
Abstract
Insertion of lipopolysaccharide (LPS) into the bacterial outer membrane (OM) is mediated by a druggable OM translocon consisting of a β-barrel membrane protein, LptD, and a lipoprotein, LptE. The β-barrel assembly machinery (BAM) assembles LptD together with LptE at the OM. In the enterobacterium Escherichia coli, formation of two native disulfide bonds in LptD controls translocon activation. Here we report the discovery of LptM (formerly YifL), a lipoprotein conserved in Enterobacteriaceae, that assembles together with LptD and LptE at the BAM complex. LptM stabilizes a conformation of LptD that can efficiently acquire native disulfide bonds, whereas its inactivation makes disulfide bond isomerization by DsbC become essential for viability. Our structural prediction and biochemical analyses indicate that LptM binds to sites in both LptD and LptE that are proposed to coordinate LPS insertion into the OM. These results suggest that, by mimicking LPS binding, LptM facilitates oxidative maturation of LptD, thereby activating the LPS translocon.
Similar content being viewed by others

Introduction
Gram-negative bacteria surround and protect their cytoplasm with a multilayered envelope formed by an inner membrane (IM), an outer membrane (OM) and a thin peptidoglycan layer, sandwiched in between. Both lipid bilayers carry out crucial functions, including nutrient uptake and energy metabolism, as well as cytoplasm detoxification and protection against noxious chemicals, thereby promoting adaptability to a wide range of niches1. The OM forms the foremost barrier to cellular access and therefore, in the context of bacterial multidrug resistance, the OM is a particularly attractive drug target for the development of new antimicrobials2,3.
Lipopolysaccharide (LPS) is a major structural component that accumulates in the external leaflet of the OM lipid bilayer. LPS typically contains six saturated acyl chains, surface displayed carbohydrates and negatively charged phosphate groups that are stabilized by divalent cations. This therefore structures a densely-packed layer that shields the cell from lipophilic molecules, detergents and antimicrobial compounds. LPS is also a modulator of protein homeostasis and peptidoglycan remodeling, regulating the overall architecture of the bacterial envelope4,5,6,7. In addition to its protective function, LPS acts as an endotoxin with potent immunogenic activity and represents a major target of the innate immune response8.
Anterograde LPS transfer across the bacterial envelope is mediated by a dedicated LPS transport (Lpt) pathway. In the enterobacterial model organism Escherichia coli, the Lpt pathway relies on the activity of 7 proteins, known as LptA-G9,10. Impairment of the Lpt pathway causes accumulation of LPS in the IM6, where it undergoes subsequent modification by colanic acid11, a polysaccharide produced upon envelope damage12. The terminal link of the Lpt pathway consists of an OM translocon that inserts LPS into the external leaflet of the OM. The OM LPS translocon, composed by an integral OM protein, LptD, and a cognate OM-associated lipoprotein, LptE, acquires a peculiar “plug-and-barrel” architecture13,14,15. LptD consists of an amino (N)-terminal periplasmic β-taco domain and a carboxy (C)-terminal 26-stranded β-barrel transmembrane domain. The β-taco domain receives LPS from a structurally analogous periplasmic protein, LptA. The C-terminal β-barrel domain defines a large internal lumen that is partly plugged by LptE14,15,16. After docking onto the LptD β-taco domain, LPS enters the OM via a mechanism involving a membrane-embedded interface between the β-taco and the β-barrel domains of LptD. This region is proximal to the β-barrel lateral gate that forms between the first and the last β-strands of the LptD transmembrane domain15,16,17,18,19.
Biogenesis of the OM LPS translocon requires the cooperation of the β-barrel assembly machinery (BAM) and the periplasmic disulfide bond formation machinery (Dsb). BAM is a heteropentamer (BamABCDE) that folds integral OM proteins, such as LptD, into membrane-spanning β-barrel structures20,21. The catalytic, integral OM protein BamA and the lipoprotein BamD are two essential BAM subunits that directly coordinate LptD and LptE assembly to form a plug-and-barrel structure22. Chaperones and proteases help preventing the accumulation of off-pathway folding intermediates that would hamper OMP biogenesis if blocked at the BAM complex23,24. Within LptD, two native disulfide bonds between pairs of non-consecutive cysteine residues interlink the β-taco and the β-barrel domains, thereby activating the LPS translocon25. This complex LptD oxidation pattern occurs via a stepwise maturation process. The first two consecutive cysteines in the β-taco domain are initially oxidized by the oxidase DsbA, generating a non-native disulfide bond. After engaging with the BAM complex for assembly of LptD together with LptE in the OM, acquisition of native disulfide bonds in LptD activates the translocon26. Typically, DsbC promotes disulfide bond shuffling in secretory proteins27, however the role of this enzyme in LptD oxidative folding has been debated25,28.
The development of Lpt-targeted drugs requires detailed understanding of the process of LPS transport to the OM. Here we report the discovery of LptM (formerly YifL) as a component of the OM LPS translocon. LptM binds the membrane-embedded portion of the translocon, making contacts with sites in both LptD and LptE that are proposed to mediate LPS insertion into the OM. Thus, LptM stabilizes an active conformation of the OM LPS translocon promoting its assembly by the BAM complex and oxidative maturation. LptM is a component of the Lpt pathway that controls activation of the OM LPS translocon by mimicking substrate binding.
Results
The OM LPS translocon stably interacts with LptM
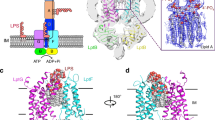
We discovered that the E. coli OM LPS translocon, LptDE, interacts with an uncharacterized lipoprotein, formerly known as YifL, that we have renamed LptM (LptD oxidative maturation-associated lipoprotein). We identified LptM upon expression and Ni-affinity purification of the LptDEHis complex (harboring a C-terminally poly-histidine tagged variant of LptE) using the mild, non-ionic detergent n-dodecyl-β-D-maltopyranoside (DDM). The elution fraction was analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) followed by Coomassie brilliant blue staining, revealing efficient co-purification of LptD along with the bait protein LptEHis (Fig. 1a, lane 1). Analysis of the same elution fraction by blue native (BN)-PAGE showed that the OM LPS translocon migrates with an apparent molecular weight of ~140 kDa (Fig. 1a, lane 3). To verify the protein content of this major gel band, we employed trypsin digestion and MALDI-TOF/TOF tandem mass spectrometry (MS). Surprisingly, in addition to LptD and LptE, this analysis identified LptM, an uncharacterized lipoprotein of ~6 kDa (Supplementary Fig. 1a–c). To explore the role of this factor, we purified the LptDE complex from a strain lacking the yifL/lptM gene. Although the yields of LptEHis and LptD resolved by SDS-PAGE were unchanged (Fig. 1a, lanes 1 and 2), the intensity of the blue native 140 kDa band was reduced (Fig. 1a, lanes 3 and 4). Part of purified LptEHis and LptD originated a slightly slower-migrating BN gel band that was most prominent in the sample lacking LptM. The accumulation of a slower-migrating form of the LptDE complex could be indicative of an effect of LptM on the native organization of the OM LPS translocon.

a The envelope fractions of wild-type (WT, lptM+) and ΔlptM transformed with pLptDEHis cells were solubilized using the mild detergent DDM and subjected to Ni-affinity purification. The purified OM LPS translocon was analyzed by SDS-PAGE (lanes 1 and 2) and BN-PAGE (lanes 3 and 4) followed by Coomassie Brilliant Blue staining. Data are representative of three independent experiments. b DDM-solubilized LptMHis purified from ΔlptM cells transformed with pLptDEMHis was analyzed by SDS-PAGE and Coomassie Brilliant Blue staining. Data are representative of three independent experiments. c Native mass-spectrometry analysis of LptDEHis and of LptDEMHis purified as in (a) and (b), respectively. A schematic diagram of each complex is shown for each spectrum; LptD in gray, LptE in yellow, LptM in purple. Top: LptDEHis formed a heterodimeric complex of 108,425 ± 11 Da (LptD, theoretical mass of the mature protein = 87,080 Da; LptEHis, theoretical mass of the mature and tri-acylated protein = 21,348 Da). Bottom: LptDEMHis formed a heterotrimeric complex of 114,325 ± 14 Da (LptD, theoretical mass of the mature protein = 87,080 Da; LptE, theoretical mass of the mature and tri-acylated protein = 20,248 Da; LptMHis, theoretical mass of the mature and tri-acylated protein = 6,988 Da). The mass difference between the two complexes is 5,900 Da ± 25 Da, corresponding to the mass of mature tri-acylated LptM 5888 Da. d ΔlptM cells transformed with pLptDEMHis derivative (*) plasmids that carry pBpa at distinct positions in LptD were subjected to UV irradiation as indicated prior to DDM-solubilization and Ni-affinity purification of LptMHis. Upon SDS-PAGE of the elution fractions, proteins were revealed by Coomassie Brilliant Blue staining or Western blotting using an antiserum against LptA. Data are representative of three independent experiments. e Logoplot representation of the LptM amino acid sequence in Enterobacteriaceae. The plot was obtained from the multiple alignment of 37 amino acid sequences of LptM in representative genomes of a restricted Enterobacteriaceae family reported in Supplementary Fig. 4 (see also Supplementary Information). The location of PF13627 is indicated above the logoplot.
To address whether LptM can stably interact with the OM LPS translocon, a C-terminally poly-histidine tagged version of E. coli LptM (LptMHis) was overproduced together with LptD and LptE. Ni-affinity chromatography revealed efficient purification of the OM LPS translocon subunits along with the bait protein (Fig. 1b). Most notably, native MS demonstrated that LptD, LptE and LptMHis formed a complex which is larger by 5900 Da ± 25 Da compared to the LptDEHis heterodimer (Fig. 1c). This mass difference fits well with the theoretical molecular weight of mature tri-acylated LptM, 5888 Da. Together, these results indicate that LptM stably associates with LptDE forming a 1:1:1 heterotrimeric complex.
Next, we asked whether, by associating with LptDE, LptM integrates the transenvelope Lpt pathway, including the periplasmic LPS chaperone LptA that bridges the IM and OM Lpt complexes. An interaction of LptD with LptA can be monitored by site-directed photocrosslinking, introducing the photoactivatable amino acid analog para-benzoyl-phenylalanine (pBpa)29 in place of Y63 at the N-terminal region of the LptD β-taco domain, which interacts with LptA13. We observed that the population of LptD that stably associates with LptMHis could efficiently crosslink LptA in proximity of LptD Y63 (Fig. 1d, lanes 1 and 2). In a control experiment, pBpa was introduced in place of Y347, a position buried in the lumen of the LptD β-barrel, which inefficiently crosslinked LptA (Fig. 1d, lanes 3 and 4). In addition, LptA and, albeit to a lower extent, LptB were co-isolated along with DDM-solubilized LptMHis (Supplementary Fig. 2a). Of note, the overproduction of LptMHis had no detectable effect on bacterial growth nor on the OM permeability barrier to the large molecular weight antibiotic vancomycin (Supplementary Fig. 2b), suggesting that its association with the Lpt pathway did not interfere with LPS transport and OM homeostasis. We conclude that LptM stably interacts with the OM LPS translocon LptDE, thus integrating the transenvelope Lpt pathway.
LptM is a lipoprotein conserved in Enterobacteriaceae
The gene lptM codes for a small lipoprotein of ~6 kDa. The LptM lipobox cysteine residue is followed by a glycine residue (Fig. 1e), suggesting that the protein is transported to the OM30, in agreement with our finding that LptM interacts with the OM complex LptDE. LptM includes the annotated Pfam domain, PF13627 (Prokaryotic lipoprotein-attachment site), overlapping with the protein lipobox and a short N-terminal region of the mature protein. This motif was used to search lptM putative orthologs in a group of 2927 bacterial genomes (comprising at least one genome per bacterial family of those present in the bacterial Genome Taxonomy Database, GTDB)31. lptM-like genes were found in ~79% of Gamma- and 57% of Alpha-proteobacteria, and were virtually absent in non-proteobacterial genomes (Supplementary Fig. 3a–d, see also Supplementary Methods). The degree of conservation of chromosomal neighboring genes surrounding lptM in Proteobacteria suggests inheritance of this gene from a common ancestor. Thus, the identified lptM genes can be considered orthologs. The amino acid sequence length downstream PF13627 is generally short with less than 70 amino acids (Supplementary Fig. 3a). Both the N-terminal PF13627 and the C-terminal moiety of the protein are conserved in a recently redefined Enterobacteriaceae family (Fig. 1e and Supplementary Fig. 4), whereas other Proteobacteria harbor a lipoprotein containing the N-terminal PF13627 motif followed by a C-terminal region that is poorly conserved with that of E. coli LptM (Supplementary Fig. 3e; see also Supplementary Methods). Taken together, our taxonomic analysis strongly suggests that distinct sub-regions of LptM from Enterobacteriaceae are functionally important.
LptM promotes efficient assembly of the OM LPS translocon at the BAM complex
The BAM complex plays a key role in the assembly of LptD and LptE as a two-protein plug-and-barrel complex. We noticed that purification of DDM-solubilized LptMHis co-isolated, to some degree, BAM subunits (Supplementary Fig. 2a, lane 4), raising the hypothesis that LptM could be assembled with the LPS translocon during its biogenesis at the BAM complex. We thus assessed whether LptM would be required for BAM-mediated assembly of LptD together with LptE. To test this hypothesis, first we generated a double deletion strain lacking lptM and bamB, which encodes a subunit of the BAM complex that is particularly critical for LPS translocon biogenesis32. ΔbamB is sensitive to detergents, such as SDS, and to large molecular weight antibiotics, such as vancomycin, which are normally excluded from the OM of wild-type cells. To compare the phenotypes of single and double lptM and bamB deletion strains, we used low concentrations of vancomycin or SDS that had only a partial inhibitory effect on the growth of ΔbamB and that had no effect on ΔlptM (Fig. 2a, frames 3-6). The sensitivity of the double deletion strain to both SDS and vancomycin was considerably accentuated compared to ΔbamB, especially at 42 °C (Fig. 2a, frames 4 and 6). This detrimental synergistic effect reveals a genetic association of lptM with bamB, pointing to a possible role of LptM in LptDE assembly.

a Drop dilution growth test of wild-type E. coli and the indicated derivative strains deleted of bamB, lptM or both under the indicated conditions. vanc, vancomycin; SDS, sodium dodecyl sulfate. b The DDM-solubilized and purified LPS translocon obtained from lptM+ or ΔlptM cells harboring pLptDEHis was analyzed by Coomassie Brilliant Blue staining to visualize the bait protein (lanes 1 and 2) used for Western blotting with the indicated antisera to identify co-eluted proteins (lanes 3-6). Load: 1.8%; Elution: 100%. Data are representative of three independent experiments.
Mutations that impair the folding of LptD delay LPS translocon assembly into the OM, thereby prolonging its time of residence at the BAM complex22,33. We reasoned that, if LptM was indeed important for efficient assembly of LptDE, the deletion of lptM could cause accumulation of the LPS translocon at the BAM complex. To address this hypothesis, LptDEHis was produced either in wild-type (lptM+) or in ΔlptM cells and affinity purified, yielding similar amounts of the bait protein (Fig. 2b, lanes 1 and 2). Western blot analysis of the elution fractions revealed that LptM was highly enriched in the LptDEHis elution fraction obtained from lptM+ cells (Fig. 2b, lane 5). Strikingly, the BAM subunits BamD and BamE were also detected in the elution fraction obtained from ΔlptM cells but not in that obtained from lptM+ cells, indicating a prolonged interaction of LptDEHis with the BAM complex in the absence of LptM (Fig. 2b, lanes 5 and 6). We conclude that LptM is required for efficient release of the OM LPS translocon by the BAM complex.
Inactivation of lptM impairs LptD oxidative maturation
We asked whether LptM influences the oxidative maturation of LptD that occurs concomitantly to LPS translocon assembly by the BAM complex. LptD contains four cysteine residues at position 31, 173, 724 and 725 of its amino acid sequence, hereafter named C1 to C4, respectively (Fig. 3a). Upon transport into the periplasm by the Sec machinery, the two most N-terminal cysteine residues C1 and C2 are oxidized by DsbA, leading to formation of an oxidation intermediate (LptDC1-C2). Folding of LptDC1-C2 into a β-barrel structure that surrounds LptE13,22 is followed by the isomerization of the C1-C2 disulfide bond to the C2-C4 bond and further oxidation of the remaining C1 and C3 cysteines25,26. Hereafter, we will refer to the mature, fully oxidized form of LptD (LptDC1-C3,C2-C4) as LptDOx. At least one disulfide bond between non-consecutive cysteines (C1-C3 or C2-C4) is required for proper LPS translocon function, whereas the earlier assembly intermediate, LptDC1-C2, is functionally defective25,26,28. The mechanisms mediating LptD disulfide bond formation and isomerization, which are critical for LPS translocon activation, remain only partially understood25,28,34.

a Schematic representation of mature LptD primary sequence indicating the position and oxidation state of Cys residues in LptDRed (top, left), LptDC1-C2 (middle, left) or fully oxidized LptDOx (bottom, left). A typical migration pattern corresponding to different oxidative states of LptD by non-reducing SDS-PAGE is represented on the right. b The total protein contents of wild-type (lptM+) and ΔlptM strains transformed with an empty vector pCtrl, and the complementation strain ΔlptM transformed with pLptMHis were separated by non-reducing SDS-PAGE and analyzed by Western blotting using the indicated antisera. **Indicates a non-identified protein band. Data are representative of three independent experiments. c The total protein contents of wild-type and the indicated mutant strains were separated by reducing or non-reducing SDS-PAGE and analyzed by Western blotting as in (b). Red, empty arrowheads indicate reduced LptD (LptDRed), whereas filled, gray arrowheads indicate LptDC1-C2. **Indicates a non-identified protein band that migrates slightly faster than LptDRed. Data are representative of three independent experiments. d Purification of LptEHis from ΔlptM cells that overproduced LptDEHis (lane 1) or from wild-type cells that overproduced LptDMEHis (lane 2) or ΔlptM cells that overproduced the indicated LptD mutant variant of LptDEHis (3-5). *Refers to the LptD Cys-to-Ser variant: wild-type LptDCCCC, LptDCCSS, LptDCSCS or LptDSCSC, as specified on the top of each gel lane. Data are representative of three independent experiments. The signals of any LptD forms in lanes 1 and 2 were quantified. The amount of intermediate forms (LptDRed + LptDC1-C2 + LptDC2-C4) was normalized to the overall amount of all LptD forms (LptDRed + LptDC1-C2 + LptDC2-C4 + LptDox). Data are represented as means ± s.e.m. (n = 3 independent experiments). Source data are provided as Source Data file. e LptDEHis containing either wild-type (LptDCCCC) or LptDCCSS co-overproduced together with LptM or alone in wild-type or ΔlptM cells were Ni-affinity purified and resolved by BN-PAGE, prior to Coomassie Brilliant Blue staining. *Indicates the plasmid-encoded LptD variant, wild-type LptDCCCC, LptDCCSS, as specified on the top of each gel lane. Data are representative of three independent experiments.
The oxidation states of LptD can be determined by non-reducing SDS-PAGE, as LptDC1-C2 migrates slightly faster than reduced LptD (LptDRed), whereas LptDOx migrates slower than both LptDC1-C2 and LptDRed (Fig. 3a)25,26. Strikingly, our analysis of endogenous LptD in wild-type and ΔlptM cells revealed that the level of LptDOx was reduced in ΔlptM and fully restored in a derivative complementation strain harboring plasmid-encoded LptMHis (Fig. 3b). Of note, ΔlptM cells accumulated some low amount of a faster-migrating form of LptD. To identify this LptD form, we analyzed the migration of LptD produced by strains lacking single components of the periplasmic oxidative folding machinery. The lack of the oxidase DsbA caused the accumulation of the reduced form, LptDRed (Fig. 3c, lane 8)25, whereas elimination of the thiol-quinone oxidoreductase DsbB and the reductase DsbG had no obvious effect on LptD oxidation under these conditions (Fig. 3c, lanes 9 and 11). In contrast, the lack of DsbC and of its electron donor DsbD caused the accumulation of LptDC1-C2 (Fig. 3c, lanes 10 and 12). The LptD migration pattern obtained with the deletion of lptM is similar to that obtained with ΔdsbC and ΔdsbD, suggesting that ΔlptM accumulates the LptDC1-C2 intermediate (Fig. 3b, lane 2; see also Fig. 3c lane 14). Furthermore, these results indicate that a ΔlptM strain fails to efficiently form mature LptDOx. Lack of the periplasmic chaperone/proteases BepA, which can degrade an early off-pathway intermediate of LptD assembly23,24, did not alter the pattern of LptD bands in ΔlptM (Fig. 3c, lanes 13-16), suggesting that BepA does not function synergistically with LptM nor degrades LptD assembly intermediates in ΔlptM, under our experimental conditions.
To further analyze the LPS translocon maturation defect caused by the absence of LptM, we overproduced LptD and LptEHis either with or without LptM and performed Ni-affinity purifications. As a reference, we also purified translocon mutant forms containing three combinations of Cys-to-Ser pair substitutions in LptD (LptDCCSS, LptDCSCS and LptDSCSC). With the overproduction of LptM, nickel-affinity purification of LptEHis yielded ~95% of LptD in its mature oxidized state, LptDOx (Fig. 3d, lane 2, and quantifications). In contrast, a significantly lower amount of LptDOx was obtained in the absence of LptM (Fig. 3d, lane 1). In this sample, approximately one third of LptD was either reduced or only partially oxidized (Fig. 3d, lane 1, and quantifications). These LptD oxidation intermediate forms included LptDC1-C2 (migration similar to that in lane 3 [LptDCCSS]) and a LptD form containing a non-consecutive disulfide bond that migrates slower than LptDOx, seemingly corresponding to LptDC2-C4 (Fig. 3d, compare lane 1 to lanes 4 [LptDCSCS] and 5 [LptDSCSC]). We also found that the complex containing LptDCCSS migrates slower on BN gel compared to the complex containing wild-type LptD (Fig. 3e, lanes 1 and 2). These results suggest that, compared to the LPS translocon containing LptDOx, the presence of LptDC1-C2 retards translocon migration on BN gels, providing a plausible explanation for the differential BN-PAGE separation patterns of LptDE purified from wild-type cells (containing mostly LptDOx) or from ΔlptM cells (containing also partially oxidized LptD forms) (Fig. 1a, lanes 3 and 4, and Fig. 3e, lanes 3 and 4). We conclude that LptM facilitates LptD oxidative maturation, whereas its inactivation causes the accumulation of intermediate oxidation forms, most prominently LptDC1-C2 (Fig. 3b, c and e).
LptM functions synergistically with DsbA during LptD oxidative maturation
The observation that ΔlptM accumulates the LptDC1-C2 oxidation intermediate suggests that LptM is not strictly required for DsbA activity. Yet LptM could have a different role in LptD oxidative maturation, thus acting synergistically with DsbA. To test this hypothesis, we generated a double deletion strain lacking lptM and the cysteine oxidase gene dsbA by using a P1 phage transduction protocol. On LB agar plates, the ΔlptM ΔdsbA strain formed slightly smaller colonies compared to its parental single deletion strains (Fig. 4a, left). Furthermore, the double deletion strain showed an enhanced sensitivity to both vancomycin and SDS (Fig. 4a, center and right), indicative of a pronounced OM permeability barrier defect that was not observed with the single deletion strains. This result implies that, as hypothesized, LptM acts synergistically with DsbA in promoting proper oxidative maturation of LptD.

a Drop dilution growth test of E. coli wild-type and the indicated derivative strains lacking lptM, dsbA or both under different conditions as indicated. vanc, vancomycin; SDS, sodium dodecyl sulfate. b LPS was extracted from the indicated strains, resolved by SDS-PAGE and silver stained. Results are representative of four independent experiments. c Drop dilution growth test of the indicated derivative strains lacking lptM, dsbC or both transformed with pDsbC on media lacking or supplemented with 0.02% arabinose (ara). d Drop dilution growth test of lptM+ or ΔlptM strains transformed with the indicated plasmids pLptD*EHis or pLptD*MEHis on media lacking or supplemented with 100 μM IPTG. * indicates variants of plasmid-encoded LptD, the wild-type LptDCCCC or the LptDCCSS variant, as specified on the top of each dilution test. e Schematic representation of the LptD oxidative maturation pathway. In wild-type cells the four cysteines of LptD are oxidized stepwise: LptDRed is first oxidized to form LptDC1-C2. Disulfide bond shuffling in the latter generates LptDC2-C4. A final event of oxidation forms a disulfide between C1 and C3, thus generating LptDOx. LptM acts synergistically with the disulfide bond formation machinery in facilitating proper oxidation of LptD. In the absence of LptM (ΔlptM, red arrow), a considerable fraction of LptD accumulates as off-pathway oxidation intermediates, most prominently LptDC1-C2. In this mutant, the assembly of LptD together with LptE stalls at the BAM complex and DsbC becomes essential for cell viability, suggesting that disulfide bond isomerization helps to rescue LPS translocon assembly.
We also investigated whether the detrimental effect caused by the lptM dsbA double deletion was related to impaired LPS transport. LPS extracted from the wild-type and the single deletion strains showed similar SDS-PAGE and silver staining patterns (Fig. 4b, lanes 2-4), whereas slower migrating forms of LPS were obtained from the double lptM dsbA deletion strain (Fig. 4b, lane 1). A mass increment of LPS reveals a modification that can be triggered as a consequence of impaired anterograde transport along the Lpt pathway, causing LPS accumulation at the IM and modification with colanic acid6,11. Most importantly, the observed LPS alteration in the lptM dsbA double deletion strain corroborates our evidence that LptM promotes LPS translocon biogenesis, thereby facilitating efficient LPS transport to the OM.
In the absence of DsbC, LptM is crucial for cell viability
To gain further insights into how LptM functions together with the oxidative folding machinery, we sought to generate the double deletion strains ΔlptM dsbC::kan or ΔdsbC lptM::kan. However, we could not combine the deletion of lptM with that of dsbC by using P1 phage transduction (see Methods for data quantification), suggesting that the double deletion of lptM and dsbC is synthetically lethal. Indeed, a strain lacking both chromosomal dsbC and lptM, and encoding DsbC under the control of an arabinose-inducible promoter, PBAD, could grow efficiently only in the presence of the inducer (Fig. 4c), indicating that dsbC and lptM are nearly essential upon inactivation of the other. We conclude that in the absence of LptM, LptD disulfide bond isomerization by DsbC becomes critical.
To gain deeper understanding of the consequences related to impaired LptD disulfide bond isomerization, we challenged cells with the overproduction of LptE and wild-type LptD (LptDCCCC) or LptDCCSS, i.e. a form of LptD that can be converted to LptDC1-C2 but that cannot undergo subsequent disulfide bond shuffling as it contains serine residues in place of C3 and C4. Notably, the overproduction of LptDCCSS impaired growth of ΔlptM but not of cells overproducing also LptM (Fig. 4d), implying that the accumulation of LptDC1-C2 in the absence of LptM is detrimental. Given the prolonged residence of LptD and LptE at the BAM complex in ΔlptM cells (Fig. 2b) and that LptDC1-C2 is the LptD oxidation intermediate that engages with the BAM complex22,26, it appears reasonable to infer that the detrimental effect caused by LptDCCSS overproduction in the absence of LptM is related to the accumulation of an off-pathway intermediate at the BAM complex, which may interfere with the correct functioning of this essential machinery. Taken together, our results suggest that, in ΔlptM, DsbC helps rescuing LptD off-pathway oxidation intermediates that fail to assemble correctly at the BAM complex (Fig. 4e).
LptM interacts with the OM embedded portion of the LPS translocon
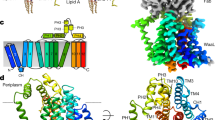
The structures of LptDE from different organisms have been determined14,15,16,18. To investigate the structural basis of how LptM interacts with LptDE, we used AlphaFold2-multimer to build a predicted model of the complex35,36,37. The top-ranking structures reveal a considerable amount of contact between LptM and LptDE (Fig. 5a and Supplementary Fig. 5), with the N-terminal cysteine of LptM consistently placed in close proximity of the flexible hinge separating the periplasmic and membrane domains of LptD16,38 that is adjacent to the LptD β-barrel lateral gate (Fig. 5a).

a View of top ranking AlphaFold2 model of LptDE-LptM. LptD is shown as gray cartoon, LptE as yellow cartoon, and the LptM backbone is shown as purple spheres. b Zoom in on the LptD hinge between the β-taco and β-barrel in either the LptDE (top) or LptDE-LptM (bottom) systems. Image from a snapshot following 500 ns of MD simulation. Several interactions are made between the β-taco and β-barrel in LptDE but broken when LptM is present (purple spheres and green sticks), including between the residues shown. c View of the LptD lateral gate with quantification of hydrogen (H)-bond number from 3 × 500 ns simulations for the LptDE and LptDE-LptM systems using gmx hbond. Representative H-bonds are shown as dashed red lines, as computed using VMD. d Differential deuterium uptake between the LptDE translocon in the presence or absence of LptM after statistical analysis with Deuteros, showing in blue regions that are significantly protected upon LptM binding. e View of the AlphaFold2 model plus modeled LptM tri-acylation, showing the position of the LptM N-terminus and tri-acylation (purple spheres and green sticks, respectively) in relation to the LptD 212-240 peptide, which is protected from deuteration by LptM in the HDX-MS experiments. Residues from the 212-240 peptide which are in contact with LptM (<0.3 nm) are shown as lines.
We next investigated how LptM might influence the conformational dynamics of the OM LPS translocon using atomistic molecular dynamics (MD) simulations of our AlphaFold2-multimer model. The presence of the LptM N-terminus close to the LptD hinge was compatible with cysteine pairs C1/C3 and C2/C4 (all reduced) being in disulfide bond distances over a simulation time of 500 ns, whereas C1 and C2 remained more distant from each other (Supplementary Fig. 6a and Supplementary Data 1). Furthermore, the positioning of the N-terminal LptM cysteine means that, when tri-acylated, the acyl tails are positioned at the interface of the LptD β-taco and β-barrel domains (Fig. 5b). This region presents several hydrophobic residues and was proposed to form a membrane-facing hole through which the acyl chains of LPS would get inserted into the OM19,39. From our simulations, the presence of LptM at this position breaks several connections of the β-taco with the β-barrel domain (Fig. 5b), which has an impact on the angle of orientation of the LptD β-taco domain (Supplementary Fig. 6b) and causes destabilization of the N-terminal loop of LptD that precedes the β-taco domain (Supplementary Fig. 6c). LptM also affects the LptD β-barrel lateral gate, reducing the number of H-bonds formed between β-strands 1 and 26 at their periplasm-facing side (Fig. 5c). This is in line with a previously proposed model of LptD lateral gate opening18 and suggests that LptM may reduce the energetic barrier necessary for β-strand unzipping at the gate15,38.
Aside from these interactions, our model also predicts that LptM inserts into the lumen of the LptD β-barrel, establishing several interaction points with both LptD and LptE. These include a salt bridge formed between LptM K23 and LptD E275 (Supplementary Fig. 6d), which are highly conserved in Enterobacteriaceae (Supplementary Fig. 7, left panel). In addition, the hook-shaped C-terminal region of LptM makes contact to LptE, as well as the inward folded LptD loop 4 that is critical for efficient LptD assembly at the BAM complex22,26,33. It should be noted, however, that the AlphaFold2 prediction confidence score decreases specifically for the C-terminal moiety of LptM, making less certain its localization in the ternary LptDEM complex (Supplementary Fig. 5C). In addition, our MD simulations predicted that the LptM C-terminal region is conformationally dynamic (Supplementary Fig. 6c), suggesting that it may coordinate multiple interactions.
Our structural model highlights that LptM mostly interacts with the OM-embedded portion of the LPS translocon. Several experiments corroborated this prediction. First, we purified LptMHis in cells that express, in addition to endogenous wild-type LptD, also a truncated form of LptD lacking the N-terminal β-taco domain, which can still interact with LptE14,15,16. In line with our AlphaFold2-multimer model, we found that the interaction of LptM with the LPS translocon does not require the LptD β-taco domain (Supplementary Fig. 8a, lane 2). Next, we employed site-directed photocrosslinking to probe the environment surrounding LptM in the attempt to map its interactions with the OM LPS translocon. To this end, pBpa was introduced at 6 different positions of the LptMHis sequence. Upon affinity purification and western-blotting with LptM-specific antibodies, a number of UV-induced crosslink adducts were detected (Supplementary Fig. 8b). The incorporation of pBpa at position L22 and, to a lower extent, Y27 led to the formation of a UV-dependent adduct of ~100 kDa that contained LptD (Supplementary Fig. 8b, c), which is in line with the AlphaFold2-multimer prediction that the conserved LptM N-terminus interacts with LptD. Two major crosslink adducts of an apparent molecular weight of ~50 kDa were obtained with pBpa introduced in the C-terminal half of LptM (Supplementary Fig. 8b). These adducts corresponded to an interaction of LptM with the abundant OM protein OmpA ( ~ 37 kDa) (Supplementary Fig. 8d). As OmpA was not detected upon native purification of LptMHis (Supplementary Fig. 2a), we speculate that the observed LptM-OmpA crosslink adducts are due to spurious association of the overproduced bait with this abundant OM protein. The inefficient crosslinking of LptM C-terminal moiety with the OM LPS translocon suggests that this segment of LptM may acquire different conformations probably mediating short-lived interactions with LptD and LptE. To further probe for the presence of LptM in the lumen of the LptD β-barrel adjacent to LptE, we introduced pBpa at two amino acid positions in LptE: K70 that faces the periplasm oriented toward LptM and A83 in a β-strand of LptE, which faces the LptD β-barrel lumen occupied by LptM in our structural model. Both LptE positions crosslinked to LptM (Supplementary Fig. 8e) supporting our prediction that LptM resides within the portion of the lumen of the LptD β-barrel that is left unoccupied by LptE (Fig. 5a).
HDX-MS reveals a stabilization effect of LptM on LptDE conformational dynamics
We also employed hydrogen-deuterium exchange mass spectrometry (HDX-MS) to monitor the dynamics and solvent accessibility of LptD and LptE in the LPS translocon produced in the presence or absence of LptM and purified in detergent micelles. We obtained 93% and 75% sequence coverage for LptD and LptE, respectively, and observed that the deuteration was very slow for the membrane-spanning portion of the LptD β-barrel domain and much faster in its periplasmic region, and extracellular loops (Supplementary Fig. 9), as expected for an integral membrane protein40,41. Less expected was the relatively high deuteration rates obtained for LptE, as this protein buries in the internal lumen of the LptD β-barrel. Comparing the deuteration of the LPS translocon with and without LptM, we observed that LptM stabilizes distinct regions of the OM LPS translocon in a statistically significant manner (Fig. 5d and Supplementary Figs. 10–12, and Supplementary Data 2 and 3). These stabilized regions include LptE (Fig. 5d and Supplementary Figs. 10–12, and Supplementary Data 2 and 3) and a segment of LptD (212-240) corresponding to the hinge followed by β 1 and loop 1 of the β-barrel domain (Fig. 5e and Supplementary Figs. 10–12, and Supplementary Data 2 and 3). The protection of LptE is in line with the multiple contacts between LptM and LptE in the lumen of the LptD β-barrel domain predicted by our AlphaFold2-multimer model. Similarly, protection of the β-barrel N-terminal peptide (LptD 212-240) fits well with the predicted positioning of the LptM N-terminus in proximity of the LptD β-taco/β-barrel hinge and the β-barrel lateral gate. This segment would also be proximal to the acyl tails of the LptM N-terminal cysteine, which might contribute to its stabilization (Fig. 5e). Notably, a recent HDX-MS analysis of Klebsiella pneumoniae (Kp) LptDE showed that, upon LPS binding, deuteration of the N-terminal region of the LptD β-barrel in proximity to the lateral gate is reduced17. Thus, the presence of LptM produces a stabilization effect in proximity of the LptD lateral gate that is similar to that caused by LPS binding. Taken together, our structure modeling and HDX-MS results suggest an LPS-mimicking effect by LptM in the membrane embedded portion of the LPS translocon.
Deuteration of the β-taco domain was also decreased in the presence of LptM. A bimodal isotopic distribution for peptides of the β-taco domain was previously reported for Kp LptDE17. Based on this result, it was suggested that the β-strands of the β-taco domain undergo concerted closing and opening motions that occur with a kinetic slower than the rate of hydrogen/deuterium exchange (EX1 regime). For peptides of the central and C-terminal region of the β-taco domain, LPS binding favored a mixed kinetics of deuterium uptake, whereas the antimicrobial peptide thanatin, which also binds the LptD β-taco domain42, shifted the equilibrium toward a slower uptake kinetics17. We observed a similar bimodal isotopic distribution for peptides of the β-taco (LptD 70-218) of the LptDE dimer in the absence of ligands, as previously reported17 (Supplementary Fig. 12). However, at this stage, we cannot exclude the presence of two populations that do not interconvert during the labeling time period, thus generating spectra that would resemble those obtained with EX1 regime kinetics. Strikingly, the presence of LptM abrogated this bimodal regime and shifted the mixed kinetics equilibrium toward the slower uptake behavior (Fig. 5d, and supplementary Figs. 10 and 12), suggesting an effect similar to that shown for Kp LptDE in the presence of thanatin. Taken together, these results provide strong evidence that the extensive contacts predicted between LptM and distinct regions of the LPS translocon may have important regulatory roles.
Discussion
Here we report the discovery of the lipoprotein LptM as a key factor for the oxidative maturation of the OM LPS translocon. We have shown that LptM stably interacts with the OM-embedded portion of the translocon formed by LptD and LptE, promoting its assembly by the BAM complex. Our functional analysis of LptM reveals a mechanism of activation of the LPS translocon upon assembly by the BAM complex, and clarifies the role of disulfide bond isomerization by DsbC during LptD oxidative maturation. Furthermore, our biochemical and structural analyses indicate that LptM integrates the Lpt transenvelope pathway by associating with LptDE and influences domains of the OM translocon reported to be crucial in coordinating LPS transport.
We provide three major lines of evidence demonstrating the critical role of LptM in the assembly of the OM LPS translocon. First, LptM interacts with the LptD β-barrel domain and LptE. This finding is fully supported by our biochemical assays, native-MS analysis, AlphaFold2 modeling, atomistic MD simulations and HDX-MS kinetics. Second, LptM improves the efficiency of LPS translocon assembly at the BAM complex. Third, LptM is required for proper oxidation of LptD and efficient anterograde transport of LPS. Our AlphaFold2-multimer model and atomistic MD simulations provide important clues as to how LptM promotes LptD maturation. LptM stably associates via its N-terminal region with the membrane-embedded portion of the LPS translocon, making contacts with both LptD and LptE. Furthermore, although with lower confidence, the LptM C-terminal region is also predicted to interact with the LPS translocon. Notably, the LptD assembly defect caused by the deletion of lptM (impaired oxidative maturation and prolonged residence at the BAM complex) is somewhat reminiscent of that reported for the variant encoded by the partial loss-of-function allele lptD421343,44. This allele encodes a form of LptD, LptDΔD330-D352, deleted of the segment comprising part of β 7 and of the inward-folded extracellular loop 4 of the β-barrel domain, which is critical for LPS translocon assembly at the BAM complex and its activation14,15,16. LptDΔD330-D352 accumulates as a C1-C2 intermediate that resides for a prolonged period of time at the BAM complex26,33, as also obtained for wild-type LptD upon deletion of lptM. This observation is consistent with a model where LptM facilitates the correct folding of LptD promoting its maturation to a fully oxidized form. However, in contrast to the LptDC1-C2 intermediate produced by the lptD4213 allele that is partly degraded by the chaperone/protease BepA23, the LptDC1-C2 intermediate produced in the absence of LptM does not appear to be influenced by this periplasmic factor.
Formation of native disulfide bonds in LptD is carefully controlled and occurs only after the β-barrel domain of LptD engages with the BAM complex to assemble together with LptE into the OM22,26. Despite the fact that native disulfide bonds are crucial for LPS translocon activation, DsbA is not essential for viability, indicating that spontaneous oxidation sustains formation of a sufficient amount of properly oxidized LptD34. Our results indicate that, with the inactivation of the oxidative folding machinery, LptM is particularly critical for efficient LPS translocon activity. Thus, LptM acts independently of, but synergistically with, the oxidative folding machinery in promoting LPS translocon activation. As indicated by our structural modeling and HDX-MS analysis, LptM binds and stabilizes regions of the OM-embedded portion of the translocon. By doing so, LptM stabilizes a conformation of the translocon that can most effectively be oxidized to form native disulfide bonds, even in the absence of a functional oxidative folding machinery (Fig. 4a and c).
Our results show that LptM is not required to generate the LptDC1-C2 oxidation intermediate, as this accumulates in its absence (Fig. 3b and d). Instead, LptM plays an important role after this first LptD oxidation event mediated by DsbA. Different views exist on the mechanisms that mediate disulfide bond isomerization in LptDC1-C2. Experimental evidence that DsbC can form an intermolecular disulfide bond with LptD suggested that this enzyme could be involved in the rearrangement of its cysteine oxidation patterns28. Yet, the observation that DsbC is not essential for cell viability casted doubts on its role during functional activation of the LPS translocon25. Our results clearly indicate that DsbC is crucial for viability in cells that lack LptM. In these cells, LptD and LptE reside at the BAM complex for a prolonged period of time compared to cells that express LptM, which is indicative of a stalled assembly process. Stalling of a LptD assembly intermediate at the BAM complex might have catastrophic effects on the biogenesis of the OM, as it can potentially interfere not only with the maturation of the LPS translocon but also with the ability of the BAM complex to insert other proteins into the OM. Our results imply that DsbC plays a role in rescuing LptD off-pathway intermediates that accumulate in ΔlptM (Fig. 4e), thus preventing deleterious consequences on OM biogenesis. In line with this scenario, overproduction of the non-isomerizable mutant form LptDCCSS in ΔlptM is highly detrimental, suggestive of a higher propensity of ΔlptM to generate LptDC1-C2 off-pathway folding intermediates. Taken together, we conclude (i) that LptM acts downstream of the first oxidation event mediated by DsbA and (ii) that, in the absence of LptM, LptD oxidation intermediates that fail to assemble into an active translocon require rescuing by DsbC (Fig. 4e). These considerations support a mechanistic model where LptM stabilizes a conformation of the LPS translocon in which LptD can efficiently complete oxidative maturation.
Finally, our results suggest that LptM, besides being crucial for the correct assembly of the LPS translocon into the OM, can also regulate its function. LptM binds the periplasm-facing luminal region of the plug-and-barrel LPS translocon structure. The N-terminus of LptM interacts with a region of the translocon in proximity of the LptD lateral gate. Intriguingly, our structural model shows that the acyl tails at the N-terminus of LptM would insert into the membrane-facing hole that forms at the interface between the β-taco and the β-barrel domains of LptD reducing the hydrogen-bonding of the lateral gate. This result was validated by our HDX-MS analysis, showing that LptM reduced deuteration of the LptD lateral gate. The stabilization effect of LptM on the lateral gate of LptD is similar to that of LPS-binding observed for Kp LptD17. The interaction of LptM with LptE, which participate in binding LPS45, further supports a possible regulatory function. Taken together, our findings show that LptM binds region of the translocon that are crucial in coordinating LPS transport into the OM. This observation suggests that LptM stabilizes an active conformation of the LPS translocon by mimicking its natural substrate. Importantly, LptM also rigidifies the periplasmic β-taco domain of LptD, suggesting tight control of the initial event of LPS docking onto the translocon17. We postulate that, during LPS transport, the initial event of LPS binding to the β-taco domain increases translocon dynamics and progressively promotes LPS insertion into the OM via the LptD membrane-facing hole and β-barrel lateral gate. A possible scenario, which will warrant further analyses, is that LPS substitutes LptM in the membrane-embedded portion of the translocon prior to breaching the OM.
The broad conservation of the N-terminal LptM PF13627 protein motif in Proteobacteria and, most notably, the conservation of full-length LptM in Enterobacteriaceae suggest that the role of LptM in LPS translocon biogenesis can be generalized to other bacteria. The two essential surface-exposed machineries, the OM LPS translocon and the BAM complex, are emerging as attractive targets for the development of novel antimicrobial compounds2,3. In this context, by discovering LptM as a LPS translocon component that is assembled together with LptD and LptE at the BAM complex, our findings provide a key piece of information that can be important for the development of OM-targeting drugs.
Methods
Bacterial strains and growth conditions
The E. coli strains used in this study are listed in Table 1. All strains are derived from BW2511346. Gene deletions were obtained by P1 phage transduction using a P1 phage lysate of the corresponding Keio collection strain47. Double gene deletions were obtained by performing a second P1 phage transduction after excising the kanamycin-resistance cassette from the first mutated locus using the heat-curable plasmid pCP2048. Transduction of wild-type and ΔlptM cells with a ΔdsbC P1 phage lysate gave rise to 137 and 0 colonies, respectively, whereas wild-type and ΔdsbC cells supplemented with the ΔlptM P1 phage lysate gave rise to 570 and 0 colonies, respectively. Thus, to build a dsbC lptM double deletion strain, the ΔlptM strain was first transformed with pDsbC, harboring the dsbC gene under the control of the arabinose-inducible PBAD promoter. This dsbC diploid strain was subjected to P1 phage transduction to delete chromosomal dsbC. The produced DsbC depletion strain was grown in the presence of 0.02% arabinose.
E. coli strains were cultured on M9 minimal medium49, lysogenic broth (LB) liquid media or LB agar plates, and supplemented with the following antibiotics: 100 μg ml−1 ampicillin, 50 μg ml−1 kanamycin, 30 μg ml−1 chloramphenicol. Serial dilution assays were conducted on LB agar plates supplemented with 5 μg ml−1 or 60 μg ml−1 vancomycin, or 0.2% (w/v) SDS, or 100-400 μM Isopropyl β-D-1-thiogalactopyranoside (IPTG), as indicated.
Plasmid construction
Plasmids and oligonucleotides are listed in Table 2 and Supplementary Table 1, respectively. Plasmid pDsbC was obtained by cloning dsbC under an arabinose inducible promoter in the pBAD33 vector50. All other plasmids were derived by cloning genes of interest downstream of an IPTG inducible promoter in the pTrc99a vector. pLptMHis, pLptDEHis, pLptDMEHis, pLptDEMHis and pDsbC were constructed by overlap extension PCR cloning. Site-directed mutagenesis was used to replace specific codons in lptM, lptE and lptD with an amber codon for incorporation of the unnatural amino acid analog p-benzoyl-L-phenylalanine (pBpa) by amber suppression29. Site-directed mutagenesis was also used to replace cysteine- with serine-encoding codons in lptD (pLptDEHis or pLptDMEHis). pLptDβ-barrelEMHis encoding a version of LptD that lacks amino acids 25-205 (corresponding to the β-taco domain) was constructed by inverse PCR on pLptDEMHis.
Taxonomic analysis of LptM
The procedure for the taxonomic analysis of LptM is detailed in the Methods section of the Supplementary Information file. The logoplot of the multiple alignment of LptM amino acid sequences was obtained using ggseqlogo51.
Cell fractionation and isolation of protein complexes upon solubilisation with a mild detergent
Whole cell lysates were prepared from cells cultured to mid-exponential phase at 37 °C. Where indicated, cells were supplemented with IPTG for 1.5 h prior to collection. Cells were pelleted by centrifugation, lysed in Laemmli sample buffer (Bio-Rad) and subjected to boiling. Protein affinity purification upon solubilisation of the crude envelope fraction with a mild detergent (native conditions) was conducted as previously described49. Briefly, when the cell cultures reached mid-exponential phase (OD600 = 0.5) protein expression was induced by supplementing 200 μM IPTG for 1.5 h prior to cell collection by centrifugation. Cells were resuspended in 20 mM Tris-HCl pH 8 containing an EDTA-free protease inhibitor cocktail (Roche). For the purification of protein samples that had to be analyzed both by reducing and non-reducing SDS-PAGE, the cell resuspension buffer was further supplemented with 50 mM iodoacetamide (Sigma) to alkylate free thiol groups of Cys residues. Resuspended cells were mechanically disrupted using a cell disruptor (Constant Systems LTD) set to 0.82 kPa. The obtained cell lysate was clarified by centrifugation at 6000 x g, 4 °C for 15 min. The crude envelope fraction was collected by subjecting the supernatant to ultracentrifugation at 100,000 x g, 4 °C for 30 min. To perform affinity purification of protein complexes, the crude envelope fraction was solubilized with 50 mM Tris-HCl pH 8, 150 mM NaCl, 20 mM imidazole supplemented with EDTA-free protease inhibitor (Roche) and 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Merck). After a clarifying spin to remove insoluble material, solubilized proteins were incubated with Protino Ni-NTA resin (Machery-Nagel) for 2 h at 4 °C. After extensive washes of the column with 50 mM Tris-HCl pH 8, 150 mM NaCl, 50 mM imidazole and 0.03% (w/v) DDM, bound proteins were eluted with a similar buffer containing 800 mM imidazole and 10% (w/v) glycerol. Aliquots of the elution fractions were snap-frozen in liquid nitrogen for storage at −80 °C or directly analyzed by SDS or blue native gel electrophoresis.
MALDI-TOF/TOF mass-spectrometry
Excised bands from BN-PAGE were washed by 25 mM NH4HCO3, pH 7.8-acetonitrile 50:50 (v/v), and digested by trypsin. MS and MS/MS experiments were carried out using α-cyano-4-hydroxycinnamic acid matrix at a concentration of 6 mg ml−1 in 50 % (v/v) acetonitrile-0.1% trifluoroacetic acid. Analyses of trypsin digested samples were performed on a MALDI TOF/TOF, in reflector positive mode. Parameters were set to source and grid voltages to 20 and 14 kV, respectively, power laser from 2000 to 3500, extraction delay time, 200 ns; shoot number, 5000. Acquisition range was between 800 and 3500 m/z. Spectra were analyzed using Data Explorer software (Applied Biosystems). MS/MS spectra were acquired using a MS/MS positive acquisition method, with 1 kV positive operating mode, and a CID off mode. The MS/MS spectra were examined and sequenced based on assignment of the N-terminal b-ion and C-terminal y-ion series.
Native Mass Spectrometry
Prior to native MS analysis, 100 μl LptDEHis (7 μM) and LptDEMHis (10 μM) samples were desalted in 200 mM ammonium acetate, pH 7.4 supplemented with 0.03% (w/v) DDM using ultrafiltration with MWCO 30 kDa Vivacon 500 (Sartorius) and concentrated to ~10-20 μM. Samples were analyzed on a SYNAPT G2-Si mass spectrometer (Waters, Manchester, UK) running in positive ion mode and coupled to an automated chip-based nano-electrospray source (Triversa Nanomate, Advion Biosciences, Ithaca, NY, USA). The voltages applied to the chip, the sample cone and the ion energy resolving quadrupole were set to 1.8 kV, 200 V and −1.0 V, respectively. Proteins were activated in the collision cell with 200 V trap collision energy and an argon flow of 8 ml min−1. The instrument was calibrated with a 2 mg ml−1 cesium iodide solution in 50% isopropanol. Raw data were acquired with MassLynx 4.1 (Waters, Manchester, UK) and analyzed manually.
Automated Hydrogen-Deuterium eXchange coupled to Mass Spectrometry (HDX-MS)
HDX-MS experiments were performed on a Synapt-G2-Si mass spectrometer (Waters Scientific, Manchester, UK) coupled to a Twin HTS PAL dispensing and labeling robot (LEAP Technologies, Carborro, NC, USA) via a NanoAcquity system with HDX technology (Waters, Manchester, UK). Briefly, 5.2 μl of protein at 20 μM were diluted in 98.8 μl of protonated (for peptide mapping) or deuterated buffer (20 mM MES pH 6.5, 200 mM NaCl) and incubated at 20 °C for 0, 0.5, 5, 10 and 30 min. 99 μl of this mixture were then transferred to vials containing 11 μL of pre-cooled quenching solution (500 mM glycine at pH 2.3). A maximally deuterated control was obtained to estimate the amount of deuterium back-change, by incubating 15 μL of LptDE at 50 μM in 285 μL d4-Urea (Sigma) 8 M pH 4.5. After 24 h, 300 μL D2O (Eurisotop) were added to reduce the urea concentration and 5 μL DCl 10% (Eurisotop) were added to reduce pH to 2.5. Deuterium uptakes obtained for this maximally deuterated control are represented in triplicate at t = 30 min in Supplementary Fig. 12, and Supplementary Data 2 and 3. Back-exchange values are shown in Supplementary Fig. 13. After 30 s quench, 105 μL of the mixture were injected to a 100 μL loop. Proteins were digested on-line with a 2.1 mm×30 mm EnzymateTM BEH Pepsin column (Waters Scientific, Manchester, UK). Peptides were desalted for 2 min on a C18 pre-column (Acquity UPLC BEH 1.7 μm, VANGUARD) and separated on a C18 column (Acquity UPLC BEH 1.7 μm, 1.0 mm × 100 mm) by a linear gradient (2% to 40% acetonitrile in 13 min). Experiments were run in triplicates and protonated buffer was injected between each triplicate to wash the column and avoid cross over contamination. Peptide identification was performed with ProteinLynx Global SERVER (PLGS, Waters, Manchester, UK) based on the MSE data acquired on the non-deuterated samples. Peptides were filtered in DynamX 3.0 with the following parameters: peptides identified in at least 3 out of 5 acquisitions, 0.3 fragments per amino-acid, intensity threshold 1000. The Relative Deuterium Uptakes were not corrected for back exchange. Deuteros 2.0 software52 was used for data visualization and statistical analysis. The online web application HDX-Viewer53 and PyMOL (The PyMOL Molecular Graphics System, Version 2.3.0 Schrödinger, LLC) were used to represent the HDX-MS data on either the LptDE and LptDEM models.
The native MS and HDX-MS data has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository54 with the dataset identifier PXD041774. A description summary of the HDX data55 is reported in Supplementary Table 2.
LPS extraction and silver staining
E. coli strains were cultured till OD600 = 0.5. 1.5 ml aliquots of cultures were withdrawn to harvest cells by centrifugation. LPS was extracted following an hot aqueous-phenol extraction56. Briefly, cell pellets were resuspended in 200 μl blue buffer (50 mM Tris-HCl pH 6.8, 2% (v/v) β-mercaptoethanol, 2% (w/v) SDS, 10% glycerol and a pinch of bromophenol blue). After boiling for 15 min and cooling at room temperature for a similar time, samples were treated with 0.5 mg ml−1 proteinase K overnight at 59 °C. A first extraction was conducted by applying to the sample 200 μl of ice-cold water-saturated phenol and heating at 65 °C for 15 min with agitation. After cooling to room temperature, 1 ml diethyl ether was applied followed by agitation by vortexing for 15 s and centrifugation at 20,600 x g. The heavier blue LPS-containing phase was withdrawn and ~1/10 of the obtained LPS fraction was resolved by SDS-PAGE before direct visualization by Silver staining (SilverQuest Silver Stain, Invitrogen).
Gel electrophoresis Coomassie staining and Western blotting
Proteins samples were prepared in Laemmli sample buffer (Bio-Rad) lacking β-mercaptoethanol (non-reducing conditions) or supplemented with β-mercaptoethanol (reducing conditions). Proteins were separated by home-made SDS gels (10% acrylamide in Bis-Tris pH 6.4 buffer, subjected to electrophoresis using either MES or MOPS buffer) or home-made blue native 6-13% acrylamide gradient gel49. Where indicated, gels were stained with Coomassie brilliant blue R250. The signal intensities of protein bands were quantified using the Multi Gauge software (Fujifilm). To perform Western blots, protein gels were blotted onto PVDF membranes (Merck). Upon blocking with skim milk, membranes were incubated with epitope-specific rabbit polyclonal antisera or with an anti-polyhistidine horseradish peroxidase-conjugated monoclonal antibody (TaKaRa). Immunodetection was revealed by using a Clarity Western ECL blotting substrate (BioRad) and detected using a LAS 4000 (Fujifilm) apparatus. Antisera were raised in rabbits against peptides or full proteins from Escherichia coli (LptM, dilution 1:1000; BamA, 1:1000; BamD, 1:1000; BamE, 1:1000; SurA, 1:1000; Skp, 1:1000; OmpA, 1:1000; CyoA, 1:1000; LptD, 1:5000; LptE, 1:10000; LptA, 1:1000; LptB, 1:10000) or Saccharomyces cerevisiae (F1beta, 1:1000). Horseradish peroxidase conjugated anti-polyhistidine (TaKaRa product n. 631210, 1:2000) and horseradish peroxidase conjugated anti-rabbit IgG (Sigma product n. A6154, 1:10000) were purchased. The rabbit polyclonal antiserum against LptD was a gift of Dr. J.F. Collet (UC Louvain, Belgium) and the rabbit polyclonal antisera against LptE, LptA and LptB were gifts of Dr. A. Polissi (University of Milan, Italy).
Protein model building
Models of LptDE and LptDEM were built using AlphaFold2, running AlphaFold Multimer v1.0 via ColabFold35,36,37. Models were built using the E. coli sequences: residues 26-784 of P31554 for LptD, residues 19-193 of P0ADC1 for LptE, and residues 20-67 of P0ADN6 for LptM (previously YifL). Five rounds were run for each prediction, with the highest ranked model used for follow-up analysis. All AlphaFold2 LptDE and LptDEM models are available for download at https://osf.io/xpfjc/, along with PAE, coverage and plDDT plots. Note that the C-terminus of LptE was truncated after Thr174 for all images used in the manuscript.
Site-directed photocrosslinking
ΔlptM cells were co-transformed with pEVOL-pBpF and plasmids expressing Lpt components with pBpa at the indicated positions (see Figure Legends and Table 2). Cells transformed with pLptMHis and LptDMEHis derivative plasmids harboring amber codons respectively in lptM and lptE ORFs were cultured in M9 minimal media, whereas cells transformed with pLptDEMHis harboring amber codons in the lptD ORF were cultured in LB. At mid-exponential phase, cells were supplemented with 1 mM pBpa and 200 μM IPTG for 1.5 h. Two identical cell culture aliquots were withdrawn, and one was kept on ice protected from light, whereas the other was subjected to UV irradiation (Tritan 365 MHB, Spectroline) for 10 min on ice. Envelope fractions were prepared from both non-treated and UV irradiated cells. To monitor the crosslinks adducts of pBpa in LptM or LptE, cells were solubilized with 20 mM Tris-HCl, pH 8, 12% (w/v) glycerol, 4% (w/v) SDS, 15 mM EDTA and 2 mM PMSF. After removing non-solubilized material by centrifugation, the supernatants were either directly resolved by SDS-PAGE (Load) or diluted in RIPA buffer (50 mM Tris/HCl pH 8, 150 mM NaCl, 1% [v/v] NP-40, 0.5% [w/v] sodium deoxycholate, 0.1% [w/v] SDS) and subjected to Ni-affinity purification as previously described49 (Elution). To monitor the crosslink adducts of pBpa in the population of LptD associated to LptMHis, we solubilized the envelope fraction with DDM and conducted Ni-affinity purification under native conditions as described above.
Molecular dynamics simulations
The top ranking AlphaFold2 models for LptDE and LptDE-LptM were used to seed MD simulations. The models were built into simulation systems using CHARMM-GUI57,58. Protein atoms were described with the fixed charge CHARMM36m force field59,60. The C-terminus of LptE was truncated after Thr174 to remove contacts with the LptD barrel, and because the lDDT was low for this region. The N-terminal cysteine residues of LptM and LptE were tri-palmitoylated. Side chain pKas were assessed using propKa3.161, and side chain side charge states were set to their default, apart from Glu263 and Asp266 of LptD, and Lys103 of LptE, which were all set to neutral. The proteins were built into asymmetric membranes, comprising 6:3:1 palmitoyl-oleoyl phosphatidylethanolamine (POPE), palmitoyl-oleoyl phosphatidylglycerol (POPG), and cardiolipin in the inner leaflet, with LPS in the outer leaflet. The E. coli Lipid A core with two 3-deoxy-α-D-manno-octulosonic acid units was chosen as a representative LPS molecule. The membranes were solvated with TIP3P waters and neutralised with K+, Cl- and Ca2+ to 150 mM. This choice of force field and water model should be sufficient to model the internal protein dynamics needed for the study59. System boxes were ~11 × 11 x 13.5 nm, with ~170,000 atoms (see Supplementary Table 3). Each system was minimized and equilibrated according the standard CHARMM-GUI protocol. Production simulations were run in the NPT ensemble, with temperatures held at 303.5 K using a velocity-rescale thermostat and a coupling constant of 1 ps, and pressure maintained at 1 bar using a semi-isotropic Parrinello-Rahman pressure coupling with a coupling constant of 5 ps62,63. Short range van der Waals and electrostatics were cut-off at 1.2 nm. Simulations were run to 500 ns and in triplicate for each system using independent initial starting velocities.
All simulations were run in Gromacs 2020.1 (https://doi.org/10.5281/zenodo.7323409)64. Data were analyzed using Gromacs tools (including residue distances, H-bond number, RMSF and angle analysis) and visualized in VMD65. Plots were made using Matplotlib https://ieeexplore.ieee.org/document/4160265.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sources data are provided within this paper. The genomes used in this studies, their accession codes and corresponding hyperlinks are listed in the Source Data file. MALDI-TOF/TOF data and quantifications of coomassie-stained gel protein bands in Fig. 3d are available in the Source Data file. Un-cropped gels are available in Supplementary Figs. 14 and 15 of the Supplementary Information file. Native- and HDX-MS data are available via the PRIDE partner repository under the accession code PXD041774. Structural models are available via the Open Science Framework under the accession code xpfjc. Source data are provided with this paper.
References
Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656 (2003).
Sousa, M. C. New antibiotics target the outer membrane of bacteria. Nature 576, 389–390 (2019).
Robinson, J. A. Folded synthetic peptides and other molecules targeting outer membrane protein complexes in gram-negative bacteria. Front. Chem. 7, 45 (2019).
Morè, N., et al. Peptidoglycan remodeling enables escherichia coli to survive severe outer membrane assembly defect. mBio. https://doi.org/10.1128/mBio.02729-18 (2019).
Klein, G. & Raina, S. Regulated assembly of LPS, its structural alterations and cellular response to LPS defects. Inter. J. Mol. Sci. https://doi.org/10.3390/ijms20020356 (2019).
Sperandeo, P. et al. Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of escherichia coli. J. Bacteriol. 190, 4460–4469 (2008).
Lima, S. et al. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science 340, 837–841 (2013).
Bertani, B. & Ruiz, N. Function and biogenesis of lipopolysaccharides. EcoSal Plus https://doi.org/10.1128/ecosalplus.ESP-0001-2018 (2018).
Sperandeo, P., Martorana, A. M. & Polissi, A. The Lpt ABC transporter for lipopolysaccharide export to the cell surface. Res. Microbiol. 170, 366–373 (2019).
Okuda, S. et al. Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat. Rev. Microbiol. 14, 337–345 (2016).
Meredith, T. C. et al. Modification of lipopolysaccharide with colanic acid (M-antigen) repeats in escherichia coli. J. Biol. Chem. 282, 7790–7798 (2007).
Wall, E., Majdalani, N. & Gottesman, S. The complex rcs regulatory cascade. Annu. Rev. Microbiol. 72, 111–139 (2018).
Freinkman, E., Chng, S. S. & Kahne, D. The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc. Natl. Acad. Sci. USA. 108, 2486–2491 (2011).
Qiao, S. et al. Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 511, 108–111 (2014).
Dong, H. et al. Structural basis for outer membrane lipopolysaccharide insertion. Nature 511, 52–56 (2014).
Botos, I. et al. Structural and functional characterization of the LPS transporter LptDE from gram-negative pathogens. Struct. (Lond. Engl. 1993) 24, 965–976 (2016).
Fiorentino, F. et al. Dynamics of an LPS translocon induced by substrate and an antimicrobial peptide. Nat. Chem. Biol. 17, 187–195 (2021).
Botte, M. et al. Cryo-EM structures of a LptDE transporter in complex with pro-macrobodies offer insight into lipopolysaccharide translocation. Nat. Commun. 13, 1826 (2022).
Gu, Y. et al. Lipopolysaccharide is inserted into the outer membrane through an intramembrane hole, a lumen gate, and the lateral opening of LptD. Struct. (Lond. Engl. 1993) 23, 496–504 (2015).
Ricci, D. P. & Silhavy, T. J. Outer membrane protein insertion by the β-barrel assembly machine. EcoSal Plus https://doi.org/10.1128/ecosalplus.ESP-0035-2018 (2019).
Ranava, D., et al. Bacterial machineries for the assembly of membrane-embedded β-barrel proteins. FEMS Microbiol. Lett.https://doi.org/10.1093/femsle/fny087 (2018).
Lee, J. et al. Characterization of a stalled complex on the β-barrel assembly machine. Proc. Natl. Acad. Sci. USA 113, 8717–8722 (2016).
Soltes, G. R., et al. Distinctive roles for periplasmic proteases in the maintenance of essential outer membrane protein assembly. J. Bacteriol. https://doi.org/10.1128/jb.00418-17 (2017).
Narita, S. et al. Protease homolog BepA (YfgC) promotes assembly and degradation of β-barrel membrane proteins in escherichia coli. Proc. Natl. Acad. Sci. USA 110, E3612–E3621 (2013).
Ruiz, N. et al. Nonconsecutive disulfide bond formation in an essential integral outer membrane protein. Proc. Natl. Acad. Sci. USA 107, 12245–12250 (2010).
Chng, S. S. et al. Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science 337, 1665–1668 (2012).
Gleiter, S. & Bardwell, J. C. Disulfide bond isomerization in prokaryotes. Biochim. Biophys. Acta 1783, 530–534 (2008).
Denoncin, K. et al. The protein-disulfide isomerase DsbC cooperates with SurA and DsbA in the assembly of the essential β-barrel protein LptD. J. Biol. Chem. 285, 29425–29433 (2010).
Chin, J. W. et al. Addition of a photocrosslinking amino acid to the genetic code of escherichiacoli. Proc. Natl. Acad. Sci. USA 99, 11020–11024 (2002).
Yamaguchi, K., Yu, F. & Inouye, M. A single amino acid determinant of the membrane localization of lipoproteins in E. coli. Cell 53, 423–432 (1988).
Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004 (2018).
Ruiz, N. et al. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121, 307–317 (2005).
Lee, J., et al. Formation of a β-barrel membrane protein is catalyzed by the interior surface of the assembly machine protein BamA. eLife https://doi.org/10.7554/eLife.49787 (2019).
Meehan, B. M., et al. The disulfide bond formation pathway is essential for anaerobic growth of escherichia coli. J. Bacteriol. https://doi.org/10.1128/jb.00120-17 (2017).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Evans, M. O. N. et al. Protein complex prediction with AlphaFold-Multimer. BioRxiv https://doi.org/10.1101/2021.10.04.463034 (2022).
Lundquist, K. P. & Gumbart, J. C. Presence of substrate aids lateral gate separation in LptD. Biochim. Biophys. Acta Biomembr. 1862, 183025 (2020).
Li, X. et al. Trapped lipopolysaccharide and LptD intermediates reveal lipopolysaccharide translocation steps across the escherichia coli outer membrane. Sci. Rep. 5, 11883 (2015).
Donnarumma, D. et al. Native state organization of outer membrane porins unraveled by HDx-MS. J. Proteome Res. 17, 1794–1800 (2018).
Javed, W. et al. Structural Insights into the Catalytic Cycle of a Bacterial Multidrug ABC Efflux Pump. J. Mol. Biol. 434, 167541 (2022).
Vetterli, S. U. et al. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Sci. Adv. 4, eaau2634 (2018).
Wu, T. et al. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121, 235–245 (2005).
Sampson, B. A., Misra, R. & Benson, S. A. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genet. 122, 491–501 (1989).
Malojčić, G. et al. LptE binds to and alters the physical state of LPS to catalyze its assembly at the cell surface. Proc. Natl. Acad. Sci. USA 111, 9467–9472 (2014).
Grenier, F., et al. Complete genome sequence of Escherichia coli BW25113. Genome Announc. https://doi.org/10.1128/genomeA.01038-14. (2014).
Baba, T., et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. https://doi.org/10.1038/msb4100050 (2006).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645 (2000).
Ranava, D., et al. Lipoprotein DolP supports proper folding of BamA in the bacterial outer membrane promoting fitness upon envelope stress. eLife https://doi.org/10.7554/eLife.67817 (2021).
Guzman, L. M. et al. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 (1995).
Wagih, O. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 33, 3645–3647 (2017).
Lau, A. M. et al. Deuteros 2.0: peptide-level significance testing of data from hydrogen deuterium exchange mass spectrometry. Bioinformatics 37, 270–272 (2021).
Bouyssié, D. et al. HDX-Viewer: interactive 3D visualization of hydrogen-deuterium exchange data. Bioinformatics 35, 5331–5333 (2019).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–d552 (2022).
Masson, G. R. et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 16, 595–602 (2019).
Davis, M. R., Jr. & Goldberg, J. B. Purification and visualization of lipopolysaccharide from gram-negative bacteria by hot aqueous-phenol extraction. J. Vis. Exp. https://doi.org/10.3791/3916 (2012).
Lee, J. et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413 (2016).
Jo, S., Kim, T. & Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PloS One 2, e880 (2007).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Best, R. B. et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012).
Søndergaard, C. R. et al. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7, 2284–2295 (2011).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52 7182–7190, (1981).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Abraham, M., Murtola, T., Schulz, R., Pall, S., Smith, J., Hess, B., Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX https://doi.org/10.1016/j.softx.2015.06.001 (2015).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 27–38 (1996).
Acknowledgements
Research in R.I.’s lab was funded by the CNRS (ATIP-Avenir grant to R.I.), the China Scholarship Council fellowships to Y.Y. and H.C. and the FRM postdoctoral fellowship to D.R. HDX-MS experiments were supported by the French Ministry of Research (Investissements d’Avenir Program, Proteomics French Infrastructure, ANR-10-INBS-08 to O. B.-S.) and the Région Midi Pyrénées to O.B.-S. Research in P.J.S.’s lab was funded by Wellcome (208361/Z/17/Z) and BBSRC (BB/P01948X/1, BB/R002517/1 and BB/S003339/1). This project made use of time on ARCHER2 and JADE2 granted via the UK High-End Computing Consortium for Biomolecular Simulation, HECBioSim (http://hecbiosim.ac.uk), supported by EPSRC (grant no. EP/R029407/1). This project also used Athena and Sulis at HPC Midlands + , which were funded by the EPSRC on grants EP/P020232/1 and EP/T022108/1. We thank the LMGM, Toulouse, for access to the E. coli Keio collection. We thank the IPBS, Toulouse, for access to the MALDI-TOF/TOF mass spectrometer. We thank the University of Warwick Scientific Computing Research Technology Platform for computational access.
Author information
Authors and Affiliations
Contributions
Y.Y. and R.I. conceived the research. Y.Y., H.C., V.M., D.R. and RI performed the genetic and biochemical analyses; R.A.C. and P.J.S. performed structural modeling and molecular dynamics simulations; Y.Q. performed phylogenetic analyses; J.M. performed native-MS and HDX-MS analyses with the help of C.F.; A.C.-S. performed the MALDI-TOF analysis with the help of C.A.; Y.Y., H.C., R.A.C., V.M., Y.Q., A.C.-S., J.M. and R.I. prepared the figures; R.A.C., Y.Q., P.J.S. J.M. and R.I. wrote the manuscript with contributions from all other authors; Y.Q., P.J.S., J.M. and R.I. supervised the work; O.B.-S., P.J.S., J.M. and R.I. acquired funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Y., Chen, H., Corey, R.A. et al. LptM promotes oxidative maturation of the lipopolysaccharide translocon by substrate binding mimicry. Nat Commun 14, 6368 (2023). https://doi.org/10.1038/s41467-023-42007-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-42007-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
