
Abstract
It is generally assumed that all bacteria must have at least one rRNA operon (rrn operon) on the chromosome, but some strains of the genera Aureimonas and Oecophyllibacter carry their sole rrn operon on a plasmid. However, other related strains and species have chromosomal rrn loci, suggesting that the exclusive presence of rrn operons on a plasmid is rare and unlikely to be stably maintained over long evolutionary periods. Here, we report the results of a systematic search for additional bacteria without chromosomal rrn operons. We find that at least four bacterial clades in the phyla Bacteroidota, Spirochaetota, and Pseudomonadota (Proteobacteria) lost chromosomal rrn operons independently. Remarkably, Persicobacteraceae have apparently maintained this peculiar genome organization for hundreds of millions of years. In our study, all the rrn-carrying plasmids in bacteria lacking chromosomal rrn loci possess replication initiator genes of the Rep_3 family. Furthermore, the lack of chromosomal rrn operons is associated with differences in copy numbers of rrn operons, plasmids, and chromosomal tRNA genes. Thus, our findings indicate that the absence of rrn loci in bacterial chromosomes can be stably maintained over long evolutionary periods.
Similar content being viewed by others
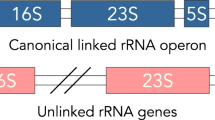


Introduction
Bacterial chromosomes are traditionally distinguished from plasmids by the fact that chromosomes encode essential genes1,2. The canonical essential genes include 16S, 23S, and 5S rRNA genes, which usually constitute an rRNA operon (rrn operon). All bacteria had been believed to have at least one rrn operon on their chromosomes, even if additional rrn operons could be found on extrachromosomal replicons3,4,5,6,7,8. Theoretical and experimental studies suggest that a population consisting of individuals with sole rrn operon only on the plasmid are unstable9 and a second copy of essential genes cannot be maintained stably on plasmids10, respectively.
This widespread belief was challenged by the unexpected finding of a plant-associated bacterium Aureimonas ureilytica (family Aurantimonadaceae, order Rhizobiales, class Alphaproteobacteria), which carries its sole rrn operon on a high copy number plasmid without partition system genes11. This finding attracted considerable attention in terms of the diversity and evolution of bacterial genomes and plasmids12,13,14,15,16,17. However, whether A. ureilytica is a unique, exceptional species and if there are essential prerequisites that allow rrn operons to transfer from chromosomes to plasmids have not been resolved. In addition, because other Aureimonas species have been revealed to have chromosomal rrn operons, how long bacteria without chromosomal rrn operons can avoid extinction over evolutionary timescales remains unclear. Regarding the uniqueness of A. ureilytica, a recent study incidentally found that an ant-associated bacteria Oecophyllibacter saccharovorans (family Acetobacteraceae, order Rhodospirillales, class Alphaproteobacteria) carries its sole rrn operon on a plasmid18. We thus envisioned a systematic study to identify more bacterial species without chromosomal rrn operons to provide clues to those fundamental questions.
In this study, by combining bioinformatic analysis and genome sequencing, we investigated if bacteria without chromosomal rrn operons evolved repeatedly and could be maintained for an evolutionarily long term. We found that bacteria without chromosomal rrn operons independently evolved at least four times in three phyla. Most notably, the family Persicobacteraceae was revealed to have lost its chromosomal rrn operons likely >492 million years ago (MYA), demonstrating that bacteria without chromosomal rrn operons can avoid extinction over geological timescales.
Results
Bioinformatic analysis found multiple bacteria that likely lost chromosomal rrn operons
To search for bacterial genomes whose rrn operons reside exclusively on plasmids, we downloaded 86,822 genomes from NCBI RefSeq19. This dataset contained numerous (90.7%) draft genomes, whose contigs were classified as neither chromosomes nor plasmids. One signature of plasmid contigs is the presence of a rep gene, which encodes the plasmid replication initiator Rep protein, as in the case of rrn plasmids of A. ureilytica11. Thus, we selected genomes whose annotated rrn operons were exclusively on contigs that encoded rep genes, using tBLASTn searches with 156 Rep protein sequences20. To enrich genomes that have rrn operons only on plasmids, we excluded genomes if one of their rrn operons was on a contig longer than 35 kb or encoded essential single-copy genes (Supplementary Fig. 1). We confirmed that our method made neither false-positive nor false-negative using the A. ureilytica genomes11,21 and RefSeq complete genomes. Aside from the A. ureilytica genomes, our analysis identified three additional genomes that potentially have rrn operons exclusively on plasmids with rep genes: genomes of Persicobacter spp. JZB09 and CCB-QB2 (family Persicobacteraceae; order Cytophagales, class Cytophagia, phylum Bacteroidota), whose 16S rRNA genes were 99% identical to those of Persicobacter diffluens; and Treponema saccharophilum DSM2985 (family Spirochaetaceae; order Spirochaetales, class Spirochaetia, phylum Spirochaetota).
Persicobacter spp. JZB09 and CCB-QB2 genomes (GCF_001308105.1 and GCF_001274635.1) were sequenced using PacBio RSII, and their assembly statuses were complete and scaffold, respectively22. Both genomes contained three rrn operons, which were arranged in tandem on a single ~30-kb contig with rep genes (Fig. 1a). Those Persicobacter genomes were of particular interest because genomes of related species in the same Persicobacteraceae family have not been sequenced yet, and thus the loss of chromosomal rrn operons might be widely conserved across related species.

a–d Maps of the rrn plasmids of a Persicobacteraceae species, b Treponema saccharophilum, c Aureimonas ureilytica11, and d Oecophyllibacter saccharovorans18. Solid and dotted inner arcs show rrn operon with or without tRNA genes, respectively. rRNA and tRNA genes are shown by black arrows. rep, parAB, bioABFCD, and sig genes encode plasmid replication initiators, partitioning proteins, biotin synthetic proteins, and sigma factors, respectively. Genes whose products belong the same function are presented in the same colors. Note that repA1A2A3 of Persicobacter diffluens and repA1’A2’ of Persicobacter psychrovividus have no sequence similarity. e Comparative synteny map of rrn plasmids of the Persicobacteraceae species. f, g Phylogenetic trees of rRNA genes and conserved single-copy protein-coding genes of f Persicobacteraceae species and g T. saccharophilum and their related species. Pink indicates bacteria without chromosomal rrn operons. Light blue indicates bacteria with chromosomal rrn operons. Numbers in parentheses are numbers of chromosomal rrn operons. Asterisks indicate genomes whose assembly levels are scaffold. Bootstrap values of >50% are shown. Scale bars indicate substitution numbers per site. See Supplementary Data 1–3 for strain names, conserved single-copy protein-coding genes, and results of topological test, respectively.
The T. saccharophilum genome (GCF_000255555.1) was sequenced by Genome Analyzer II and 454 GS FLX. Its rRNA genes were found only at both ends of a single 8.4-kb contig, suggesting that its rrn operon is present only on this circular plasmid. Other Treponema genomes whose assembly statuses were complete or chromosome had chromosomal rrn operons. Thus, T. saccharophilum would have recently lost its chromosomal rrn operons around the time of speciation.
De novo genome sequencing revealed that bacteria lost chromosomal rrn operons at least four times independently
To confirm that the genomes of P. diffluens and T. saccharophilum genuinely have no rrn operons in their chromosomes, we obtained their type strains (P. diffluens NBRC 15940T and T. saccharophilum JCM 32279T) and experimentally determined their genomes through hybrid assembly using PacBio RSII and HiSeq sequencers (Table 1 and Supplementary Table 1). Neither of those genomes had rrn operons in their chromosomes. Like the two complete/scaffold Persicobacter genomes, the genome of P. diffluens was confirmed to have three rrn operons that are arranged in tandem on a single ~30-kb plasmid harboring rep genes (Fig. 1a). In addition, the T. saccharophilum genome was confirmed to have an rrn operon on a single 8.4-kb plasmid with a rep gene (Fig. 1b). Therefore, we concluded that P. diffluens and T. saccharophilum are species that lost rrn operons from their chromosomes. In other words, the bacterial domain lost chromosomal rrn operons at least four times independently in the Bacteroidota, Spirochaetota, and Pseudomonadota phyla (i.e., A. ureilytica (Fig. 1c) and O. saccharovorans (Fig. 1d)).
Next, we determined genomes of species related to P. diffluens in family Persicobacteraceae: P. pyschrovividus, Aureibacter tunicatorum, and Fulvitalea axinellae (Table 1 and Supplementary Table 1). Their complete genomes revealed that they do not have rrn operons in their chromosomes but have two rrn operons in tandem on their smallest plasmid (13.7–25.9 kb) (Fig. 1a). Moreover, the rrn operons of all the six Persicobacteraceae genomes shared a common structure of rrs (16S rRNA)-trnl (tRNAIle)-trna (tRNAAla)-rrl (23S rRNA)-rrf (5S rRNA) except for one operon on the Persicobacter genomes, strongly suggesting that they share a single evolutionary origin (Fig. 1a, e). This is the first discovery that rrn operons are absent from bacterial chromosomes at the taxonomic family level.
Persicobacteraceae lost their chromosomal rrn operons hundreds of millions of years ago
The plasmid rrn operons of the Persicobacteraceae species and T. saccharophilum were probably transferred from their chromosomal rrn operons or through horizontal gene transfers from distant species. To determine the evolutionary origins of their rrn operons, we reconstructed and compared the rRNA gene trees and genomic trees based on single-copy protein-coding genes (Fig. 1f, g and Supplementary Data 1, 2). In both cases, the two trees exhibited topological similarities (Supplementary Data 3), suggesting that their plasmid rrn operons were transferred from their chromosomes and not horizontally transferred from distant clades. The plasmid rrn operons of A. ureilytica and O. saccharovorans were also transferred from the chromosomes11,23.
The chromosomal origins of the plasmid rrn operons allow us to estimate the timings of the transfer of their rrn operons to plasmids and their loss from chromosomes using genomic data. The genomic phylogenomic trees of the four clades were obtained from GTDB or inferred using GTDB-Tk24,25, and their divergence times were estimated using RelTime26 (Fig. 2a–d, Supplementary Fig. 2, Supplementary Data 4). The chromosomal rrn operons were estimated to have been lost 492-651 MYA, 0-221 MYA, 69.3-166 MYA, and 1.27-91.2 MYA in the ancestors of Persicobacteraceae species, T. saccharophilum, A. ureilytica, and O. saccharovorans, respectively, under the assumption that the chromosomal rrn operons were lost at the common ancestor of each clade (see Discussion). The outstanding ancient origin of the Persicobacteraceae ancestor was particularly notable for two reasons: First, it challenges the widely accepted belief that essential genes cannot be maintained stably on plasmids for an evolutionarily long term9. Second, comparing bacteria without chromosomal rrn operons between the Persicobacteraceae family and the other three clades will enable us to investigate how rrn operons transferred from chromosomes to plasmids and subsequently plasmids matured in the long term.

Genomic phylogenetic trees and estimated divergence times of a Persicobacteraceae species, b T. saccharophilum, c A. ureilytica, and d O. saccharovorans. Pink indicates bacteria without chromosomal rrn operons. Gray bars show confidence intervals of RelTime estimates which contain the actual time with 94% probability90. Purple branches are estimated timing of gain of rrn plasmids and loss of chromosomal rrn operons. Species and genus names follow GTDB taxonomy, but names of bacteria without chromosomal rrn operons follow IJSEM for consistency with the main text. Phylogenetic trees including calibration points are shown in Supplementary Fig. 2. e Numbers of gene gain and loss events in ancestors that lost chromosomal rrn genes in the four clades. f Numbers of Rep_3-family genes in genomes of related species of bacteria without chromosomal rrn operons. g Numbers of plasmids (or extrachromosomal contigs) that carry Rep_3-family with or without rrn operons in bacteria without chromosomal rrn operons. Pink indicates plasmids (or extrachromosomal contigs) carrying Rep_3 in bacteria without chromosomal rrn operons.
rrn plasmids of bacteria without chromosomal rrn operons always had Rep_3-family genes
As mentioned above, we used plasmid replication initiator rep genes as a signature of plasmids. The 156 representative Rep proteins20 contained 17 families in Pfam27 whose primary function is plasmid replication (Supplementary Data 5).
We found that all rrn plasmids of the four bacterial clades had Rep_3-family genes (Fig. 1a–d). Furthermore, we inferred all gene gain (birth, horizontal gene transfer, or duplication) and loss events in the ancestors that lost chromosomal rrn operons using PastML28. This analysis showed that no genes other than Rep_3-family genes were consistently gained or lost in the ancestors of the four clades (Fig. 2e). Evolutionary analysis using CAFE29 did not identify additional gene-number expansion and contraction events common to those four ancestors. These lines of evidence strongly suggested that Rep_3-type plasmids represent essential prerequisites behind the losses of chromosomal rrn operons.
Phylogenetic analysis of the Rep_3-family genes estimated that those on several rrn plasmids were horizontally transferred from distant clades (such as different classes) to their ancestors (Supplementary Fig. 3). We also noted that related species of the four clades were not frequent hosts of Rep_3-family genes (Fisher’s exact test p < 0.05, Fig. 2f). Thus, we assume that transfer of rrn operons from chromosomes to plasmids would have enabled those ancestors to maintain Rep_3-type plasmids, possibly because of the essentiality of the rrn operons. Notably, in the Persicobacteraceae, we found cases that Rep_3 transferred between the rrn plasmid and other plasmids (open stars in Supplementary Fig. 3a, c, e, Fig. 1e). Moreover, the Rep_3 family was observed in all plasmids mainly as clades (Fig. 2g, diamond symbols in Supplementary Fig. 3a–d), while no other Rep families were found in plasmids (Fig. 2g). These results suggest that the Rep_3-family genes were intracellularly transferred and expanded among plasmids so that replication initiator genes of all plasmids became Rep_3-family genes (Fig. 2g).
Rep_3-type plasmids significantly and specifically acquire rrn operons
Given that bacteria without chromosomal rrn operons always carried Rep_3-family genes on their rrn plasmids in our dataset, we hypothesized that Rep_3-family plasmids would have a propensity to carry rRNA genes. We examined the localization of rrn operons and Rep-family genes on plasmid contigs obtained from AnnoTree (GTDB r89)30. We found that rrn operons occur significantly more frequently on plasmids with Rep_3-family genes than on those with other rep genes (p = 0.002, Chi-square test) (Fig. 3a). Therefore, we argue that Rep_3-family genes have special characteristics that allow them to acquire and maintain rrn operons on a replicon.

a Proportions of rep-family genes that are encoded on plasmids with (right) and without (left) rRNA genes. Numbers at the top of the panel show the total numbers. b Proportions of rep-family genes that are encoded on plasmids with (right) and without (left) conserved single-copy genes. c Estimated relative copy numbers of the rrn plasmids and other replicons in bacteria without chromosomal rrn operons. The average values of the chromosome are one and shown as a dotted line. d Estimated effective copy numbers of rrn operons.
We next hypothesized that Rep_3-family genes can allow their replicons to acquire not only rrn operons but also other essential genes. This was because, for example, the Rep_3-family genes could make plasmids extremely stable over generations by decreasing the plasmid loss rates, so that essential genes could have been maintained on plasmids. However, we found that Rep_3-family genes less frequently co-existed with universal single-copy genes in a plasmid than the other rep genes (p < 0.001, Fig. 3b).
Rep_3-type plasmids stably maintained rrn operons due to their high-copy numbers
The fact that Rep_3-family genes can enable plasmids to specifically acquire and maintain rrn operons suggests that Rep_3-family genes may play a role in controlling plasmid copy numbers. This is because what characterizes rRNA genes compared to other essential protein-coding genes is their lack of translational-level amplification and high copy numbers. Because the rrn plasmid of A. ureilytica has a high copy number (10.9 plasmid copies per genome in stationary phase, Fig. 3c)11, we analyzed short-read sequencing data to examine if the rrn plasmid of T. saccharophilum, which also recently lost chromosomal rrn operons, also has a high copy number. The copy number of the rrn plasmid of Aureimonas sp. AU20 were differently estimated from the previous study11 (18.2 copies in the stationary phase). This difference could be attributed to discrepancies in culture techniques or methods to determine plasmid copy numbers. For example, the previous study used qPCR (rrs/rpsB), which can be affected by a PCR amplification bias. For comparison, we also estimated copy numbers of rrn plasmids with rep genes of bacteria with chromosomal rrn operons. We selected four strains whose short-read sequencing data were publicly available and that had small (<35 kb) contigs that encoded rrn operon(s) and Rep_3-family genes (Bacillus sp. OV322 and Ureibacillus xyleni JC22) or the other rep genes (Eubacterium siraeu DSM 15702 and Tistlia consotensis USBA 355) (Supplementary Fig. 4).
We estimated that Rep_3-type rrn plasmids of all the above species have high copy numbers (>10 copies, OV322 with parA gene was exceptionally ~8 copies) (Fig. 3c, Supplementary Fig. 4), whereas the rrn plasmids with other rep genes had low copy numbers (<2 copies) (Supplementary Fig. 4). These results suggested that Rep_3-family genes can maintain plasmids at high copy numbers, even with the existence of rrn operons. High-copy number plasmids can be inherited stably even by stochastic segregations31,32.
Rep_3-type rrn plasmids of Persicobacteraceae decreased copy numbers but obtained partitioning mechanisms for stable inheritance
Next, we estimated copy numbers of the rrn plasmids of the four Persicobacteraceae species, which lost their chromosomal rrn operons much earlier. Interestingly, the copy numbers of their rrn plasmids were lower (four to eight copies) than those of species that recently lost their chromosomal rrn operons (Fig. 3c). Instead, rrn operons in the Persicobacteraceae species increased their copy numbers on the plasmids to two or three (Fig. 1a). In total, the copy numbers of rrn operons per cell remained constant (Fig. 3d). To our knowledge, no previous studies have observed that evolutionary pressure actively maintained copy numbers of plasmids and rrn operons33.
Because we assumed that the high-copy numbers of the Rep_3-type rrn plasmids contributed to their stable inheritance, the copy number decrease of rrn plasmids in Persicobacteraceae may have needed to be compensated for by another mechanism for stable inheritance. Here, we found that rrn plasmids of Persicobacter spp. have parAB genes for active plasmid partitioning. On the other hand, the rrn plasmids of A. tunicatorum, F. axinellae and T. saccharophilum lack plasmid partition systems, as well as A. ureilytica and O. saccharovorans11,18. While rrn plasmids carrying parAB were synapomorphic in Persicobacter, rrn plasmids of F. axinellae and A. tunicatorum were outgroups in the 16S rRNA phylogenetic tree (Supplementary Fig. 5). Thus based on the Occam’s razor principle, we estimate that the ancestor of Persicobacteraceae originally had rrn plasmids without parAB and acquired parAB in the ancestor of Persicobacter, which was likely the first empirical data that support a prediction that a plasmid obtains a partition system after the acquisition of essential genes14.
Losses of chromosomal rrn operons led to substantial increase of chromosomal tRNA genes
Next, we investigated genomic outcomes of losses of chromosomal rrn operons over short and long terms. Bacteria without chromosomal rrn operons are those that have the highest effective copy numbers of rrn operons per cell (Fig. 3d)17,18, whereas related species of the four clades typically had only 1–5 rrn operons on their chromosomes (Fig. 1f, g and Supplementary Fig. 6).
We first focused on tRNA genes because rRNA gene numbers are known to correlate with those of tRNA genes as they constitute translation systems together34,35. As expected, tRNA gene numbers of A. ureilytica and T. saccharophilum were significantly higher than those of related species (Fig. 4a and Table 1). The tRNA gene numbers of O. saccharovorans were not significantly greater than those of related species, but this may be because O. saccharovorans and its related species are undergoing substantial genome reduction (Supplementary Fig. 7)36. Notably, tRNA gene numbers of the Persicobacteraceae species were substantially high (128–173) (Fig. 4a and Table 1).

a Comparison with related species for the four independently evolved bacterial clades: Persicobacteraceae (n = 6) and related species (n = 101), T. saccharophirum (n = 1) and related species (n = 43), A. ureilytica (n = 3) and related species (n = 18), and O. saccharovorans (n = 3) and related species (n = 126). Each dot represents a genome. b tRNA gene numbers per isoacceptor family in the Persicobacteraceae (n = 6) and related species (n = 101). tRNAIle (anticodon GAT) genes are exclusively on rrn plasmids, tRNAAla genes are on both rrn plasmids and chromosomes, and the others are exclusively on chromosomes. tRNAIle2 is an AUA codon-specific isoleucine tRNA that has a CAU anticodon. Data are presented as mean values ±standard deviations. c Minimal doubling time estimated using gRodon39. Dataset of genomes is the same as in a. d Numbers of tRNA loci (gene clusters) per cluster size (tRNA gene numbers). a, c P values were calculated using Mann–Whitney U test (one sided). n.s. not significant (p > 0.05). Source data are provided as a Source Data file.
The substantial increase of tRNA gene numbers in the Persicobacteraceae species let us analyze which tRNA genes specifically increased in abundance. As mentioned above, tRNAAla and tRNAIle genes are resident on rrn plasmids and their copy numbers are estimated to be eight to twelve copies per cell. Copy numbers of chromosomal tRNA genes per amino acid ranged from three to fifteen and those per anticodon ranged from one to fifteen (Fig. 4b and Supplementary Fig. 8a). These numbers are substantially different from those of related species with chromosomal rrn operons, one to two copies of tRNA genes per anticodon (Supplementary Fig. 8a). tRNA gene-copy number differences among amino acids (i.e., tRNA isoacceptor families) likely reflect amino-acid compositions in the protein-coding sequences in genomes37,38, because significant correlations were observed (Supplementary Figs. 8c, 9). Differences among anticodons (i.e., tRNA isodecoder families) were also attributed to codon biases34 (Supplementary Fig. 8b). Minimal doubling time estimated using codon-usage bias39 was also significantly shorter than that of related species (Fig. 4c). These results suggest that bacteria whose rrn operon is only on a plasmid were under selection pressure leading to faster growth rates and increased effective numbers of rrn operons and tRNA genes.
Finally, we investigated molecular mechanisms behind the increase in tRNA gene numbers in the Persicobacteraceae family. We found that certain tRNA genes underwent tandem duplication on chromosomes, and each genome contains 20–25 tRNA gene clusters (Fig. 4d and Supplementary Fig. 8d). The largest tRNA gene cluster of P. psychrovividus (22 tRNA genes) is comparable in size to clusters belonging to a special group known as tRNA arrays40,41,42. A notable difference between the previously reported tRNA gene arrays and those of the Persicobacteraceae species was that the latter has a smaller repertoire of anticodons, possibly because the increase of tRNA gene-copy number in the Persicobacteraceae family is still ongoing (Supplementary Fig. 8d, e). Merging of different tRNA gene clusters also likely occurred in the Persicobacteraceae family, because the numbers of tRNA gene clusters were significantly lower than those of related species (Fig. 4d, Supplementary Fig. 8f). Because tRNAIle genes (anticodon GAT) were found only within rrn operons on the plasmids of the Persicobacteraceae species, the increase in tRNA gene abundance on the chromosomes likely occurred after the transfer of tRNAIle genes to plasmids, unless tRNAIle gene-copy numbers first increased on the chromosomes and subsequently decreased.
Discussion
In this study, we found that chromosomal rrn operons were lost at least four times in the bacterial domain, where the Rep_3-family genes were revealed to be prerequisites. The long-term evolution of bacteria without chromosomal rrn operons has likely been supported by subsequent acquisition of plasmid partition systems. A notable evolutionary outcome was a substantial increase in tRNA gene numbers on chromosomes and decreased plasmid copy numbers (Fig. 5).

(1) An ancestral bacterium obtains a prerequisite Rep_3-type plasmid by horizontal gene transfer. (2) An rrn operon is translocated to the Rep_3-type plasmid. (3) Effective copy numbers of rrn operons increase on the high copy number Rep_3-type plasmid. (4) Resultant evolutionary pressure leads to loss of chromosomal rrn operons, making the rrn plasmid indispensable to the bacteria. (5) Copy numbers of rrn plasmids decrease by keeping effective copy numbers of rrn operons, the rrn plasmid acquires a partitioning system for stable inheritance, and chromosomal tRNA genes undergo tandem duplications.
By assuming that the loss of chromosomal rrn operons occurred once in the common ancestor of each clade, we estimated that the Persicobacteraceae clade has survived throughout evolution without chromosomal rrn operons for >492 million years since the Paleozoic era. Even if we relax this criterion to allow three independent losses of chromosomal rrn operons within the Persicobacteraceae clade, the loss is estimated to have occurred at 124–492 MYA (i.e., in the common ancestor of the genus Persicobacter) based on the shared genomic and plasmid structures in Persicobacter (Fig. 2a). We also argue that the unique plasmid and genomic characteristics of the four Persicobacter species strongly suggest that their rrn plasmids have evolved under the same evolutionary pressure as that on the rrn operons. This, in turn, supports the hypothesis under which the loss of their chromosomal rrn operons occurred in their common ancestor. Although there are debates on the accuracy of divergence time estimation in prokaryotes43, our data prove that bacteria without chromosomal rrn operons can avoid extinction for an evolutionarily long term.
The convergent evolution of chromosomal rrn-operon losses strongly suggests that more clades lost chromosomal rrn operons in the bacteria domain. Above all, draft genomes did not allow us to list all bacteria without chromosomal rrn operons. Our bioinformatic search depended on the existence of rep genes, but there exist plasmids without rep genes20. In addition, our filtering criteria excluded plasmids that are large and/or have essential genes. Bioinformatic tools for detecting plasmid contigs may be used for detecting more cases44. Moreover, considering the fast increase in the number of publicly available high-quality metagenome-assembled genomes and single amplified genomes45,46, it would soon be possible to search for vast numbers of uncultured bacteria for those without chromosomal rrn operons. For example, a recent long-read metagenomic study reconstructed a metagenome-assembled genome of A. ureilytica without chromosomal rrn operon47. In addition, a strain with a Rep_3-type high-copy rrn plasmid may lose chromosomal rrn operons in the future according to the present results.
The molecular mechanisms by which Rep_3-family genes maintain rrn plasmids at high copy numbers are of interest, and the molecular biology of Rep_3-family proteins merits exploration. We envision that, once their molecular mechanisms are clarified, Rep_3-family genes may be used as a synthetic biology tool for controlling plasmid functions. Plasmids with Rep_3-family genes are also known to encode iterons near the replication origins48, which are direct repeats that control plasmid copy numbers by binding to Rep_3 family protein49. Thus, iterons and Rep_3 family protein might also play some role in maintaining the high copy numbers of rrn plasmids. While the order of events was inferred by the Occam’s theorem (Fig. 5), the order of the Rep_3 stabilization and the rrn-operon acquisition is not clear. We argue that the rrn-operon acquisition may be the first because more Rep_3 hosts would have lost the chromosomal rrn operons if the Rep_3 stabilization was the first. Another limitation of this study would be that the observed plasmid copy numbers may be different from those in native conditions. Meanwhile, copy numbers of rrn operons, tRNA genes, and codon biases are indicators of growth rates, where the number of tRNA genes correlates with the amino-acid composition of CDSs34,37,38,39,50,51. The speciality of the Persicobacteraceae clade may be due to differences in the time elapsed since the increase of the copy numbers of rrn operons; tRNA gene-copy numbers have been under strong evolutionary pressure to increase their copy numbers because of the increase of rrn-operon copy numbers.
The Persicobacteraceae species may also be of general interest in the context of plasmid biology, especially for those interested in plasmid evolution. Evolution of a specific plasmid can seldom be tracked for a very long time because of few conserved regions, high numbers of repetitive sequences, high rates of gene exchange, and many structural variants52,53. In the context of genome evolution, these species may also provide a unique platform to investigate the evolution of bacterial tRNA gene-copy numbers. During the long-term evolution of the Persicobacteraceae family, both partition system and biotin synthesis genes were transferred to their rrn plasmids (Fig. 1a). This may be because the Persicobacteraceae family needed to adopt rrn plasmids to produce more biotin, or because gene transfers between a chromosome and a stably inherited plasmid can occur by chance.
Finally, we may inquire why rrn operons have not returned to chromosomes again. One possible explanation is that other genomic characteristics (e.g., tRNA gene-copy number) that already adapted to the high-copy number plasmid rrn operons did not allow losses of plasmid rrn operons. It may also be possible that having rrn operons exclusively on plasmids provided evolutionary advantages, although the isolation sources of the four clades were diverse (marine organism or soil, Persicobacteraceae; rumen, T. saccharophylum; phyllosphere and air, Aureimonas ureilytica; and insect guts, O. saccharovorans). In addition to advantages previously proposed11, one hypothesis would be that, because some antibiotics target rRNAs and rRNA genes on different plasmid copies can harbor sequence diversity, high-copy number rrn plasmids may have been beneficial against toxin attacks54. Another hypothesis would be that, because transcription-replication clashes can act as a barrier to bacterial replication55, rrn operons only on plasmids can make transcription and replication separated.
Methods
Genome dataset and search for genomes without chromosomal rrn operons
GBFF files of 86,822 bacterial genomes were downloaded from NCBI RefSeq on May 30, 2017. Among them, 406,111 sequences (contigs) encoded rRNA genes. Contigs coding Rep-family genes were selected by tBLASTn searches with 156 Rep sequences20 at a threshold e value < 10−5, set to obtain hits for Rep genes on the rrn plasmid of A. ureilytica. Then, we selected genomes with a contig that encoded more than one full-length rRNA genes, encoded Rep-family genes, were <35 kb, and encoded no essential single-copy genes (bac120, GTDB-Tk v.0.3.2 identify25). The presence of full-length rRNA genes was based on RefSeq gene annotations. Genomes that had more than one contig that encoded rRNA genes were removed.
Accession numbers of Persicobacter spp. CCB-QB2 and JZB09 genomes were GCF_001274635.1 and GCF_001308105.1, respectively.
Sample preparation, genome sequencing, assembly, and annotation
Supplementary Table 1 presents a summary of seven genomes sequenced in this study. These strains were provided by the Japan Collection of Microorganisms (JCM), NITE Biological Resource Center (NBRC), and Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ). Strains of T. saccharophilum, T. bryantii, and Persicobacteraceae and related species were incubated using Treponema saccharophilum medium (DSMZ 323), Treponema bryantii medium (DSMZ 159), and Marine Broth medium (Difco, Tokyo, Japan), respectively. Cells in stationary phase were collected and total DNA was extracted using DNeasy PowerSoil Kit (QIAGEN) in accordance with the manufacturer’s instructions.
Genomes were sequenced using PacBio RSII (Pacific Biosciences) and HiSeq X ten (2 × 150 bp). PacBio RSII libraries were size-selected by BluePippin (6–50 kb for A. tunicatorum, P. diffluens, T. saccharophilum, and T. bryantii, and 17–50 kb for F. axinellae and P. psychrovividus). Primer sequences of HiSeq reads were trimmed using Trimmomatic v.0.3356. Hybrid assembly was conducted using Unicycler v.0.4.8-beta (normal mode)57, and/or HGAP v.2.2.058 provided in DDBJ annotation Pipeline with Pilon v.1.2359. Finishing was performed using GenomeMatcher60, minimap2 v.2.0-r191-dirty61, and Bandage v.0.8.162. Completeness of the genomes was assessed using BUSCO v.4.0.263 on the Cytophagales_odb10, Spirocheatales_odb10, Rhizobiales_odb10, and Rhodospirillales_odb10 orthologue datasets. Although the assembly status of Persicobacter diffluens was scaffold, its completeness was comparable with complete genomes of other Persicobacteraceae species. Circular contigs were labeled as chromosomes or plasmids (Supplementary Table 1).
Genome annotation was performed using DFAST v.1.1.064 and gene prediction was using Prodigal v.2.6.365. Barrnap v.0.8 (https://github.com/tseemann/barrnap) and tRNAscan-SE v.2.0.666 were used for rRNA and tRNA gene prediction. KofamScan v.1.1.0 (with KOfam v.2020-01-0667) and InterProScan v.5.24-6368 (with Pfam v.31.027 and TIGRFAMs v.15.069) were used for functional annotation of CDSs. For Persicobacter spp., rep genes were numbered according to sequence similarity and synteny as shown in Fig. 1e.
Phylogenetic analysis
In the phylogenetic analyses based on concatenated rRNA genes (rrs, rrl, rrf) and single-copy genes of the Persicobacteraceae species and T. saccharophilum and their related species, 17 and 19 genomes were used, respectively (Supplementary Data 1). Nucleotide sequences of the rRNA genes were aligned using MAFFT v.7.27370 and curated using TrimAl v.1.2rev5971 (strict mode for Treponema, and gappyout mode for Cytophagales). Partitioned analysis of the rRNA genes with the best substitution models for each alignment was conducted using IQ-TREE v2.0.3 and ModelFinder72,73,74. The alignments of 4467 (1512, 2848, 107) or 4558 (1529, 2919, 110) nucleotides, respectively, were subjected to phylogenetic tree reconstruction using IQ-TREE v2.0.3 with the best substitution model and 1000 bootstrap replicates. The similarity between the topologies of core genes and rRNA genes was tested statistically by likelihood tests with IQ-TREE75,76,77,78,79.
Phylogenetic analysis based on amino-acid sequences of single-copy core genes was performed using bcgtree v.1.0.10, which uses hidden Markov models and performs a partitioned maximum-likelihood analysis80, and RAxML v.8.2.981 with bootstrap trial set to 1000. In total, 85 or 88 CDSs were used, respectively.
For phylogenetic analysis of Rep genes including Rep_3-family genes (PF01051), we used BLASTp (ncbi-blast-2.10.0+, e value < 10−5, query coverage >0.5, identity >30%) against nr database by querying 93 amino-acid sequences of Rep_3-family genes from genomes without chromosomal rrn operons, and obtained bacterial 2,723 sequences. The representative sequences of 1413 clusters generated using CD-HIT v.4.8.1 (95% identity)82 (Supplementary Data 5) were aligned using MAFFT and curated using TrimAl (gappyout) and pgelimdupseq v.2.0.2016.09.06 (208 positions). The alignment was subjected to phylogenetic tree reconstruction using FastTree v.2.1.1083.
Phylogenetic tree visualization was performed with iTOL v.684, and NCBI taxonomy was assigned using ETE 385.
Divergence time estimation
We obtained g__Aureimonas and g__Treponema_D phylogenetic trees from GTDB v.89.024. Phylogenetic trees of the Persicobacteraceae species and O. saccharovorans were reconstructed using GTDB-Tk v.0.3.2 gtdbtk de_novo_wf25. These phylogenetic trees consisted of bacteria without chromosomal rrn operons, related species, calibration species for divergence time estimation, outgroups, and those for stability of phylogenetic trees shown in Supplementary Data 6. The related species were f__Cyclobacteriaceae for Persicobacteraceae, g__Treponema_D for T. saccharophilum, g__Aureimonas for A. ureilytica, and f__Acetobacteraceae for O. saccharovorans. Divergence time was estimated using RelTime-Branch Lengths26,86 for the GTDB or GTDB-Tk phylogenetic trees. In RelTime, branch-specific relative rates are estimated by applying equal elapsed time periods of separation of two sister lineages from their most recent common ancestor26 and were used for estimating branching time of bacteria87. Calibration constraints were adopted from a previous study88.
Ancestral gene-content reconstruction
We obtained subtrees consisting of bacteria with chromosomal rrn operons and related species from the phylogenetic trees used for divergence time estimation and constructed gene-family tables using Pfam27. PastML v.1.9.2428 was used to predict gains/losses of gene families using presence/absence tables. CAFE v4.2.1 was used to predict expansion and contraction of gene numbers of gene families that were predicted to be present at the root29. Analyses using KEGG67 and TIGRFAMs69 were also performed.
Dataset of plasmids encoding Rep-family genes
The 156 Rep proteins20 were contained in 32 Pfam families (e value < 10−5, Supplementary Data 7). Among them, 17 families have functions clearly related to plasmid replication and are in AnnoTree30 with GTDB r89.0. We obtained 21,716 contigs that had those Rep-family genes from 9728 genomes. We removed chromosomes that had Rep-family genes by filtering genomes containing single contigs, and the largest contigs in complete genomes. We also calculated numbers of universal single-copy genes by the identify command of GTDB-Tk and removed contigs >one unique gene for contigs whose lengths were <54 kb, more than four unique genes for contigs whose lengths were 54 kb or more and <800 kb, and more than nine unique genes for contigs whose lengths were 800 kb or more. The annotation of rRNA genes was performed using Barrnap v.0.8.
Estimation of plasmid copy numbers
Plasmid copy numbers were calculated by mapping short reads to genomes and dividing average coverage depths of plasmids by those of chromosomes. For T. saccharophilum and the Persicobacteraceae species, HiSeq reads used for genome assembly were retrieved. For Aureimonas sp. AU20, we conducted sequencing using HiSeq X Ten (2 × 150 bp). We obtained genomes and short-read data for Bacillus sp. OV322 (GCF_900112495.1 and SRR4235142), Ureibacillus xyleni JC22 (GCF_900217795.1 and SRR6007419), Eubacterium siraeu DSM 15702 (GCF_000154325.1 and SRR15171209.1), and Tistlia consotensis USBA 355 (GCF_900177295.1 and SRR5194480) from SRA and NCBI RefSeq. These strains were selected from our in-house dataset entries that contained rrn operons in the chromosome and a small plasmid (<35 kb). Whether the contigs were chromosomal or not was assessed by Bandage62. The threshold for distinguishing chromosomal and plasmid contigs was the same as above. Coverage depths were calculated using Bowtie2 v.2.2.789 and bbmap v.38.34 (https://www.osti.gov/biblio/1241166).
tRNA clusters and codon bias S
A group of tRNA genes was considered a cluster if the tRNA genes were adjacent to each other at ≤500 bp. Spearman’s correlation coefficient between the numbers of tRNA genes on chromosomes and the amino-acid compositions of all CDSs were calculated. Codon bias S34 was calculated using a python script by referring to R-package v.2.24.0 sscu. The growth-rate potential was estimated using gRodon R-package v. 2.3.039. Forty highly expressed genes were selected according to a previous study34 and used for calculating codon bias S and growth-rate potential. Statistical significance was assessed using Mann-Whitney U-tests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequences reported in this paper have been deposited in the DNA Data Bank of Japan (DDBJ) database, http://www.ddbj.nig.ac.jp (accession numbers are listed in Supplementary Table 1). Other data generated in this study are provided in the Supplementary Data and Source Data files. Source data are provided in this paper.
References
Egan, E. S., Fogel, M. A. & Waldor, M. K. Divided genomes: negotiating the cell cycle in prokaryotes with multiple chromosomes. Mol. Microbiol. 56, 1129–1138 (2005).
Harrison, P. W., Lower, R. P., Kim, N. K. & Young, J. P. Introducing the bacterial ‘chromid’: not a chromosome, not a plasmid. Trends Microbiol. 18, 141–148 (2010).
Suwanto, A. & Kaplan, S. Physical and genetic mapping of the Rhodobacter sphaeroides 2.4.1 genome: presence of two unique circular chromosomes. J. Bacteriol. 171, 5850–5859 (1989).
Michaux, S. et al. Presence of two independent chromosomes in the Brucella melitensis 16M genome. J. Bacteriol. 175, 701–705 (1993).
Rodley, P. D., Römling, U. & Tümmler, B. A physical genome map of the Burkholderia cepacia type strain. Mol. Microbiol. 17, 57–67 (1995).
Yamaichi, Y., Iida, T., Park, K. S., Yamamoto, K. & Honda, T. Physical and genetic map of the genome of Vibrio parahaemolyticus: presence of two chromosomes in Vibrio species. Mol. Microbiol. 31, 1513–1521 (1999).
Kunnimalaiyaan, M., Stevenson, D. M., Zhou, Y. & Vary, P. S. Analysis of the replicon region and identification of an rRNA operon on pBM400 of Bacillus megaterium QM B1551. Mol. Microbiol. 39, 1010–1021 (2001).
Battermann, A., Disse-Krömker, C. & Dreiseikelmann, B. A. functional plasmid-borne rrn operon in soil isolates belonging to the genus Paracoccus. Microbiology 149, 3587–3593 (2003).
Tazzyman, S. J. & Bonhoeffer, S. Why there are no essential genes on plasmids. Mol. Biol. Evol. 32, 3079–3088 (2014).
Wein, T et al. Essential gene acquisition destabilizes plasmid inheritance. PLoS Genet. https://doi.org/10.1371/journal.pgen.1009656 (2021).
Anda, M. et al. Bacterial clade with the ribosomal RNA operon on a small plasmid rather than the chromosome. Proc. Natl Acad. Sci. USA 112, 14343–14347 (2015).
diCenzo, G. C. & Finan, T. M. The divided bacterial genome: structure, function, and evolution. Microbiol. Mol. Biol. Rev. 81, e00019–17 (2017).
Antipov, D. et al. A. plasmidSPAdes: assembling plasmids from whole genome sequencing data. Bioinformatics 32, 3380–3387 (2016).
Hülter, N. et al. An evolutionary perspective on plasmid lifestyle modes. Curr. Opin. Microbiol. 38, 74–80 (2017).
Hall, J. P. J., Brockhurst, M. A. & Harrison, E. Sampling the mobile gene pool: innovation via horizontal gene transfer in bacteria. Philos. Trans. R. Soc. B 372, 20160424 (2017).
Hall, J. P. J., Botelho, J., Cazares, A. & Baltrus, D. A. What makes a megaplasmid? Philos. Trans. R. Soc. 377, 20200472 (2021).
Espeio, R. T. & Plaza, N. Multiple ribosomal RNA operons in bacteria; their concerted evolution and potential consequences on the rate of evolution of their 16S rRNA. Front. Microbiol. 9, 1232 (2018).
Chua, K. O. et al. Plasmid localization of sole rrn operon in genomes of Oecophyllibacter saccharovorans (Acetobacteraceae). Plasmid 114, 102559 (2021).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Shintani, M., Sanchez, Z. K. & Kimbara, K. Genomics of microbial plasmids: classification and identification based on replication and transfer systems and host taxonomy. Front. Microbiol. 6, 242 (2015).
Midha, S. et al. Genomic resource of rice seed associated bacteria. Front. Microbiol. 6, 1551 (2016).
Furusawa, G., Lau, N. S., Suganthi, A. & Amirul, A. A. A. Agarolytic bacterium Persicobacter sp. CCB‐QB2 exhibited a diauxic growth involving galactose utilization pathway. Microbiologyopen 6, e00405 (2017).
Chua, K. O. et al. Oecophyllibacter saccharovorans gen. nov. sp. nov., a bacterial symbiont of the weaver ant Oecophylla smaragdina. J. Microbiol. 58, 988–997 (2020).
Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004 (2018).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927 (2019).
Tamura, K. et al. Estimating divergence times in large molecular phylogenies. Proc. Natl Acad. Sci. USA 109, 19333–19338 (2012).
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412:D419 (2021).
Ishikawa, S. A., Zhukova, A., Iwasaki, W. & Gascuel, O. A fast likelihood method to reconstruct and visualize ancestral scenarios. Mol. Biol. Evol. 36, 2069–2085 (2019).
Han, M. V., Thomas, G. W. C., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Mendler, K. et al. AnnoTree: visualization and exploration of a functionally annotated microbial tree of life. Nucleic Acid Res. 47, 4442–4448 (2019).
Nordström, K. & Austin, S. J. Mechanisms that contribute to the stable segregation of plasmids. Annu. Rev. Genet. 1, 37–69 (1989).
Million-Weaver, S. & Camps, M. Mechanisms of plasmid segregation: have multicopy plasmids been overlooked? Plasmid 75, 27–36 (2014).
Kembel, S. W., Wu, M., Eisen, J. A. & Green, J. L. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput. Biol. 8, e1002743 (2012).
Sharp, P. M., Bailes, E., Grocock, R. J., Peden, J. F. & Sockett, R. E. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acid Res. 33, 1141–1153 (2005).
Lee, A. M.-P., Bussema, C. & Schmidt, T. M. rrnDB: documenting the number of rRNA and tRNA genes in bacteria and archaea. Nucleic Acid Res. 37, D489–D493 (2008).
Li, L. et al. Whole-genome sequence analysis of Bombella intestini LMG 28161T, a novel acetic acid bacterium isolated from the crop of a red-tailed bumble bee, Bombus lapidaries. PLoS One 11, e0165611 (2016).
Yamao, F., Andachi, Y., Muto, A., Ikemura, T. & Osawa, S. Levels of tRNAs in bacterial cells as affected by amino acid usage in proteins. Nucleic Acids Res. 19, 6119–6122 (1991).
Kanaya, S., Yamada, Y., Kudo, Y. & Ikemura, T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: Gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 238, 143–155 (1999).
Weissman, J. L., Hou, S. & Fuhrman, J. A. Estimating maximal microbial growth rates from cultures, metagenomes, and single cells via codon usage patterns. Proc. Natl. Acad. Sci. USA 118, e2016810118 (2021).
Puerto-Galan, L. & Vioque, A. Expression and processing of an unusual tRNA gene cluster in the cyanobacterium Anabaena sp. PCC 7120. FEMS Microbiol. Let. 337, 10–17 (2012).
Tran, T. T. T., Belahbib, H., Bonnefoy, V. & Talla, E. A comprehensive tRNA genomic survey unravels the evolutionary history of tRNA arrays in prokaryotes. Genome Biol. Evol. 8, 282–295 (2016).
Morgado, S. M. & Vicente, A. C. P. Beyond the Limits: tRNA array units in Mycobacterium genomes. Front. Microbiol. 9, 1042 (2018).
Wang, S. & Luo, H. Dating Alphaproteobacteria evolution with eukaryotic fossils. Nat. Commun. 12, 3324 (2021).
Andreopoulos, W. B. et al. Deeplasmid: deep learning accurately separates plasmids from bacterial chromosomes. Nucleic Acids Res. 50, e17 (2022).
Alneberg, J. et al. Genomes from uncultivated prokaryotes: a comparison of metagenome-assembled and single-amplified genomes. Microbiome 6, 173 (2018).
Nayfach, S. et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 39, 499–509 (2021).
Masuda, S. et al. Uncovering plant microbiomes using long-read metagenomic sequencing. bioRxiv https://www.biorxiv.org/content/10.1101/2023.03.20.533568v1# (2023)
Castro-Jaimes, S., Guerrero, G., Bello-López, E. & Cevallos, M. A. Replication initiator proteins of Acinetobacter baumannii plasmids: an update note. Plasmid 119-120, 102616 (2022).
Konieczny, I., Bury, K., Wawrzycka, A. & Wegrzyn, K. Iteron plasmids. Microbiol. Spectrum. 2, 2.6.14 (2014).
Vieira-Silva, S. & Rocha, E. P. C. The systemic imprint of growth and its uses in ecological (meta)genomics. Plos Genet. https://doi.org/10.1371/journal.pgen.1000808 (2010).
Roller, B. R. K., Stoddard, S. F. & Schmidt, T. M. Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat. Microbiol. 1, 16160 (2016).
Orlek, A. et al. Plasmid classification in an era of whole-genome sequencing: application in studies of antibiotic resistance epidemiology. Front. Microbiol. 8, 182 (2017).
Weisberg, A. J. et al. Unexpected conservation and global transmission of agrobacterial virulence plasmids. Science 368, eaba5256 (2020).
Rodriguez-Beltran, J. et al. Multicopy plasmids allow bacteria to escape from fitness trade-offs during evolutionary innovation. Nat. Ecol. Evol. 2, 873–881 (2018).
Merrikh, H., Machón, C., Grainger, W. H., Grossman, A. D. & Soultanas, P. Co-directional replication–transcription conflicts lead to replication restart. Nature 470, 554–557 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017).
Chin, C. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9, e112963 (2014).
Ohtsubo, Y., Ikeda-Ohtsubo, W., Nagata, Y. & Tsuda, M. GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinformatics 9, 376 (2008).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Simão, F. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Tanizawa, Y., Fujisawa, T. & Nakamura, Y. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34, 1037–1039 (2018).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14 (2019).
Aramaki, T. et al. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Haft, D. H. et al. TIGRFAMs: a protein family resource for the functional identification of proteins. Nucleic Acids Res. 29, 41–43 (2001).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Chernomor, O., von Haeseler, A. & Minh, B. Q. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 65, 997–1008 (2016).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Kishino, H., Miyata, T. & Hasegawa, M. Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J. Mol. Evol. 31, 151–160 (1990).
Kishino, H. & Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J. Mol. Evol. 29, 170–179 (1989).
Shimodaira, H. & Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114 (1999).
Strimmer, K. & Rambaut, A. Inferring confidence sets of possibly misspecified gene trees. Proc. R. Soc. Lond. B. 269, 137–142 (2002).
Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508 (2002).
Ankenbrand, M. J. & Keller, A. bcgTree: automatized phylogenetic tree building from bacterial core genomes. Genome 59, 783–791 (2016).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 (2007).
Huerta-Cepas, J., Serra, F. & Bork, P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638 (2016).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Yang, Y., Zhang, Y., Capiro, N. L. & Yan, J. Genomic characteristics distinguish geographically distributed Dehalococcoidia. Front. Microbiol. 11, 546063 (2020).
Battistuzzi, F. U. & Hedges, S. B. Eubacteria. In: The Timetree of Life (ed Hedges, S. B. & Kumar, S.) 106–115 (Oxford University Press, 2009).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Tao, Q., Tamura, K., Mello, B. & Kumar, S. Reliable confidence intervals for RelTime estimates of evolutionary divergence times. Mol. Biol. Evol. 37, 290–290 (2020).
Acknowledgements
We thank Motomu Matsui, Seishiro Aoki, Joel Nitta, and Ken Kuroki for their helpful comments, and Naomi Sakurai for helping in culturing the anaerobic bacteria. This study was supported by JSPS KAKENHI Grant Numbers 18J00444 (to M.A.), 16H06279 (to W.I. and A.T.), 19H05688, and 22H04925 (to W.I.) and JST Grant Number JPMJCR19S2 (to W.I.). M.A. was supported by JSPS Research Fellowships.
Author information
Authors and Affiliations
Contributions
M.A. and W.I. designed the research; M.A. did the genome finishing; M.A. performed experiments; M.S., M.O., M.T. cultured anaerobic bacteria; A.T. performed genome sequencing; M.A., S.Y. and S.C. analyzed data; W.I. supervised the study; and M.A. and W.I. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jeronimo Rodriguez-Beltran and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Anda, M., Yamanouchi, S., Cosentino, S. et al. Bacteria can maintain rRNA operons solely on plasmids for hundreds of millions of years. Nat Commun 14, 7232 (2023). https://doi.org/10.1038/s41467-023-42681-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-42681-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
