
Abstract
All-solid-state lithium-sulfur batteries offer a compelling opportunity for next-generation energy storage, due to their high theoretical energy density, low cost, and improved safety. However, their widespread adoption is hindered by an inadequate understanding of their discharge products. Using X-ray absorption spectroscopy and time-of-flight secondary ion mass spectrometry, we reveal that the discharge product of all-solid-state lithium-sulfur batteries is not solely composed of Li2S, but rather consists of a mixture of Li2S and Li2S2. Employing this insight, we propose an integrated strategy that: (1) manipulates the lower cutoff potential to promote a Li2S2-dominant discharge product and (2) incorporates a trace amount of solid-state catalyst (LiI) into the S composite electrode. This approach leads to all-solid-state cells with a Li-In alloy negative electrode that deliver a reversible capacity of 979.6 mAh g−1 for 1500 cycles at 2.0 A g−1 at 25 °C. Our findings provide crucial insights into the discharge products of all-solid-state lithium-sulfur batteries and may offer a feasible approach to enhance their overall performance.
Similar content being viewed by others


Introduction
The increasing number of countries committing to net-zero emissions has sparked a greater demand for economically feasible, highly energy-dense, and intrinsically safe energy storage systems1,2. All-solid-state lithium-sulfur batteries (ASSLSBs) have emerged as a promising energy storage solution because they possess several distinct advantages compared to traditional electrochemical energy storage systems such as lithium-ion batteries (LIBs). First, ASSLSBs utilize abundant, evenly distributed, and cost-effective sulfur as the active material3,4. Second, the high specific capacity of sulfur (1672 mAh/g) and lithium metal (3860 mAh/g) offers a theoretical specific energy of 2600 Wh/kg, which is much higher than traditional LIBs3,4. Third, ASSLSBs replace the flammable liquid electrolyte with a non-flammable inorganic solid-state electrolyte (SSE), mitigating the thermal runaway concerns inherent to traditional liquid electrolyte-based batteries5,6. All-solid-state configurations also eliminate the polysulfide shuttle effect, a phenomenon that is notorious for plaguing the development of liquid lithium-sulfur batteries (LSBs)7,8,9.
Due to their numerous advantages, tremendous efforts have been dedicated to the development of ASSLSBs, particularly using sulfide-based solid-state electrolytes (SSEs) because of their high room-temperature ionic conductivity and low mechanical moduli10,11,12. However, despite ongoing efforts, ASSLSB technology remains nascent, and several challenges prevent it from surpassing the specific energy capabilities of current LIBs and LSBs. Some of these challenges include: (1) low electronic/ionic conductivity of S/Li2S resulting in poor active material utilization; (2) severe volume changes of sulfur (~78%) upon (de)lithiation causing physical contact losses and poor reversible redox; (3) SSE degradation leading to the formation of less conductive interphases that increase interfacial resistance and hinder electron/ion transport in the S composite electrode; and (4) lithium dendrite growth that causes short-circuiting and substantially diminishes battery lifetime as a result3,7,8,13. Various strategies have been adopted to address the challenges mentioned above and improve the specific energy of ASSLSBs, such as nanosizing S/Li2S to establish more triple-phase boundaries and improve active material utilization14,15, forming solid solutions to improve redox reversibility16,17, and suppressing lithium dendrite growth by interface modification18,19,20.
While these strategies have proven fruitful, a key obstacle hindering the development of ASSLSBs stems from conceptual ambiguity surrounding their underlying redox mechanisms. Initial research employing in situ transmission electron microscopy explored the evolution of Li2S in ASSLSBs, revealing a three-step lithiation process and direct conversion from S8 to Li2S, without the formation of other sulfur species21. Another study investigated the decomposition behavior of Li2S highlighting that the decomposition of Li2S is governed by Li+ ion conductivity rather than electronic conductivity22. A recent study investigating the electrochemical reaction pathway of ASSLSBs reported the presence of a Li2S2 intermediate phase during the conversion from S8 to Li2S23. These studies have set important precedents and resulted in a richer understanding of the fundamental redox mechanisms of ASSLSBs. However, the intricate interplay between the discharge products and the electrochemical behavior of ASSLSBs, encompassing crucial aspects such as initial discharge capacity, cycling stability, and reversibility, remains insufficiently explored but stands as a pivotal prerequisite for driving the advancement of ASSLSB technology.
In this study, we first interpret the active material utilization of ASSLSBs reported in recent literature20,24,25,26,27,28,29,30,31,32 to postulate a mixed discharge product consisting of lithium sulfide (Li2S) and lithium disulfide (Li2S2). Using X-ray absorption spectroscopy (XAS), we reveal the existence of Li2S2, and confirm the premise of a mixed discharge product as a result. To achieve direct chemical identity of Li2S2 and provide further evidence of its existence, we use time-of-flight secondary ion mass spectrometry (ToF-SIMS)33, which has superior chemical selectivity via the detection of diagnostic ions34,35 and/or exploration of the relationships36 among relevant ions. ToF-SIMS has previously been used to depth profile S- in cycled sulfur cathodes of liquid Li-S batteries37. We demonstrate that ToF-SIMS can be used to detect Li2S2 and differentiate it from Li2S, which can provide information of the relationship between the discharge products of ASSLSBs. Furthermore, using first principles calculations, we demonstrate that Li2S2 exhibits better redox kinetics than Li2S, which suggests that the reversibility and cycling stability of ASSLSBs can be improved by inducing a Li2S2-dominant discharge product. As a proof-of-concept, we manipulate the lower cutoff voltage to induce a Li2S2-dominant final discharge product and ensure stable, long-term cycling performance. To further improve the conversion efficiency of ASSLSBs, a trace amount of lithium iodide (LiI) is incorporated into the S composite electrode to facilitate the electrochemical oxidation of Li2S2/Li2S during charge. As a result, ASSLSBs are fully reversible, and deliver a reversible capacity of 979.6 mAh g−1 for 1500 cycles under a high specific current of 2.0 A g−1, representing unrivaled cycling behavior for elemental sulfur positive electrodes in an all-solid-state configuration. To demonstrate their practical viability, LiI-incorporated ASSLSBs are tested at −10 and 60 °C, delivering stable cycling stability. High active material loading ASSLSBs also deliver areal capacities above 4.0 mAh cm−2. This work provides valuable insights into the discharge product of ASSLSBs, and demonstrates a feasible approach toward achieving fully reversible, all-climate ASSLSBs with high capacity, long lifetime, and improved safety.
Results and discussion
Postulating a Li2S2/Li2S mixed discharge product
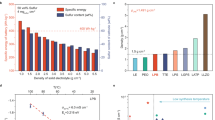
Figure 1a depicts the theoretical discharge curve of an ASSLSB showing a discharge capacity of 1672 mAh g−1 that corresponds to an active material utilization of 100%. The initial discharge capacities of ASSLSBs reported in recent literature often falls below 1400 mAh g−1, as summarized in Fig. 1b20,24,25,26,27,28,29,30,31,32. It should be noted that these values include capacity contribution that comes from sulfide SSE decomposition38,39,40. The single plateau observed in the charge/discharge curves of ASSLSBs has been widely attributed to a single-phase solid-solid conversion from S8 to Li2S, where the discharge product consists solely of Li2S. Theoretically, if this premise holds true, the reported discharge capacities of ASSLSBs in literature should approach or even surpass sulfur’s theoretical value (i.e.,1672 mAh g−1), particularly when considering sulfide SSE decomposition.

a Voltage profile showing the theoretical discharge curve of ASSLSBs. b Initial discharge capacities of ASSLSBs recently reported in literature. c Voltage profile of an ASSLSB tested using different lower limit potentials. d Computational Gibbs-free formation energies of Li2S2 and Li2S per atom in vacuum (red line) and on the LiI(100) surface (blue line). e Simplified activation diagram illustrating the energy landscape of Li2S2 (red line) and Li2S (blue line) oxidation in the adsorbed phase on the LiI surface.
Low discharge capacities observed in ASSLSBs can be interpreted three ways. First, sulfur’s poor electronic/ionic conductivity results in a large quantity of unreacted sulfur that remains after discharge. While unreacted sulfur leads to low discharge capacity, it seems unlikely that only a very small amount of sulfur can participate in redox considering the large fraction of SSE and conductive additives that are typically used to fabricate sulfur composite electrodes (Supplementary Table 1). Second, solid-solid conversion from S to Li2S is restricted by a lower potential limit that is set too high. To verify this reason, we investigated the electrochemical behavior of ASSLSBs at different lower potential limits (Fig. 1c). The theoretical capacity of sulfur cannot be reached even when the discharge potential goes down to −0.2 V (Li-In/Li+). This result demonstrates that the lower potential is not the main reason for low discharge capacities observed in ASSLSBs. Third, the final discharge product of ASSLSBs is a mixture of Li2S2 and Li2S. It is well known that the conversion of S8 to Li2S2 contributes 50% theoretical capacity (836 mAh g−1) and subsequent conversion of Li2S2 to Li2S contributes another 50% capacity (836 mAh g−1). The premise of a Li2S2/Li2S mixed discharge product is reasonable considering the plethora of ASSLSBs studies that report discharge capacities between 836 mAh g−1 and 1672 mAh g−1.
Another recurrent feature observed in the literature regarding ASSLSBs is their poor electrochemical reversibility, particularly following the initial discharge cycle15,20,41. This phenomenon has been attributed to the irreversible formation of Li2S, where the stable antifluorite structure of Li2S necessitates high activation potentials, typically approaching 4 V (versus Li+/Li), to facilitate the electrochemical oxidation (or delithiation) of Li2S back to S8 during the charging process42,43,44,45. We conducted density functional theory (DFT) calculations to investigate the influence of Li2S2 and Li2S on the reversibility of ASSLSBs (Supplementary Note 1). The calculated formation energies of Li2S2 and Li2S were approximately −1.01 eV/atom and −1.59 eV/atom, respectively (Fig. 1d). These results indicate that Li2S2 exhibits better redox activities compared to Li2S. However, it is important to note that the formation energy of Li2S2 remains considerably lower than that of S8. This suggests that both Li2S2 and Li2S hinder the electrochemical reversibility of ASSLSBs. Previous studies have demonstrated the use of lithium iodide (LiI) to effectively enhance the electrochemical oxidation of Li2S, thereby achieving fully reversible ASSLSBs16,46,47. Indeed, our DFT calculations reveal that the molecular conversion of Li2S2/Li2S to S8 on the LiI(100) surface requires a lower activation barrier compared to the process in vacuum (Fig. 1d, e). These results suggest that LiI can facilitate the electrochemical oxidation of not only Li2S but also Li2S2, thereby improving the reversibility of ASSLSBs as a result. Further discussion regarding the DFT calculations and the role of LiI in promoting the electrochemical oxidation of Li2S2/Li2S is provided in Supplementary Note 1.
In the following section, we thoroughly investigate the discharge products of ASSLSBs and evaluate the impact of incorporating LiI to address the poor reversibility of ASSLSBs. For this study, two types of S composite electrodes were prepared and tested: one without LiI (S/LGPS/CNT) and one with LiI (S/LGPS/CNT/LiI). Structural characterization and analysis of the S composite electrodes with and without LiI are presented in Supplementary Fig. 1–7 and Supplementary Note 2.
Probing the discharge products of all-solid-state Li-S batteries
Confirming the premise of a Li2S2/Li2S mixed discharge product is essential for providing insights into the reaction mechanism of ASSLSBs. Because Li2S2 exists as a meta-stable phase, determining its existence requires characterization techniques that are element-specific and chemically sensitive48. Synchrotron X-ray absorption spectroscopy (XAS) has been effectively used to identify and study various lithium polysulfide intermediates (i.e., Li2S2) in liquid/semi-liquid Li-S batteries45,49,50. In this study, XAS is used to determine the final discharge product of ASSLSBs by probing sulfur evolution at different discharge/charge states.
Figure 2a shows the S K-edge X-ray absorption near-edge structure (XANES) spectra of S composite electrode without LiI (i.e., S/LGPS/CNT) at the pristine, fully discharged (100% DOD), and fully charged (100% SOC) state. The S K-edge XANES spectra of the S/LGPS/CNT composite before discharge (i.e., pristine state) shows two broad features at 2473.0 eV and 2480.0 eV, which correspond to elemental sulfur51,52,53. After discharge, three features at 2474.1, 2476.8 and 2484.4 eV emerge, which denote the partial formation of Li2S51,54,55. Interestingly, a pre-edge feature appears at 2471.3 eV, which has previously been characterized as Li2S2 (Fig. 2b)50,51,56. After charge, the Li2S and Li2S2 features become weaker but are still present in the spectra, which indicates the irreversible transformation from Li2S2/Li2S to S. The XANES spectra of the LiI-incorporated composite at the pristine, fully discharged (100% DOD), and fully charged (100% SOC) state is plotted in Fig. 2c, d. After full charge, the Li2S and Li2S2 features become less prominent, and the features around 2473.0 eV and 2480.0 eV dominate again, resembling the pristine state (i.e., before discharge). This result suggests that LiI, even in trace quantities, plays a critical role in facilitating the electrochemical oxidation of Li2S2/Li2S during charge. The reversibility difference between the ASSLSBs at different discharge/charge states with and without LiI is further illustrated by the XANES spectra shown in Fig. 2e, f and Supplementary Fig. 8.

a, b S K-edge XANES spectra of S composite electrodes without LiI. c, d S K-edge XANES spectra of LiI-incorporated S composite electrodes. e S K-edge XANES spectra of S scomposite electrode without LiI at different discharge/charge states. f S K-edge XANES spectra of LiI-incorporated S compossite electrode at different discharge/charge states. g Ion intensity ratio of Li3S2+/Li3S+ for pure Li2S powder and the 100% DOD and 100% SOC products of ASSLSBs. The error bars represent the standard deviation of the measured intensity ratio and are produced using five independent measurements. DOD and SOC represent depth of discharge and state of charge, respectively.
As for chemical analyses of Li2S2 and Li2S, X-ray photoelectron spectroscopy (XPS) has been used previously to investigate the chemical composition of Li2S2 and Li2S in liquid Li-S batteries57,58. The detection of a S 2p3/2 peak at 162.2 eV is attributed to Li2S2 due to its binding energy’s proximity to the reference sample of Na2S2 (162.0 eV). We utilized XPS to complement the XANES results and confirm the presence of the Li2S2 phase. However, as illustrated in Supplementary Fig. 9, a discernible Li2S2 peak was not found. Previous studies that investigate ASSLSBs using XPS show similar results, where no distinct Li2S2 peak is evident in the XPS spectra32,59. Chemical similarity and overlapping peaks of Li2S2 and Li2S pose challenges in accurately identifying Li2S2 using traditional XPS analysis. Additionally, the difficulty in isolating Li2S2 as a reference sample further complicates the analysis.
Therefore, to directly determine the chemical identity of Li2S2 and gather additional supporting evidence for its existence, we used time-of-flight secondary ion mass spectroscopy (ToF-SIMS), which demonstrates superior chemical selectivity compared to XPS, enabling effective differentiation between Li2S and Li2S2. Both negative and positive secondary ion mass spectra were collected from pure Li2S powder (as reference) and the S composite electrodes with and without the addition of LiI. Ions related to the S composite electrodes include Li±, S2−, S3−, LiS−, LiS2−, Li2S+, Li3S+ and Li3S2+. Li3S+ and Li3S2+ are the most useful ions in differentiating Li2S and Li2S2 because they correspond to positive ions with the addition of a Li+ to the molecules, i.e., [Li2S + Li]+ and [Li2S2 + Li]+, respectively. While Li3S+ should be generated from both Li2S and Li2S2, Li3S2+ is more likely generated from Li2S2. This ion fragmentation pattern was confirmed by looking at the positive ions of the pure Li2S powder, where there was little to no Li3S2+ detected. The positive secondary ion mass spectra in the mass range showing Li3S+ and Li3S2+ are shown in Supplementary Fig. 10. Also shown in Supplementary Fig. 10 are the detection of Li2I+ and LiI2− from the S composite electrodes with LiI added.
Shown in Fig. 2g are the ratios of the intensity of Li3S2+ against Li3S+, which can be used to compare the relative portion of Li2S2 in the mixture of Li2S and Li2S2. For the pure Li2S powder, the ratio between Li3S2+ and Li3S+ is practically zero as little to no Li3S2+ is detected. For S composite electrode samples LiI 100% DOD, LiI 100% SOC, No LiI 100% DOD and No LiI 100% SOC, their ratios of Li3S2+/Li3S+ are 10.2% ± 1.5%, 3.0% ± 0.4%, 9.6% ± 0.9% and 3.4% ± 0.2%, respectively. Therefore, the ToF-SIMS results confirm that there is significantly more Li2S2 in the fully discharged (100% DOD) products of ASSLSBs than in the fully charged (100% SOC) ones, with or without the addition of LiI. With the addition of LiI, the fully charged product of ASSLSBs shows a slightly reduced ratio in comparison with that without the addition of LiI.
Inducing a Li2S2-dominant discharge product to enhance performance
After confirming the existence of a Li2S2/Li2S mixed discharge product, we devise an integrated strategy to enable high performance ASSLSBs by: (1) manipulating the lower potential to induce a Li2S2-dominant final discharge product and (2) incorporating a trace amount of LiI to facilitate the electrochemical oxidation of Li2S2/Li2S. In principle, this approach should significantly enhance the reversibility and cycling stability of ASSLSBs. As a proof of concept, ASSLSBs with and without LiI were evaluated using a Li-In negative electrode and LGPS as the SSE interlayer (Fig. 3a). The lower voltage limit was set at 0.6 V (vs. Li-In/Li+) to limit Li2S formation and obtain a Li2S2-dominant discharge product. ToF-SIMS analysis reveals that the intensity of Li3S+ ions decreased considerably in the cell discharged to 0.6 V compared to the one discharged to −0.2 V, which suggests that a Li2S2-dominant discharge product can be obtained by limiting the lower voltage threshold (Supplementary Fig. 11). Although promoting a discharge product with an Li2S2-dominant phase comes at the expense of the initial discharge capacity, doing so enhances both the reversibility and cycling stability of ASSLSBs (Supplementary Fig. 12). This is because Li2S2 is more electrochemically active than Li2S, and the volumetric expansion of Li2S2 is comparatively smaller, at ~60%, compared to Li2S which expands by approximately 78%. As for fixing the optimum quantity of LiI to facilitate the electrochemical oxidation of Li2S2/Li2S, we determined that a minimum of 6 wt% LiI was necessary to attain fully reversible ASSLSBs when setting the lower voltage limit to 0.6 V (vs. Li-In/Li+). Lowering the LiI content to 3 wt% resulted in ASSLSBs that could only be charged to approximately 86% of discharge capacity (Supplementary Fig. 13).

a Schematic illustration showing all-solid-state lithium-sulfur battery configuration. b Voltage profile showing the reversibility of ASSLSBs with and without LiI in the first cycle at 0.2 A g−1 and 25 °C. c Rate performance comparison of ASSLSBs with and without LiI at different specific currents from 0.2 to 6.0 A g−1 and 25 °C. d Cycling performance of ASSLSBs at 0.3 A g−1 and 25 °C. e Continued cycling of ASSLSBs at 2.0 A g−1 and 25 °C.
The ASSLSB without LiI loses approximately 18% of its initial discharge capacity after charging while the LiI-incorporated ASSLSB is fully reversible and exhibits a smaller electrode polarization (Fig. 3b). These results suggest that LiI plays a critical role in facilitating the electrochemical oxidation of Li2S2 and the small fraction of irreversibly formed Li2S after initial discharge, which coincide well with the DFT calculations presented in Fig. 1e, f. A galvanostatic intermittent titration technique (GITT) was used to estimate the Li+ diffusion coefficient (DLi) and evaluate the reaction kinetics of the ASSLSBs with and without LiI. The average DLi value for the LiI-incorporated ASSLSB during both the discharge and charge process is 4.83 × 10−13 cm−2 s−1, which is much higher than its counterpart (Supplementary Fig. 14). The LiI-incorporated ASSLSBs exhibits a reversible capacity of 100% during the GITT test, while the ASSLSB without LiI can only charge back to approximately 75%, further demonstrating the effectiveness of LiI for improving the reversibility of ASSLSBs. Rate performance of the ASSLSBs with and without LiI was investigated over a specific current range of 0.2 to 6.0 A g−1 as shown in Fig. 3c The LiI-incorporated ASSLSBs delivers a discharge capacity of 933, 1027.4, 996.4, 978.9, 938.2, 760.8, 467.8, and 303.7 mAh g-1 at specific current 0.2, 0.4, 0.6, 0.8, 1.0, 2.0, 4.0, and 6.0 A g−1, respectively, recovering to 1222.4 mAh g−1 as the specific current is restored back to 0.2 A g−1. The ASSLSB without LiI delivers much lower discharge capacities in the subsequent cycles, due to the poor reversibility of the cell after initial discharge. The LiI-incorporated ASSLSBs also demonstrate much better stability to specific current changes and exhibit a smaller electrode polarization compared to the ASSLSB without LiI (Supplementary Fig. 15). These results suggest that LiI plays a vital role in enhancing charge transfer kinetics within the S composite electrode.
Figure 3d shows the cycling performance of the LiI-incorporated ASSLSB cycled between 0.6–2.4 V (vs. Li-In/Li+) at 0.3 A g−1 and 25 °C. A reversible capacity of 1496.9 mAh g−1 is obtained after 250 cycles. The gradual capacity increase observed during cycling is likely a result of two factors. First, sulfur undergoes an activation process in the initial cycles, as a large electrode polarization results in low active material utilization. As cycling continues, the electrode polarization decreases, resulting in higher active material utilization and increasing capacity. Indeed, the electrode polarization of the LiI-incorporated ASSLSB decreases from 0.721 V in the 1st cycle to 0.682 V in the 200th cycle (Supplementary Fig. 16). Similar behavior has been reported in other sulfur-based cathodes60,61,62. Second, thiophosphate SSEs such as LGPS possess a narrow electrochemical stability window (e.g., 1.71 − 2.14 V vs. Li+/Li), and decompose in the operating voltage range of ASSLSBs as a result63. The decomposition products of LGPS are electrochemically active, and contribute to the reversibility capacity of the cell13. These two phenomena can help explain the gradual capacity increase observed in Fig. 3d. To demonstrate long-term cycling, LiI-incorporated ASSLSBs were further cycled at a high specific current of 2.0 A g−1, delivering a stable capacity of 1069.4 mAh g−1 for over 1200 cycles and a reversible capacity of 979.6 mAh g−1 for over 1500 cycles (Fig. 3e). To our knowledge, the reported cycling behavior is the best to date for elemental sulfur cathodes in an all-solid-state configuration (Supplementary Fig. 17).
All-climate all-solid-state Li-S batteries
Developing ASSLSBs that can operate within a wide temperature range is crucial for enabling applications such as electric aviation, electric vehicles, and spaceflight64. Thus, LiI-incorporated ASSLSBs were further evaluated at high and low temperature to evaluate their practical viability. When tested at 60 °C, the cell shows much lower overpotential, and delivers a high initial discharge capacity of 1136.8 mAh g−1 (Fig. 4a). Interestingly, another discharge plateau appears at approximately 1.4 V (vs. Li-In/Li+). At 25 °C, the stepwise transition from elemental sulfur to high order polysulfides, low order polysulfides, and finally Li2S is not obvious in a solid-state configuration, as a high conversion barrier results in sluggish conversion kinetics. However, charge transfer within the S composite electrode is improved under more favorable conditions such as at elevated temperature, allowing for stepwise sulfur redox to occur. This likely gives rise to a distinct discharge plateau in the voltage profile at 60 °C, corresponding to the formation of intermediate sulfur species. A similar phenomenon has been observed in ASSLSBs that incorporate selenium into the sulfur cathode and solid-state lithium-selenium batteries, which is logical considering the high conductivity of selenium62,65.

a Voltage profile of ASSLSB tested at 60 °C, with enlarged discharge profile shown between 1.3–1.5 V (vs. Li-In/Li+). b Cycling performance of ASSLSB at 0.4 A g−1 and 60 °C. c Voltage profile of ASSLSB with active material loading of 3 mg cm−2. d Cycling performance of high loading ASSLSBs at 60 °C. e Voltage profile of ASSLSB tested at 0.2 A g−1 and −10 °C. f Cycling performance of ASSLSBs at 0.2 A g−1 and −10 °C.
The cycling stability of the LiI-incorporated ASSLSB at 60°C and 0.4 A g−1 is shown in Fig. 4b. The cell delivers a reversible capacity of 1323.6 mAh g−1 for over 400 cycles, demonstrating stable cycling stability at 60 °C. The capacity of the high temperature cell is much higher than the one tested at 25 °C, which corresponds to a greater quantity of Li2S that is formed after discharge. At high temperature, charge transfer kinetics within the S composite electrode improves, and sulfur redox can occur more efficiently as a result. Consequently, the solid-solid conversion of Li2S2 to Li2S is less hindered, leading to higher initial discharge capacities.
ASSLSBs with active material loadings for of 3 and 12 mg cm−2 were tested to evaluate the practical viability of the cells, as shown in Fig. 4c and Supplementary Fig. 18, respectively. Both cells are fully reversible and sustain areal capacities around 3.0 mAh cm−2 for 50 cycles (Fig. 4d). The cycling stability of the 12 mg cm−2 loading cell is poor compared to the 3 mg cm−2 cell, which is likely due to severe volume change of sulfur during (de)lithiation. Volume change induces contact loss between the active material, SSE, carbon, which increases internal cell resistance, and considerably limits cycling stability as a result.
Figure 4e shows the voltage profile of the LiI-incorporated cell tested at −10 °C. The overpotential increases considerably, which can be attributed to slow charge transfer kinetics within the S composite electrode at low temperature. Still, the cell is fully reversible and exhibits a relatively high initial discharge capacity of 336 mAh g−1, maintaining a stable capacity for over 100 cycles (Fig. 4f). These results demonstrate the effectiveness of the catalytic incorporation of LiI for achieving fully reversible all-climate ASSLSBs with high active material loading.
Understanding the discharge products of electrochemical energy storage systems such as metal-air and metal-sulfur batteries has proven crucial for enhancing key performance metrics such as active material utilization, specific capacity, cycle life, and reversibility66,67,68,69. In this regard, elucidating the intricate relationship between the discharge products and the electrochemical behavior of ASSLSBs is critical but remains inadequately studied thus far. In this study, X-ray absorption spectroscopy was employed to reveal that the discharge product of ASSLSBs is not exclusively composed of Li2S but rather a mixture of Li2S and Li2S2. Time-of-flight secondary ion mass spectrometry was utilized to validate the presence of Li2S2 by detecting its characteristic ion, Li3S2+, and to quantify the relative proportion of Li2S2 in the Li2S and Li2S2 mixture using the ion intensity ratio of Li3S2+/Li3S+. Density functional theory calculations were employed to showcase that while Li2S2 exhibits superior redox kinetics compared to Li2S, both species hinder the reversibility of ASSLSBs. Building upon these findings, an integrated strategy was proposed to enhance the reversibility and cycling stability of ASSLSBs. This approach involved: (1) manipulating the lower cutoff potential of ASSLSBs to promote the formation of Li2S2-dominant discharge product and (2) incorporating a trace amount of LiI into the S composite electrode to improve the electrochemical oxidation of Li2S2 and Li2S. As a result, ASSLSBs delivered a reversible capacity of 979.6 mAh g−1 for 1500 cycles at 2.0 A g−1 at 25 °C and demonstrated stable cycling stability across a wide temperature range (−10, 25, and 60 °C). Furthermore, high active material loading ASSLSBs were tested and achieved areal capacities exceeding 3.0 mAh cm−2, demonstrating the practical viability of this approach. In summary, this work utilizes advanced analytical techniques to probe the discharge products of ASSLSBs, yielding valuable insights into their electrochemical behavior and resulting in strategies that can be widely adopted to achieve fully reversible, all-climate ASSLSBs with high capacity, long lifetime, and enhanced safety.
Methods
Preparation of sulfur composite electrodes
A mixture of sulfur powder (Sigma-Aldrich), Li10GeP2S12 (MSE supplies), and carbon nanotubes (Sigma-Aldrich) with a weight ratio of 36:40:24 was transferred into a 50 mL agate ball-milling jar filled with 40 g of 5 mm zirconia balls under an Ar atmosphere (H2O < 0.1 ppm, O2 < 0.1 ppm). The mixture was ball-milled using a high-speed ball-milling machine at 200 rpm for 4 h. The same procedure was used to prepare the LiI-incorporated S composite electrodes.
Materials characterization
Powder X-ray diffraction (XRD) patterns were recorded on a Bruker AXS D8 Advance instrument with Cu Kα radiation (λ = 1.5406 Å). The sample holder was covered with Kapton tape to prevent air exposure. Raman spectra were obtained on a HORIBA Scientific LabRAM HR Raman spectrometer system (532.4 nm laser). Thermogravimetric analysis was performed using a thermal analyzer (Diamond TG, PerkinElmer, USA) under a nitrogen atmosphere using a heating rate of 5 °C min−1. Scanning electron microscopy (SEM) images were recorded using a FE-SEM (S4800, Hitachi high-technologies) equipped with an energy-dispersive X-ray spectroscopy (EDS) system. XAS was carried out at the Canadian Light Source (CLS). Sulfur K-edge XAS was collected using total electron yield (TEY) mode on the Soft X-ray Microcharacterization beamline (SXRBM) at the CLS. To achieve a good signal to noise ratio, an ambient table setup was used at the SXRMB beamline. The chamber was filled with helium gas to reduce absorption and scattering at low energies. The S composite electrodes and pure Li2S powder pressed on an aluminum foil were examined using TOF-SIMS IV (ION-TOF GmbH, Germany) equipped with a BiMn cluster liquid metal ion source. A pulsed 25 keV Bi3+ primary cluster ion beam was used to generate secondary ions from the topmost 1–3 nm of the sample surface. Ion mass spectra, i.e., intensities of ions against mass to charge ratio (m/z), were collected at three spots in an area of 200 × 200 µm2. A pulsed, low energy electron flood was used to neutralize the sample so that insulating samples can be measured. Positive secondary ion mass spectra were calibrated by Li+, CH3+ and C3H5+, while negative ones by Li−, CH− and S−. The mass resolutions of CH3+ and C3H5+ were 4000 and 5200, respectively, while the mass resolutions of CH and C2H were 3400 and 4000, respectively. For comparison purposes, the spectra shown in Supplementary Fig 10 were normalized to their total ion intensities. X-ray photoelectron spectroscopy (XPS) testing was conducted using a monochromatic Al Kα source (1486.6 eV) in a Kratos AXIS Nova Spectrometer. The Ar-filled glovebox was connected to the XPS machine to avoid exposure to air.
First principles calculations
All the first principles calculations were carried out in the DFT framework implemented in the VASP package70. The projector augmented-wave pseudopotentials were used to describe the interaction between ions and electrons, and the exchange-correlation effects were treated using the Perdew–Burke–Ernzerhof (PBE) functional under the generalized gradient approximation (GGA)71. A Monkhorst–Pack k-point grid of 3 × 3 × 1, and a kinetic energy cut-off of 600 eV was used to optimize all surface calculations. The LiI surface was created from a 3 × 3 × 3 supercell and a vacuum of 15 Å was used to avoid interaction between images. For surface calculations, the van der Waals (vdW) correction function proposed by Grimme was utilized72. All the atoms were optimized until the total energies converged to below 10−4 eV and the forces acting on atoms were less than 10−2 eV/Å. The adsorption energy (EA) was calculated using the expression EA = Esurface+adsorbate – (Esurface + Eadsorbate). The formation energy (Ef) was calculated using the expression Ef = Exy – (Ex + Ey), where x and y are pristine elements forming compound xy. The structures were visualized using the VESTA package73.
Electrochemical testing/characterization
ASSLSBs were assembled inside an Ar-filled glovebox and tested using model cells. First, 120 mg of Li10GeP2S12 was placed into a polytetrafluoroethylene (PTFE) die with a diameter of 10 mm and pressed at 1 ton. The thickness of the SSE layer was approximately 1 mm. Next, approximately 1.5 ~ 2 mg of S composite electrode powder was dispersed onto the Li10GeP2S12 side and pressed at 3 tons. The mass loading of S was approximately 0.65 mg cm−2. Finally, a Li-In alloy was placed on the bare Li10GeP2S12 side and pressed at 1 ton. The Li-In alloy was prepared by pressing a piece of In foil (ϕ 10 mm, thickness 0.1 mm) and a piece of Li foil (ϕ 10 mm, thickness 20 μm) together under ~60 MPa for 5 min. All the batteries were tested under an external pressure of ~150 MPa. ASSLSBs were tested within the voltage range of 0.6–2.4 V (vs. Li-In/Li+) using a Land cycler (Wuhan, China). Battery testing at 25 °C was conducted in a designated battery testing lab equipped with a temperature control system to ensure accurate temperature conditions. For testing at −10 °C, a freezer manufactured by Thermo Fisher Scientific was used to create the desired low temperature environment. For testing at 60 °C, a convection oven manufactured by Thermo Fisher Scientific was employed to achieve the required high temperature conditions. All cells underwent a resting period and were allowed to equilibrate for 12 h to ensure that they reached the target temperatures and stabilized before the actual testing took place. The galvanostatic intermittent titration technique (GITT) was performed using a constant specific current of 0.2 A g−1 for 20 min followed by a relaxation period of 2 h during the charge/discharge process in the first cycle.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on request.
References
Tian, Y. et al. Promises and challenges of next-generation “Beyond Li-ion” batteries for electric vehicles and grid decarbonization. Chem. Rev. 121, 1623–1669 (2021).
Choi, J. W. & Aurbach, D. Promise and reality of post-lithium-ion batteries with high energy densities. Nat. Rev. Mater. 1, 16013 (2016).
Yang, X., Luo, J. & Sun, X. Towards high-performance solid-state Li–S batteries: from fundamental understanding to engineering design. Chem. Soc. Rev. 49, 2140–2195 (2020).
Yang, X., Li, X., Adair, K., Zhang, H. & Sun, X. Structural design of lithium–sulfur batteries: from fundamental research to practical application. Electrochem. Energy Rev. 1, 239–293 (2018).
Manthiram, A., Yu, X. & Wang, S. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2, 16103 (2017).
Zhao, Q., Stalin, S., Zhao, C.-Z. & Archer, L. A. Designing solid-state electrolytes for safe, energy-dense batteries. Nat. Rev. Mater. 5, 229–252 (2020).
Xing, C. et al. Regulating liquid and solid-state electrolytes for solid-phase conversion in Li–S batteries. Chem 8, 1201–1230 (2022).
Lei, D. et al. Progress and perspective of solid-state lithium–sulfur batteries. Adv. Funct. Mater. 28, 1707570 (2018).
Kim, J. T., Hao, X., Wang, C. & Sun, X. Cathode materials for single-phase solid-solid conversion Li-S batteries. Matter 6, 316–343 (2023).
Wang, C. et al. All-solid-state lithium batteries enabled by sulfide electrolytes: from fundamental research to practical engineering design. Energy Environ. Sci. 14, 2577–2619 (2021).
Kato, Y. et al. High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 1, 16030 (2016).
Kamaya, N. et al. A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011).
Ohno, S. & Zeier, W. G. Toward practical solid-state lithium−sulfur batteries: challenges and perspectives. https://doi.org/10.1021/accountsmr.1c00116 (2019).
Jiang, M., Liu, G., Zhang, Q., Zhou, D. & Yao, X. Ultrasmall Li2S-carbon nanotube nanocomposites for high-rate all-solid-state lithium-sulfur batteries. ACS Appl. Mater. Interfaces 13, 18666–18672 (2021).
Yao, X. et al. High-performance all-solid-state lithium–sulfur batteries enabled by amorphous sulfur-coated reduced graphene oxide cathodes. Adv. Energy Mater. 7, 160293 (2017).
Fujita, Y. et al. Li 2 S–LiI solid solutions with ionic conductive domains for enhanced all-solid-state Li/S batteries. ACS Appl. Energy Mater. https://doi.org/10.1021/acsaem.2c00978 (2022).
Hakari, T., Hayashi, A. & Tatsumisago, M. Li2S-based solid solutions as positive electrodes with full utilization and superlong cycle life in all-solid-state Li/S batteries. Adv. Sustain. Syst. 1, 1700017 (2017).
Duan, C. et al. Realizing the compatibility of a Li metal anode in an all-solid-state Li−S battery by chemical iodine–vapor deposition. Energy Environ. Sci. 15, 3236–3245 (2022).
Chen, Z. et al. Bulk/Interfacial synergetic approaches enable the stable anode for high energy density all-solid-state Lithium–Sulfur batteries. ACS Energy Lett. 7, 2761–2770 (2022).
Pan, H. et al. Carbon-free and binder-free Li-Al alloy anode enabling an all-solid-state Li-S battery with high energy and stability. Sci. Adv 8, https://www.science.org (2022).
Yang, Z. et al. Phase separation of Li2S/S at nanoscale during electrochemical lithiation of the solid-state Lithium–Sulfur battery using in Situ TEM. Adv. Energy Mater. 6, 160006 (2016).
Wang, Z. et al. In Situ TEM observations of discharging/charging of solid-state lithium-sulfur batteries at high temperatures. Small 16, 2001899 (2020).
Cao, D. et al. Understanding electrochemical reaction mechanisms of sulfur in all‐solid‐state batteries through operando and theoretical studies **. Angewandte Chemie International Edition 62, e202302363 (2023). .
Sakuda, A., Sato, Y., Hayashi, A. & Tatsumisago, M. Sulfur‐based composite electrode with interconnected mesoporous carbon for all‐solid‐state lithium–sulfur batteries. Energy Technology 7, 1900077 (2019).
Sun, X. et al. High surface area N‐doped carbon fibers with accessible reaction sites for all‐solid‐state Lithium‐Sulfur batteries. Small 18, 2105678 (2022).
Alzahrani, A. S. et al. Confining Sulfur in porous carbon by vapor deposition to achieve high-performance cathode for all-solid-state Lithium–Sulfur batteries. ACS Energy Lett. 6, 413–418 (2021).
Han, Q. et al. Outstanding cycle stability and rate capabilities of the all-solid-state Li-S battery with a Li 7 P 3 S 11 glass-ceramic electrolyte and a core-shell S@BP2000 nanocomposite. J. Mater. Chem. A Mater. 7, 3895–3902 (2019).
Hou, L.-P. et al. Improved interfacial electronic contacts powering high sulfur utilization in all-solid-state lithium–sulfur batteries. Energy Storage Mater. 25, 436–442 (2020).
Zhu, G. L. et al. A self-limited free-standing sulfide electrolyte thin film for all-solid-state lithium metal batteries. Adv. Funct. Mater. 31, 2101985 (2021).
Zhang, Y. et al. High-performance all-solid-state lithium-sulfur batteries with sulfur/carbon nano-hybrids in a composite cathode. J. Mater. Chem. A Mater. 6, 23345–23356 (2018).
Yao, X. et al. High-energy all-solid-state lithium batteries with ultralong cycle life. Nano Lett. 16, 7148–7154 (2016).
Zhu, X. et al. Exploring the concordant solid-state electrolytes for all-solid-state lithium-sulfur batteries. Nano Energy 96,107093 (2022).
Benninghoven, A. Chemical analysis of inorganic and organic surfaces and thin films by static time-of-flight secondary ion mass spectrometry (TOF-SIMS). Angewandte Chemie Int. Ed. Engl. 33, 1023–1043 (1994).
Vickerman, J. C. & Winograd, N. SIMS—A precursor and partner to contemporary mass spectrometry. Int. J. Mass Spectrom 377, 568–579 (2015).
Starr, N. J. et al. Elucidating the molecular landscape of the stratum corneum. Proc. Natl Acad. Sci. 119, e2114380119 (2022).
Nie, H.-Y. Self-assembled monolayers of octadecylphosphonic acid and polymer films: surface chemistry and chemical structures studied by time-of-flight secondary ion mass spectrometry. Surf. Interface Anal. 49, 1431–1441 (2017).
Zhao, C. et al. Author correction: a high-energy and long-cycling lithium–sulfur pouch cell via a macroporous catalytic cathode with double-end binding sites. Nat. Nanotechnol. 16, 224–224 (2021).
Han, F., Gao, T., Zhu, Y., Gaskell, K. J. & Wang, C. A battery made from a single material. Adv. Mater. 27, 3473–3483 (2015).
Ohno, S., Rosenbach, C., Dewald, G. F., Janek, J. & Zeier, W. G. Linking solid electrolyte degradation to charge carrier transport in the thiophosphate-based composite cathode toward solid-state lithium-sulfur batteries. Adv. Funct. Mater. https://doi.org/10.1002/adfm.202010620 (2021).
Koerver, R. et al. Redox-active cathode interphases in solid-state batteries. J. Mater. Chem. A Mater. 5, 22750–22760 (2017).
Li, X. et al. Totally compatible P4S10+n cathodes with self-generated Li+ pathways for sulfide-based all-solid-state batteries. Energy Storage Mater. 28, 325–333 (2020).
Obrovac, M. N. & Dahn, J. R. Electrochemically active lithia/metal and lithium sulfide/metal composites. Electrochem. Solid-State Lett. 5, A70 (2002).
Ye, H., Li, M., Liu, T., Li, Y. & Lu, J. Activating Li 2 S as the Lithium-Containing Cathode in Lithium–Sulfur Batteries. ACS Energy Lett 5, 2234–2245 (2020).
Yang, Y. et al. High-capacity micrometer-sized Li 2S particles as cathode materials for advanced rechargeable lithium-ion batteries. J Am Chem Soc 134, 15387–15394 (2012).
Gao, X. et al. All-solid-state Lithium–Sulfur batteries enhanced by redox mediators. J. Am. Chem. Soc. jacs.1c07754. https://doi.org/10.1021/jacs.1c07754 (2021).
Wan, H. et al. Understanding LiI-LiBr catalyst activity for solid state Li 2 S/S reactions in an all-solid-state Lithium battery. Nano Lett. 21, 8488–8494 (2021).
Liu, M. et al. Quantification of the Li-ion diffusion over an interface coating in all-solid-state batteries via NMR measurements. Nat. Commun. 12, 5943 (2021).
Kerr, B. et al. Characterization of energy materials with X-ray absorption spectroscopy─advantages, challenges, and opportunities. Energy Fuels 36, 2369–2389 (2022).
Lowe, M. A., Gao, J. & Abruña, H. D. Mechanistic insights into operational lithium–sulfur batteries by in situ X-ray diffraction and absorption spectroscopy. RSC Adv. 4, 18347 (2014).
Pascal, T. A. et al. X-ray absorption spectra of dissolved polysulfides in lithium–sulfur batteries from first-principles. J. Phys. Chem. Lett. 5, 1547–1551 (2014).
Yang, X. et al. Promoting the Transformation of Li 2 S 2 to Li 2 S: significantly increasing utilization of active materials for high‐Sulfur‐loading Li–S batteries. Adv. Mater. 31, 1901220 (2019).
Feng, X. et al. Understanding the degradation mechanism of rechargeable lithium/sulfur cells: a comprehensive study of the sulfur–graphene oxide cathode after discharge–charge cycling. Phys. Chem. Chem. Phys. 16, 16931–16940 (2014).
Lin, Z. et al. High-performance lithium/sulfur cells with a bi-functionally immobilized sulfur cathode. Nano Energy 9, 408–416 (2014).
Vijayakumar, M. et al. Molecular structure and stability of dissolved lithium polysulfide species. Phys. Chem. Chem. Phys. 16, 10923–10932 (2014).
Cuisinier, M. et al. Sulfur speciation in Li–S batteries determined by operando X-ray absorption spectroscopy. J. Phys. Chem. Lett. 4, 3227–3232 (2013).
Patel, M. U. M. et al. X-ray absorption near-edge structure and nuclear magnetic resonance study of the Lithium-Sulfur battery and its components. ChemPhysChem. 15, 894–904 (2014).
Helen, M. et al. Single step transformation of sulphur to Li2S2/Li2S in Li-S batteries. Sci. Rep. 5, 12146 (2015).
Liang, X. et al. A highly efficient polysulfide mediator for lithium–sulfur batteries. Nat. Commun. 6, 5682 (2015).
Ohno, S. et al. Observation of chemomechanical failure and the influence of cutoff potentials in all-solid-state Li-S batteries. Chem. Mater. 31, 2930–2940 (2019).
Zhang, C., Lin, Y. & Liu, J. Sulfur double locked by a macro-structural cathode and a solid polymer electrolyte for Lithium-Sulfur batteries. J. Mater. Chem. A Mater. 3, 10760–10766 (2015).
Han, F. et al. High-performance all-solid-state lithium-sulfur battery enabled by a mixed-conductive Li2S nanocomposite. Nano Lett. 16, 4521–4527 (2016).
Li, X. et al. High-Performance Li–SeS x All-Solid-State Lithium Batteries. Adv. Mater. 31, 1808100 (2019).
Zhang, W. et al. Degradation Mechanisms at the Li10 GeP2 S12 /LiCoO 2 Cathode Interface in an All-Solid-State Lithium-Ion Battery. ACS Appl. Mater. Interfaces 10, 22226–22236 (2018).
Rodrigues, M.-T. F. et al. A materials perspective on Li-ion batteries at extreme temperatures. Nat. Energy 2, 17108 (2017).
Li, X. et al. Highly stable halide-electrolyte-based all-solid-state li-se batteries. https://doi.org/10.1002/adma.202200856 (2022).
Basak, S. et al. Accessing lithium–oxygen battery discharge products in their native environments via transmission electron microscopy grid electrode. ACS Appl. Energy Mater. 3, 9509–9515 (2020).
Su, Y.-S., Fu, Y., Cochell, T. & Manthiram, A. A strategic approach to recharging lithium-sulphur batteries for long cycle life. Nat. Commun. 4, 2985 (2013).
Xia, C., Black, R., Fernandes, R., Adams, B. & Nazar, L. F. The critical role of phase-transfer catalysis in aprotic sodium oxygen batteries. Nat. Chem. 7, 496–501 (2015).
Xia, C., Kwok, C. Y. & Nazar, L. F. A high-energy-density lithium-oxygen battery based on a reversible four-electron conversion to lithium oxide. Science (1979) 361, 777–781 (2018).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B. 47, 558–561 (1993).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S. S. GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Momma, K. & Izumi, F. VESTA: a three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 41, 653–658 (2008).
Acknowledgements
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC); Canada Research Chair Program (CRC); Canada Foundation for Innovation (CFI); Ontario Research Fund (ORF); Canada Light Source (CLS) at University of Saskatchewan; Interdisciplinary Development Initiatives (IDI) by Western University; and University of Western Ontario. C.W. acknowledges the Banting Postdoctoral Fellowship (BPF – 180162). J.T.K. thanks Professor Yang Zhao and Professor Yukwon Jeon for their thought-provoking discussions.
Author information
Authors and Affiliations
Contributions
J.T.K. and C.W. conceived the study and analyzed the results. J.T.K. conducted the experiments and collected data. J.T.K. wrote the manuscript. A.R. conducted the DFT calculations. S.D. helped with XANES measurements. W.L. and F.Z. helped with XRD and SEM characterization. J.L. and H.D. helped with Raman characterization. H-Y. N. helped with ToF-SIMS characterization. Y.H., J.F., and X.H. participated in the discussion of the data. X.S., C.V.S., and C.W. supervised the project. All authors have approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sufu Liu and Abhilash Karuthedath Parameswaran for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, J.T., Rao, A., Nie, HY. et al. Manipulating Li2S2/Li2S mixed discharge products of all-solid-state lithium sulfur batteries for improved cycle life. Nat Commun 14, 6404 (2023). https://doi.org/10.1038/s41467-023-42109-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-42109-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
