNPS web platform enables molecular networking and integration of the results into a broader MS ecosystem. We anticipate the SIMSEF workflow will enrich (public) MS imaging data by providing confident in-depth compound annotation by high-quality MS2 spectra paired with CCS values. This study is to stimulate developments in dataset-dependent acquisition strategies for MS imaging by delivering open source solutions.
Results
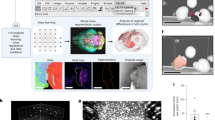
A non-target workflow for the dataset-dependent acquisition of MS2 spectra for MALDI-TIMS-MS imaging experiments was developed. The workflow is illustrated in Fig. 1. After TIMS-MS1 imaging data acquisition, the non-targeted feature detection in MZmine 3 creates a feature list, summarising all possible precursor ions in the dataset. The developed SIMSEF algorithm finds pixels for MS2 experiments (see SIMSEF—Precursor scheduling in MZmine), which are acquired using a custom Python script (see The MS2 acquisition tool and data format). After scheduling, MZmine 3 can directly launch the data acquisition on the acquisition computer. Otherwise, the schedule can be moved to the instrument computer and launched manually. Furthermore, data analysis modules allow evaluation of the acquired fragmentation experiments. Acquiring TIMS-MS1 experiments with a laser spot size smaller than the raster size preserves sample area for subsequent TIMS-MS2 experiments, in which the laser spot size is increased to the raster size, resulting in sufficient analyte ionisation and increased signal-to-noise ratios. The TIMS-MS1 imaging run is set up in flexImaging 7.2, and the imaging run geometry is saved for the subsequent TIMS-MS2 acquisition.

1, the sample is imaged in TIMS-MS1 mode. 2, non-target feature detection is applied in MZmine 3 and 3, the resulting feature list is sorted by ascending area (summed intensity) to search eligible spots for low-intensity precursor ions first. 4, every possible spot where a precursor was detected is evaluated for non-overlapping mobility isolation, spatial criteria for various collision energies, and spectral purity within the precursor m/z isolation window. The resulting precursor lists are saved to one .csv file per spot. 5, the scheduled TIMS-MS2 spectra are acquired by a Python script and a prototype instrument control software.
SIMSEF—precursor scheduling in MZmine
The MZmine 3 import for Bruker .d/tdf data was extended to import spot names and coordinates in imaging analyses. A dataset-dependent precursor scheduling algorithm was designed to ensure several quality criteria in the created MS2 spectra, including spectral and spatial considerations. Due to the ion mobility separation and quadrupole switching times in the low millisecond range, multiple precursors are scheduled within a single IMS ramp. The scheduler exploits this to queue multiple precursors in a single imaging pixel. Furthermore, the algorithm considers criteria such as minimum intensity in a pixel, expected isolation purity, distance between MS2 pixels, and ensures enough quadrupole m/z switching time. These filters aim to increase the chance for “pure” MS2 spectra, but co-isolation of isobaric and isomeric interferences remains an issue that may be flagged and analysed during downstream processing in MZmine. The SIMSEF algorithm is illustrated in Supplementary Fig. S1. The SIMSEF scheduler works with the results of the MZmine feature detection workflow, which produces mobility-resolved extracted ion images. Every feature is additionally associated with an extracted ion mobilogram and intensities in every pixel39.
Initially, the MZmine feature table is sorted from the lowest to the highest signal area, i.e., the sum of all signal intensities across the sample. Thereby, low abundant precursors are scheduled first to ensure fragmentation in their most intense pixels. This further increases the chance of acquiring enough MS2 scans for low-abundant compounds only detected in few spots throughout the image. The respective pixels for each precursor are considered from maximum to minimum intensity. Each of these TIMS-MS1 pixels is assessed individually for several quality criteria. Initially, the intensity at which a precursor was detected must exceed an intensity threshold (Minimum MS1 intensity). Then, the scheduler ensures that the mobility window of the precursor does not overlap with already scheduled precursor ions in the same pixel (see Supplementary Fig. S2a). Afterward, spatial limitations are considered to distribute MS2 scans across the sample (see Supplementary Fig. S3). Usually, non-targeted analyses have no way of optimising fragmentation energies for every precursor before the MS2 acquisition, however, this limitation is overcome by scheduling the same precursor in multiple pixels with different, user-defined collision energies. Thereby, the algorithm exploits that the number of pixels in imaging experiments is orders of magnitude higher than the number of detected features. In case multiple MS2 spectra shall be acquired per precursor, the MS2 events of the same collision energy are scheduled with a minimum distance away from each other, which is defined as the Euclidean distance by the user. However, the minimum distance is ignored for MS2 pixels of different collision energies. If a pixel matches the previous criteria, the isolation width of the quadrupole around the precursor m/z in that pixel is assessed within the mobility window of the precursor for its expected purity (Minimum purity score) (see Supplementary Fig. S2a). Once a pixel passes all quality checks, the scheduler adds the precursor to the pixel. This process is repeated until all pixels have been tested or the required number of MS2 spectra have been scheduled for the precursor. The remaining precursors are distributed by the same algorithm, but considering already scheduled MS2 pixels first. After all precursors are scheduled, the necessary information for the acquisition is exported to a project folder. The folder contains the SIMSEF schedule, a precursor list for every MS2 pixel, and collision energy information. IMS-MS imaging datasets with thousands of pixels and millions of mobility-resolved mass spectra are better processed on data processing computers with enough RAM (e.g., 64–128 GB) and multi-core processors. Therefore, the MZmine SIMSEF scheduling can be performed on another computer, and the resulting schedule can be transferred to the data acquisition computer. A graphical preview of scheduled MS2 events for every precursor simplifies the evaluation of the chosen parameters and lets the user decide if satisfactory MS2 coverage is achieved (see Supplementary Fig. S4). The source code is provided in the official MZmine GitHub (https://github.com/mzmine/mzmine3).
The MS2 acquisition tool and data format
A Python script was developed to run the acquisition list supplied by MZmine 3, utilising a vendor-supplied Python library to control the instrument. The script sets the precursor isolation width, collision energies, and mobility windows and moves the sample stage according to the coordinates and the geometry files stored during the TIMS-MS1 acquisition. The timsControl 4.1 prototype then acquires TIMS-MS2 spectra of the precursor lists, by switching the quadrupole isolation mass along the IMS ramp34. The results of one acquired TIMS ramp with 11 precursor ions are compiled in Fig. 2, depicting the extraction of TIMS-resolved precursor-specific MS2 spectra. Rule-based lipid annotation in MZmine annotated phosphatidic acid (PA) 18:0_18:1 based on lipid headgroup and fatty acyl (FA) chain fragments40,41. Here, it should be noted that it is not expected that every TIMS-MS2 pixel contains high-quality spectra for every precursor. The applied collision energy may be too high or too low for some molecular species, hence SIMSEF allows scheduling of multiple collision energies, to address this issue.

SIMSEF scheduled 11 precursor ions (magenta) for a, a TIMS-MS1 frame spectrum, here visualised as accumulated spectrum with merged mobility dimension. The corresponding acquired MS2 frame visualised as b, the total ion mobilogram showing the intensity distribution along the mobility dimension and c, the mobility-m/z heatmap of the TIMS-MS2 pixel, showing precursor m/z isolation windows (magenta) and ion mobility MS2 extraction windows for individual precursors (blue). d, an extracted TIMS-MS2 scan for the precursor m/z 701.5121 within 1.29 − 1.33 Vs/cm2 (bold blue) was annotated with the rule-based lipid annotation module and enriched by manual annotation of fragment signals. The observed headgroup fragment (m/z 152.9964) and signals for chain fragment ions (FA18:0; FA18:1) allow annotation of this precursor as PA 18:0_18:1. The applied collision energy produced high-quality fragment spectra for lipid species between m/z 400 – 850, while being too low for larger and too high for smaller molecules.
Data evaluation in MZmine
Developed MZmine 3 modules now simplify the evaluation of fragment ion spectra from SIMSEF experiments. A MALDI-MS2 grouping module links fragmentation spectra to their corresponding image features. The MS2 grouping relies on the spot names of the imaging analysis, the precursor m/z, and the mobility window in which the precursor was isolated. All three criteria must match the MS1 feature. The MS2 scans are extracted for each pixel and then optionally merged into consensus spectra with the same collision energy and across energies, taking the maximum intensity of a signal across all scans (see Fig. 2d). During the spectral merging, even low abundant spectra contribute to the final quality of the TIMS-MS2. Afterwards, existing MS2-based annotation modules in MZmine are available for compound annotation, such as spectral library matching, rule-based lipid annotation, and fragment ion-based formula prediction. Data export and direct interfaces allow integration of the results with open source tools, such as SIRIUS and the GNPS ecosystem for further compound annotation, verification, and molecular networking. Figure 3 demonstrates how MZmine facilitates inspection of raw data and annotation results in its graphical user interface (GUI). Here, the extracted image view is interactively linked to the underlying spectral raw data. Spectral mirror matches can compare two MS2 spectra either between a feature and a spectral library or between two features. In this example, rule-based lipid annotation41 and spectral matching against the GNPS library, annotated features as glutathione (a), phosphatidyl serine (PS) 18:0_18:1 (b), and PS 18:0_22:6 (c). Rule-based annotation tagged the headgroup neutral loss and both chain fragments for both lipids in accordance to the shorthand notation42. Measured CCS values of glutathione (162 Å2), PS 18:0_18:1 (286 Å2) and PS 18:0_22:6 (292 Å2) are within 3% deviation with reported literature values for other IMS analysers (glutathione: 164.24 Å2, PS 36:1: 289.4 Å2, 279.1 Å2; PS 40:6: 295.4 Å2, 284.9 Å2)43,44,45 It should be noted that the fatty acid chain composition was not specified in literature, which, including the different IMS setup, could explain the CCS discrepancies. Additional annotations for small metabolites are shown in Supplementary Fig. S5. A composition of all obtained spectral library matches is provided within the Source Data zip file merged_speclibrary_matches.pdf. Additional supporting charts are composed in features_summary.zip contained in the MassIVE dataset (see data availability). GNPS feature-based molecular networks in Fig. 4 and Supplementary Fig. S6 show clustering of compound classes by connecting features by their modified MS2 cosine similarity. Figure 4a shows all networks containing 3 or more nodes, with the largest network composed of lipids, while Fig. 4b shows a smaller sub-network of annotated and structurally related metabolites. Furthermore, an imaging specific ‘All MS/MS’ visualizer shows the extracted ion image and indicates the spots in which an MS2 spectrum was acquired. The indicated spots are colour-coded by their collision energy. The MS2 spectra are shown on the side of the visualizer, as well as the extracted ion mobilogram of the precursor ion and the mobility isolation window (see Supplementary Fig. S7).

a–c show spectral library matches to the public MoNA LC-MS/MS negative mode library. Spectral match to a reference spectrum of the metabolite glutathione (a), and the phosphatidyl serines PS 18:0_18:1 (b) and PS 18:0_22:6 (c). Multiple MS2 spectra were acquired across the tissue by MALDI-TIMS-SIMSEF. d The feature table summarises all features by their properties (e.g., m/z and CCS) and adds interactive charts linked to the underlying data structures. SMART notation for images48: Step size: 50 µm, spot size: 30 µm, identification confidence: MSI level 2, resolution: 40,000 FWHM @m/z 1,221, Time 48 min (R2c_1a), 50 min (R2c_1b), 40 min (R2c_1c), 54 min (R2c_1d).

These networks prove the gained data and annotation quality without the need to acquire additional LC-IMS-MS2 data. Nodes describe ion image features with MS2 spectra. A single representative, i.e., the most abundant MS2 spectrum, was exported for each feature. Edges connect MS2 spectra based on their modified cosine similarity (0.7–1.0) which is reflected by the edge weight. Diamond-shaped nodes are annotated by the MZmine rule-based lipid annotation module. Nodes with orange borders are annotated by spectral library matching to the MoNA, MassBank EU, MSDIAL Lipid Blast, or GNPS public libraries. Network a shows the lipid sub-network with many connections between annotated lipid species with a single fatty acyl chain substitution. Network b shows a small sub-network of annotated metabolites, such as nucleic acid diphosphates and phytic acid. The molecular networking job is available at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=9a6f6b34367f4ff69d81c9efe6aedd03.
Since SIMSEF scatters fragmentation spectra across the whole tissue instead of using only hotspot regions, the acquired MS2 information can be used to identify isomeric or isobaric compound distributions, which were not separated in IMS and m/z dimension (see Supplementary Fig. S8). Additionally, an MZmine module compares the similarity of MS2 spectra of the same collision energy for every individual feature. This can help to identify chimeric distributions, if the fragmentation pattern changes within the MS2 replicates across the tissue. In the present dataset, 68% of all MS2 pairs had an intra-row similarity of ≥0.7 (see Supplementary Fig. S9).
Performance evaluation
Table 1 shows an overview of the scheduling parameters as Purity score, Quadrupole switch time (ms), and the Minimum distance of MS2 pixels. In dataset R2c_a, 1514 features were detected with an intensity of at least 5000 in one spot, making them eligible for scheduling. Method 1 shows the experiment parameters described here (see Online Methods), while methods 2–5 were scheduled for comparison but not queued for data acquisition. In Method 1, a purity score of 0.8 was used to prohibit the scheduling of MS2 events that would lead to chimeric MS2 spectra. The quadrupole was set to a sufficient switching time of 1.65 ms. The distance between MS2 scans was set to 20 pixels, to distribute the MS2 spots across the tissue and to avoid over-scheduling in hotspot regions. Methods 2–4 altered one of these parameters, while method 5 altered all of them, as indicated by the bold cells in Table 1. Method 1 schedules 1396 of the 1514 precursors in 6477 MS2 pixels with a total of 28,916 MS2 events. A spectra coverage (\({SC}\)) parameter was calculated from the sum of all scheduled MS2 scans per precursor (\({N}_{{{{{{\rm{scheduled}}}}}},{{{{{\rm{p}}}}}}}\)) and divided by the theoretical maximum number of scheduled MS2 scans (Number of precursors (\({N}_{p}\)) multiplied by the number of spectra (\({N}_{{{{{{\rm{spectra}}}}}}}\)) and collision energies (\({N}_{{CE}}\))) (see formula 1).
The spectra coverage describes the ratio of scheduled TIMS-MS2 events which pass all quality criteria to the theoretically possible MS2 events. For the rat brain dataset R2c_1a, the \({SC}\) was 69.0% with an average of 4.5 precursors per spot. Reducing the quadrupole switch time from 1.65 ms to 1.00 ms slightly reduces the number of required MS2 pixels and increases the number of MS2 events, since more precursors are scheduled per spot. However, this can produce chimeric spectra, due to too low switch times which lead to multiple precursors in a single isolation. Reducing the minimum distance of MS2 pixels to 5 pixels also increases the number of MS2 events, as seen in Method 3. Method 4 decreases the purity score from 0.8 to 0.6, which increases the spectra coverage to 74.4%, while also increasing the number of scheduled precursors, indicating that spectral purity is an influential factor for SIMSEF scheduling. Method 5 combines the previous changes and shows an even higher coverage while requiring fewer spots and increasing the average number of precursors per spot further. Although method 5 would produce more spectra and lead to a higher coverage, the MS2 spectra would be of lower purity, distributed closer together, and contain chimeric isolations. Therefore, method 1 was selected for acquisition.
In sample R2c_1a, 28,916 MS2 events were scheduled resulting in 23,444 non-empty scans, i.e., scans with at least one signal above the electronic noise level. Rating the spectral information content, 17,887 (76%) scans contained at least 4 signals and a base peak intensity of at least 1000 (10x noise level). 12,540 (53%) of the TIMS-MS2 spectra also had a base peak-to-total ion count ratio of below 0.5, meaning that multiple signals contribute to the overall spectral intensity. This may indicate appropriate quality for metabolite annotation. 83% of features with non-empty MS2 scans had at least one MS2 spectrum that matched all criteria. A summary for all replicates is shown in Supplementary Table S2. Briefly, the quality of the acquired MS2 spectra is reproduced across all datasets. The TIC distribution of all MS2 spectra is shown in Supplementary Fig. S10. A combination of rule-based lipid annotation and spectral library matching to the MassBank EU, Lipid BLAST negative ion mode, MoNA, and GNPS reference libraries annotated 208 out of 1514 features in sample R2c_1a (14%). Replicate measurements showed similar annotation rates of 13–15%. Supplementary Fig. S11 shows an exemplary spectrum of a feature annotated as LPE 16:1 by rule-based lipid annotation. The TIMS-MS1 feature was detected with a maximum intensity of just 9500. Still, the MS2 spectra acquired by SIMSEF allowed the annotation of the FA 16:1 fragment ion. This information lacks in the TIMS-MS1-only imaging data.
Discussion
We described SIMSEF, a strategy for the dataset-dependent acquisition of MS2 spectra in IMS-MS imaging analysis. SIMSEF schedules fragmentation events across an already acquired sample in TIMS-MS1. This workflow maximises the MS2 coverage while exploiting the benefits of TIMS to schedule multiple, mobility separated precursor ions in a single image pixel. Further, the MS2 locations are selected by considering the precursor intensity, spatial distance, and expected isolation purity, to achieve optimal spectral quality and spectral repetition from various image locations. The SIMSEF algorithm was embedded into the open source software MZmine to provide a straightforward and accessible workflow. The acquisition of the scheduled experiments is executed by a Python script, controlling a timsControl 4.1 prototype. Post-acquisition data analysis workflows were implemented in MZmine, assigning the MS2 spectra to their TIMS-MS1 features and allowing a subsequent compound annotation by spectral library matching, rule-based lipid annotation, and the export to other community tools, such as SIRIUS and the GNPS ecosystem. The gained MS2 depth enables molecular networking to map the (un)known chemical space and propagate knowledge throughout the spectral networks. Thereby, the SIMSEF workflow in MZmine allows more confident compound annotation for MS imaging analysis, without requiring acquisition of multimodal datasets by LC-IMS-MS and MALDI-IMS-MS. This greatly reduces the experimental complexity of spatial metabolomics studies. The whole SIMSEF workflow is described in the MZmine documentation, including a step-by-step guide (https://mzmine.github.io/mzmine_documentation/workflows/simsef/simsef_workflow.html). SIMSEF improves the MS2 coverage and data quality in TIMS-MS imaging, but technical challenges remain in resolving interfering ions that may produce chimeric mass spectra.
Methods
Ethical statement
Rat (rattus norvegicus domestica, Wistar strain, female, 58 ± 3 days) samples in this study originated from an animal study (ZH214/16+) approved by the Swiss Federal Veterinary Office Zurich. The study was performed according to the Swiss Animal Welfare Act (TSchG, 2005) and Swiss Animal Welfare Ordinance (TSchV, 2008). Samples were collected for postmortem analysis.
Materials
Methanol (LC-MS grade) was obtained from VWR International GmbH. N-(1-naphthyl) ethylenediamine dihydrochloride (NEDC) (>99%) was obtained from Carl Roth GmbH + Co. KG. Water was purified by a Milli-Q Academic system (18.2 MΩ cm; 0.2 μm filter; Millipore).
Sample preparation
Rat brain tissue was cut into sections stored at –80 °C. Parasagittal sections of rat cerebellum were prepared using a CryoStar NX70 (Epredia) in 10 µm thickness and placed on indium tin oxide coated glass slides (70–100 Ω/sq, Sigma Aldrich). Bright field microscopic images were acquired on a BIOREVO BZ-9000 digital microscope (Keyence). A parallel cut of parasagittal cerebellar section was H&E stained (see Supplementary Fig. S12).
NEDC (7 mg/mL) was dissolved in methanol/water (7:3 v/v)46. The MALDI matrix was applied by a HTX TM-sprayer (HTX Technologies) with the following settings: 10 psi nitrogen pressure, 40 mm nozzle height, 0.120 mL/min flow rate, 75 °C spray temperature, in a ‘CC pattern’, with 1200 mm/min z-arm velocity, 10 passes, 3 mm track spacing and 0 s drying time.
SIMSEF workflow
The workflow comprises multiple steps that are described in the next sections. All steps for the MS1 and MS2 data acquisition are outlined in Supplementary Note 1. Parameters for the SIMSEF scheduling algorithm in MZmine are described in Supplementary Note 2. The general step order is:
-
1.
TIMS-MS1 image acquisition
-
2.
Data analysis and SIMSEF scheduling in MZmine
-
3.
SIMSEF-MS2 data acquisition
-
4.
Merging of TIMS-MS1 and MS2 data in MZmine
-
5.
Data analysis and compound annotation
TIMS-MS1 image acquisition
MALDI-IMS-MS negative ion mode data were acquired on a timsTOF fleX (Bruker Daltonics GmbH & Co. KG) using timsControl 4.1 and flexImaging 7.2. The imaging run was exported from flexImaging 7.2 via “Save run file as…” to make the geometry files available for MS2 measurements.
R2c_1a-d: Imaging data were acquired with a laser field size of 30 µm. The imaging raster size was 50 µm to keep material for subsequent MS2 acquisition (see Supplementary Fig. S13a). Resulting in a final image resolution of 50 µm. The laser frequency was 10,000 Hz with 100 shots and 1 burst per pixel. The laser parameters were set to ‘Custom’ (Application), a 0% (Power Boost), single (Smart Beam), enabled (Beam Scan), and 26 µm (Scan Range). The mass range was set to 100–1500 m/z, the 1/K0 range was set to 0.65–1.75 Vs/cm² and a 150 ms ramp time. Tune parameters were set to 50 V (MALDI plate offset), −70 V (Deflection delta), 500 Vpp (Funnel 1 RF), 0.0 eV (isCID), 350 Vpp (Funnel 2 RF), 350 Vpp (Multipole Vpp), 10 eV (Collision energy), 1100 Vpp (Collision cell RF), 5 eV (Ion energy), 100 m/z (Low mass), 80 µs (Transfer time), 5 µs (Pre pulse storage). TIMS parameters were set to 20 V (Δt1), 120 V (Δt2), −70 V (Δt3), −100 V (Δt4), 0 V (Δt5), −100 V (Δt6), −220 V (Collision cell in). After MS1 acquisition, data analysis was performed in MZmine 3.
TIMS-MS1 data analysis
An MZmine 3.8 batch configuration is shared through the MassIVE data repository (see Data availability). Briefly, each MS data file (.d folder) was imported to MZmine using the ‘Import MS data’ module with the advanced import enabled to perform mass detection directly for decreased analysis times. Afterwards, ion images were built for every m/z, and subsequently expanded into ion mobility dimension. The extracted ion mobilograms were resolved to features and the resulting feature list was filtered to retain features with a maximum intensity of 5000 or more. The results were used as the input for the scheduling of MS2 data acquisition using the ‘timsTOF SIMSEF imaging scheduler’ module.
SIMSEF scheduling in MZmine
The MS2 schedule was created with a 0.02–0.04 Vs/cm² precursor mobility window, 1.7 Da precursor isolation width, 5 MS2 spectra for every collision energy, 6 collision energies (20 eV, 30 eV, 40 eV, 50 eV, 60 eV, 70 eV), a minimum distance of 20 pixels between MS2 spectra of the same energy, a minimum absolute intensity of 3000 a.u. or 20%, and a minimum purity score of 0.8 in the respective TIMS-MS1 pixel. An MZmine 3.8 batch configuration is shared through the MassIVE data repository (see Data availability).
SIMSEF MS2 acquisition
The dataset was acquired using a prototypic version of timsControl 4.1. The scan mode was set to MS/MS and the laser spot size was increased to 50 µm for MS2 acquisitions. Collision RF (800 Vpp) and Transfer time (55 µs) were adapted for ions of lower m/z ions. All other instrument parameters remained unaltered.
SIMSEF post-analysis in MZmine
An MZmine 3.8 batch configuration is shared through the MassIVE data repository (see Data availability). Briefly, the data were imported and filtered for noise. Image features were grouped with their MS2 scans by the ‘Assign MALDI MS2 to features’ module. Features were annotated by spectral library matching and rule-based lipid annotation within MZmine. MS1 and MS2 mass tolerances for precursor and fragment ion matching were set to 5 ppm or 0.005 Da and 15 ppm and 0.005 Da, respectively. Afterwards, the processed feature table was exported to execute molecular networking on the GNPS web platform.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry imaging raw data generated in this study have been deposited in the MassIVE database under accession code MSV000092935. This dataset also contains the batch configuration and supporting charts in features_summary.zip. Within the Source Data zip file, merged_speclibrary_matches.pdf composes all spectral library match mirror plots. GNPS FBMN molecular networks and annotations are available at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=9a6f6b34367f4ff69d81c9efe6aedd03. Source data are provided with this paper.
Code availability
MZmine modules are available on the official MZmine GitHub (https://github.com/mzmine/mzmine3) under the MIT open source licence. Documentation and a step-by-step guide are available in the official MZmine documentation (https://mzmine.github.io/mzmine_documentation/module_docs/tools_simsef/simsef.html, https://mzmine.github.io/mzmine_documentation/workflows/simsef/simsef_workflow.html).
The source code for the MS2 acquisition tool is available on Zenodo (DOI: 10.5281/zenodo.8009939) and GitHub (https://github.com/SteffenHeu/simsef_py)47. Working binaries are available on GitHub (https://github.com/SteffenHeu/simsef_py/releases). Bruker bindings were removed in the source code. To obtain the timsControl 4.1 prototype, researchers may contact their Bruker Daltonics representative.
References
Taylor, M. J., Lukowski, J. K. & Anderton, C. R. Spatially resolved mass spectrometry at the single cell: recent innovations in proteomics and metabolomics. J. Am. Soc. Mass Spectrom. 32, 872–894 (2021).
Niehaus, M., Soltwisch, J., Belov, M. E. & Dreisewerd, K. Transmission-mode MALDI-2 mass spectrometry imaging of cells and tissues at subcellular resolution. Nat. Methods 16, 925–931 (2019).
Soltwisch, J. et al. MALDI-2 on a trapped ion mobility quadrupole time-of-flight instrument for rapid mass spectrometry imaging and ion mobility separation of complex lipid profiles. Anal. Chem. 92, 8697–8703 (2020).
Müller, M. A., Kompauer, M., Strupat, K., Heiles, S. & Spengler, B. Implementation of a high-repetition-rate laser in an AP-SMALDI MSI system for enhanced measurement performance. J. Am. Soc. Mass Spectrom. 32, 465–472 (2021).
Körber, A., Keelor, J. D., Claes, B. S. R., Heeren, R. M. A. & Anthony, I. G. M. Fast mass microscopy: mass spectrometry imaging of a gigapixel image in 34 min. Anal. Chem. 94, 14652–14658 (2022).
Bednařík, A. et al. MALDI MS imaging at acquisition rates exceeding 100 pixels per second. J. Am. Soc. Mass Spectrom. 30, 289–298 (2019).
Miki, A. et al. MALDI-TOF and MALDI-FTICR imaging mass spectrometry of methamphetamine incorporated into hair. J. Mass Spectrom. 46, 411–416 (2011).
Sommella, E. et al. MALDI mass spectrometry imaging highlights specific metabolome and lipidome profiles in salivary gland tumor tissues. Metabolites 12, 530 (2022).
Wang, M. et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 34, 828–837 (2016).
Nothias, L.-F. et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 17, 905–908 (2020).
Dührkop, K. et al. Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra. Nat. Biotechnol. 39, 462–471 (2021).
Ludwig, M. et al. Database-independent molecular formula annotation using Gibbs sampling through ZODIAC. Nat. Mach. Intell. 2, 629–641 (2020).
Dührkop, K. et al. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 16, 299–302 (2019).
Landgraf, R. R., Prieto Conaway, M. C., Garrett, T. J., Stacpoole, P. W. & Yost, R. A. Imaging of lipids in spinal cord using intermediate pressure matrix-assisted laser desorption-linear ion trap/Orbitrap MS. Anal. Chem. 81, 8488–8495 (2009).
Perdian, D. C. & Lee, Y. J. Imaging MS methodology for more chemical information in less data acquisition time utilizing a hybrid linear ion trap-orbitrap mass spectrometer. Anal. Chem. 82, 9393–9400 (2010).
OuYang, C., Chen, B. & Li, L. High throughput in situ DDA analysis of neuropeptides by coupling novel multiplex mass spectrometric imaging (MSI) with gas-phase fractionation. J. Am. Soc. Mass Spectrom. 26, 1992–2001 (2015).
Hansen, R. L. & Lee, Y. J. Overlapping MALDI-mass spectrometry imaging for in-parallel MS and MS/MS data acquisition without sacrificing spatial resolution. J. Am. Soc. Mass Spectrom. 28, 1910–1918 (2017).
Ellis, S. R. et al. Automated, parallel mass spectrometry imaging and structural identification of lipids. Nat. Methods 15, 515–518 (2018).
Trim, P. J. et al. Matrix-assisted laser desorption/ionization-ion mobility separation-mass spectrometry imaging of vinblastine in whole body tissue sections. Anal. Chem. 80, 8628–8634 (2008).
Lanekoff, I. et al. High-speed tandem mass spectrometric in situ imaging by nanospray desorption electrospray ionization mass spectrometry. Anal. Chem. 85, 9596–9603 (2013).
Skowronek, P. et al. Rapid and in-depth coverage of the (phospho-)proteome with deep libraries and optimal window design for dia-PASEF. Mol. Cell. Proteom. 21, 100279 (2022).
Meier, F., Park, M. A. & Mann, M. Trapped ion mobility spectrometry and parallel accumulation-serial fragmentation in proteomics. Mol. Cell. Proteom. 20, 100138 (2021).
Helmer, P. O., Behrens, A., Rudt, E., Karst, U. & Hayen, H. Hydroperoxylated vs dihydroxylated lipids: differentiation of isomeric cardiolipin oxidation products by multidimensional separation techniques. Anal. Chem. 92, 12010–12016 (2020).
Drakopoulou, S. K., Damalas, D. E., Baessmann, C. & Thomaidis, N. S. Trapped ion mobility incorporated in LC-HRMS workflows as an integral analytical platform of high sensitivity: targeted and untargeted 4D-metabolomics in extra virgin olive oil. J. Agric. Food Chem. 69, 15728–15737 (2021).
Fernandez-Lima, F. A., Kaplan, D. A. & Park, M. A. Note: integration of trapped ion mobility spectrometry with mass spectrometry. Rev. Sci. Instrum. 82, 126106 (2011).
Fernandez-Lima, F., Kaplan, D. A., Suetering, J. & Park, M. A. Gas-phase separation using a trapped ion mobility spectrometer. Int. J. Ion Mobil. Spectrom. 14, 93–98 (2011).
Meier, F. et al. Online parallel accumulation–serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer *. Mol. Cell. Proteom. 17, 2534–2545 (2018).
Meier, F. et al. Parallel accumulation-serial fragmentation (PASEF): multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. J. Proteome Res. 14, 5378–5387 (2015).
Meier, F. et al. diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods 17, 1229–1236 (2020).
Lesur, A. et al. Highly multiplexed targeted proteomics acquisition on a TIMS-QTOF. Anal. Chem. 93, 1383–1392 (2021).
Distler, U. et al. midiaPASEF maximizes information content in data-independent acquisition proteomics. bioRxiv, https://doi.org/10.1101/2023.01.30.526204 (2023).
Szyrwiel, L., Sinn, L., Ralser, M. & Demichev, V. Slice-PASEF: fragmenting all ions for maximum sensitivity in proteomics. bioRxiv, https://doi.org/10.1101/2022.10.31.514544 (2022).
Skowronek, P. et al. Synchro-PASEF allows precursor-specific fragment ion extraction and interference removal in data-independent acquisition. Mol. Cell. Proteom. 22, 100489 (2022).
Wolf, C. et al. Mobility-resolved broadband dissociation and parallel reaction monitoring for laser desorption/ionization-mass spectrometry—Tattoo pigment identification supported by trapped ion mobility spectrometry. Anal. Chim. Acta. 1242, 340796 (2023).
Eiersbrock, F. B., Orthen, J. M. & Soltwisch, J. Validation of MALDI-MS imaging data of selected membrane lipids in murine brain with and without laser postionization by quantitative nano-HPLC-MS using laser microdissection. Anal. Bioanal. Chem. 412, 6875–6886 (2020).
Burnum, K. E. et al. Spatial and temporal alterations of phospholipids determined by mass spectrometry during mouse embryo implantation. J. Lipid Res. 50, 2290–2298 (2009).
Hankin, J. A. & Murphy, R. C. Relationship between MALDI IMS intensity and measured quantity of selected phospholipids in rat brain sections. Anal. Chem. 82, 8476–8484 (2010).
Helmer, P. O. et al. Complementing matrix-assisted laser desorption ionization-mass spectrometry imaging with chromatography data for improved assignment of isobaric and isomeric phospholipids utilizing trapped ion mobility-mass spectrometry. Anal. Chem. 93, 2135–2143 (2021).
Schmid, R. et al. Integrative analysis of multimodal mass spectrometry data in MZmine 3. Nat. Biotechnol. 41, 447–449 (2023).
Pulfer, M. & Murphy, R. C. Electrospray mass spectrometry of phospholipids. Mass Spectrom. Rev. 22, 332–364 (2003).
Korf, A., Jeck, V., Schmid, R., Helmer, P. O. & Hayen, H. Lipid species annotation at double bond position level with custom databases by extension of the MZmine 2 open-source software package. Anal. Chem. 91, 5098–5105 (2019).
Liebisch, G. et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res. 61, 1539–1555 (2020).
Groessl, M., Graf, S. & Knochenmuss, R. High resolution ion mobility-mass spectrometry for separation and identification of isomeric lipids. Analyst 140, 6904–6911 (2015).
Zhou, Z., Tu, J., Xiong, X., Shen, X. & Zhu, Z.-J. LipidCCS: prediction of collision cross-section values for lipids with high precision to support ion mobility-mass spectrometry-based lipidomics. Anal. Chem. 89, 9559–9566 (2017).
Zheng, X. et al. A structural examination and collision cross section database for over 500 metabolites and xenobiotics using drift tube ion mobility spectrometry. Chem. Sci. 8, 7724–7736 (2017).
Wang, J. et al. MALDI-TOF MS imaging of metabolites with a N-(1-naphthyl) ethylenediamine dihydrochloride matrix and its application to colorectal cancer liver metastasis. Anal. Chem. 87, 422–430 (2015).
Heuckeroth, S. SteffenHeu/simsef_py: SIMSEF v1.0.0. Zenodo https://doi.org/10.5281/ZENODO.8009939 (2023).
Xi, Y. et al. SMART: a data reporting standard for mass spectrometry imaging. J. Mass Spectrom. 58, e4904 (2023).
Acknowledgements
K.K. and U.K. were supported by the Deutsche Forschungsgemeinschaft (DFG) – CRC 1450 – 431460824. R.S. was supported by the Czech Science Foundation (GA CR) grant 21-11563 M. C.B. was supported by the Czech Academy of Sciences PPLZ fellowship number L200552251.
Author information
Authors and Affiliations
Contributions
S.H. conceptualised and implemented the scheduling algorithm, the instrument control script, data evaluation tools in MZmine 3, prepared and acquired MALDI measurements. S.H., A.B. conceptualised the imaging MS2 application. A.B., K.K. prepared thin sections of rat brain tissue. I.D.N., K.K., C.W., C.B. provided test data during development and procedures for sample preparation. A.F. supported S.H. during the Python control script development. R.S. reviewed code, provided conceptual support. A.J., H.R. provided sheep brain tissue during method optimisation. A.J., H.R. provided the rat brain samples. R.S., U.K. supervised S.H. S.H., R.S., wrote the initial manuscript, all authors revised the final manuscript.
Corresponding author
Ethics declarations
Competing interests
A.B., A.F., I.D.N., A.K. are currently employed at Bruker Daltonics GmbH & Co. KG. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Carsten Hopf, Laura Sanchez and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heuckeroth, S., Behrens, A., Wolf, C. et al. On-tissue dataset-dependent MALDI-TIMS-MS2 bioimaging. Nat Commun 14, 7495 (2023). https://doi.org/10.1038/s41467-023-43298-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-43298-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
