
Abstract
In the quest for renewable fuel production, the selective conversion of CO2 to CH4 under visible light in water is a leading-edge challenge considering the involvement of kinetically sluggish multiple elementary steps. Herein, 1-pyrenebutyric acid is post-synthetically grafted in a defect-engineered Zr-based metal organic framework by replacing exchangeable formate. Then, methyl viologen is incorporated in the confined space of post-modified MOF to achieve donor-acceptor complex, which acts as an antenna to harvest visible light, and regulates electron transfer to the catalytic center (Zr-oxo cluster) to enable visible-light-driven CO2 reduction reaction. The proximal presence of the charge transfer complex enhances charge transfer kinetics as realized from transient absorption spectroscopy, and the facile electron transfer helps to produce CH4 from CO2. The reported material produces 7.3 mmol g−1 of CH4 under light irradiation in aqueous medium using sacrificial agents. Mechanistic information gleans from electron paramagnetic resonance, in situ diffuse reflectance FT-IR and density functional theory calculation.
Similar content being viewed by others
Introduction
The current scenario of the global energy crisis due to uncontrolled fossil fuel consumption and continuous elevation of greenhouse gases inspires us to utilize solar light as a renewable energy source for CO2 reduction reaction (CO2RR). Unfortunately, photoreduction of the CO2 molecule is arduous due to the high C = O dissociation energy of ̴ 750 kJ mol−11. CO2RR involves proton-coupled multielectron reduction process in producing CO, CH4, and higher hydrocarbons. Considering the C1-based reduced product from CO2, the formation of CH4 is thermodynamically more favorable (\({{E}}_{{{{{{{\rm{CO}}}}}}}_{2}/{{{{{{\rm{CH}}}}}}}_{4}}^{0}\) = −0.24 V vs. NHE at pH 7) compared to other C1 feedstock (\({{E}}_{{{{{{{\rm{CO}}}}}}}_{2}/{{{{{\rm{CO}}}}}}}^{0}\) = −0.53 V; \({{E}}_{{{{{{{\rm{CO}}}}}}}_{2}/{{{{{\rm{HCHO}}}}}}}^{0}\) = −0.48 V; \({{E}}_{{{{{{{\rm{CO}}}}}}}_{2}/{{{{{{\rm{CH}}}}}}}_{3}{{{{{\rm{OH}}}}}}}^{0}\) = −0.38 V, \({{E}}_{{{{{{{\rm{CO}}}}}}}_{2}/{{{{{\rm{HCOOH}}}}}}}^{0}\) = −0.61 V vs. NHE at pH 7)1,2. In contrast, from a kinetic point of view, the formation of CH4 is challenging due to the involvement of the eight-electron transfer process, which requires precise electrons drive to carry forward the overall reaction for selective product formation3. Additionally, the kinetically sluggish nature of the multielectron transfer process from photosensitizer to the catalytic site makes the process even harder. Apart from these hurdles, the production of CH4 as a CO2-reduced product is highly beneficial, as it is the main component of natural gas that provides a significant environmental benefit, producing more energy by mass than other hydrocarbons4,5. Bearing these aspects in mind, a significant effort has been directed to design robust photocatalysts that can capture and reduce CO2 to CH4 selectively and efficiently1,6,7,8,9.
The introduction of supramolecular donor–acceptor (D–A) complex by post-synthetic modification within the confined nano-space of a metal-organic framework (MOF) can eventually alter the chemical environment and overall optoelectronic properties10. Through a rational supramolecular design approach with structural regulation at a molecular level, a catalytically active coordination nanospace can be engineered in a highly stable nanoscale MOF. However, a molecular design approach with remarkable precision and exquisite control needs to be implemented to achieve such a catalytically active site in a confined nanospace. The versatile and highly amenable structural tunability of MOFs by post-synthetic modification (PSM) allows tailoring inherent properties, including semiconducting and optoelectronic properties, compared to traditional inorganic semiconductors. By virtue, the PSM of a nanospace of MOF using an electron donor moiety would allow the introduction of a suitable acceptor moiety to attain a charge transfer (CT) complex without altering the integrity of the framework. Besides, it would be an excellent choice to establish a functional photosensitizer to galvanize the electronic and optical properties. Furthermore, the D–A interaction can be exploited for channeling the electron flow based on the push-pull effect to the catalytic center to carry out catalytic reactions. Additionally, the charge transfer complex would also result in a low energy absorption band to harvest visible light to generate photo-modulated electron-hole pair with low exciton binding energy9,11. Recently, MOFs have been extensively studied as a photocatalyst for CO2RR due to their promising CO2 capture ability, high surface area, and textural properties12,13,14,15,16,17,18. However, there are a handful of reports for MOF-based photocatalysts, which are studied in an aqueous medium for visible-light-driven CO2RR19,20,21.
Previous studies on ZrIV-based MOFs showed that the Zr-oxo cluster in PCN222 (ZrIV-porphyrin MOF) and PCN136 (ZrIV-hexakis(4-carboxyphenyl)hexabenzocoronene MOF) acts as an active catalytic site, where integrated organic linkers perform as an antenna for light-harvesting to activate the metal cluster on photoirradiation22,23. However, the reduction ability is mostly limited to a two-electron reduction process to produce CO or HCOOH. Herein, we have envisioned for post-synthetic fabrication of a donor-acceptor CT complex as a visible light-harvesting unit inside the nanospace of MOF, which would enhance the electron transfer kinetics to the catalytic center for CO2 reduction in an aqueous medium. Our design principle lies with the following consideration: the low energy CT band will help in visible light-harvesting; secondly, close confinement inside the coordination nanospace will provide a high excited-state lifetime of the photogenerated electrons by decreasing the nonradiative electron–hole recombination pathway; and thirdly, the proximity of catalytic center and the light-harvesting unit will enhance the kinetics of electrons transfer which is of paramount importance for highly reduced and selective CO2 reduction product19,24,25,26,27,28.
In this context, we envisaged mesoporous MOF-808 (Zr) as a suitable platform for PSM for its well-accessible formate to exchange with 1-pyrenebutyric acid (PBA) to produce a luminescent MOF (MOF-808-PBA) with a high excited-state lifetime19,29. Moreover, the prudent choice of defect-regulated mesoporous MOF-808 (Zr) can be justified by its large pore size, presence of hierarchical meso and microporosity, very high water/chemical stability combined with available unsaturated ZrIV metal site29,30,31. Further, a supramolecular D-A assembly was introduced using noncovalent grafting of methyl viologen (MV; an electron acceptor) inside the pore of MOF-808-PBA. Integrating the D–A module inside the pore surface can create an artificial “special-pair” like the system to facilitate fast charge transfer kinetics by driving the charge-separation process to reduce CO2 beyond the two-electron reduction process32. Hence, rapid electron transfer process from PBA → MV → catalytic site (Zr-oxo cluster) and suitable band position overcome the required electron injection for CO2RR to produce a highly reduced product in aqueous medium (Fig. 1). MOF-808-PBA-MV produced 7.3 mmol g−1 of CH4 with >99% selectivity suppressing H2 evolution in aqueous medium using BNAH (1-benzyl-1,4-dihydro-nicotinamide) and TEA (triethylamine) as a sacrificial agent. Furthermore, the reaction mechanism was established by in situ diffuse reflectance FT-IR (DRIFT), electron paramagnetic resonance (EPR) studies and transient absorption (TA) spectroscopy, which were well supported by density functional theory (DFT) calculation.

Here, the D–A complex acts as a light harvester to boost the electron flow near the catalytic site in the presence of light.
Results
Synthesis and characterizations of MOF-808-PBA and MOF-808-PBA-MV
MOF-808 was synthesized by a slight modification of the reported procedure and characterized using powder X-ray diffraction (PXRD), FT-IR, NMR spectroscopy and thermogravimetric analysis (Fig. 2a and Supplementary Figs. 1–3)30,33. TEM images of as-synthesized MOF-808 showed octahedral morphology with particle sizes in the range of 300-400 nm (Fig. 2b)1. The porosity of the MOF-808 was confirmed by performing N2 adsorption measurements at 77 K, and it showed type IV adsorption isotherm according to IUPAC classification, which suggested the presence of meso and micropore (Fig. 2c)34. The Brunauer–Emmett–Teller (BET) surface area was calculated to be 889 m2 g−1 in the range between P/P0 ̴ 0.05 and 0.3. The pore size distribution using the NLDFT method showed that the micropore centered at 1.6 nm, whereas the mesopores were distributed in between 3-10 nm (Supplementary Fig. 4). Accordingly, the micro and mesopore volume was evaluated to be 0.3 and 0.78 cm3 g−1, respectively, with Vmicro/Vmeso ratio of 0.39. The origin of the mesopore can be attributed to the missing linker defect, as reported earlier30,31. The hierarchical porosity is also endorsed by HRTEM analysis (Fig. 2b). As-synthesized MOF-808 showed sponge-like morphology with a clear contrast that can be attributed to the defects-based mesopore, which was randomly distributed throughout each particle35. HRTEM images also exhibited lattice fringes with an interplanar distance of 1.66 nm which can be credited to the inherent microporosity of the framework, which is also supported by the pore size distribution analysis19.

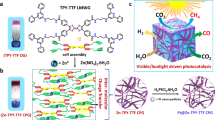
a Synthetic scheme: Construction of MOF-808-PBA as a donor module via post-synthetic linker exchange and then introducing methyl viologen (MV) as an electron acceptor to prepare MOF-808-PBA-MV. b HRTEM images of MOF-808. Magnified image shows the presence of micro and mesopore. c N2 adsorption isotherm of MOF-808, MOF-808-PBA, and MOF-808-PBA-MV at 77 K. d Solid-state UV–vis spectra of PBA, MV, MOF-808, MOF-808-PBA, and MOF-808-PBA-MV; inset showing the photograph of MOF-808-PBA (i) and MOF-808-PBA-MV (ii). e Photoluminescence spectra of MOF-808-PBA and MOF-808-PBA-MV. The inset shows a photograph of MOF-808-PBA and MOF-808-PBA-MV dispersed in MeOH under UV light. f Time-resolved luminescence decay of MOF-808-PBA and MOF-808-PBA-MV (λex = 330 nm, λcol = 480 nm).
At first, the internal pore of mesoporous MOF-808 was covalently modified with 1-pyrenebutyric acid (PBA) via solvent-assisted linker exchange (SALE) method by submerging MOF powder in PBA solution (MeOH/DMF) and the resulting luminescent MOF named as MOF-808-PBA (Supplementary Figs. 5–9)29,36. 1H-NMR spectra of digested MOF-808-PBA revealed that the formate peak integration diminished to one from five along with the presence of an additional peak related to the PBA molecule, indicating four formates of Zr6 cluster substituted by the PBA molecule (Supplementary Figs. 7 and 8)29. FT-IR spectra of MOF-808-PBA exhibited bands at ̴ 3000 and 846 cm−1 which corresponds to ν(C–H) symmetric and asymmetric stretching frequency of the PBA molecule, further confirming the successful encapsulation of PBA into the MOF pore (Supplementary Fig. 9)37. N2 adsorption measurement of MOF-808-PBA revealed a decrease in surface area from 889 to 413 m2 g−1 with a significant decrease of micropore volume to 0.148 cm3 g−1, whereas mesopore volume remained intact with Vmicro/Vmeso ratio of 0.191. This result suggested the micropore is occupied by the PBA during post-synthetic linker exchange (Fig. 2c and Supplementary Fig. 4). Here, mesoporosity helped the diffusion of the PBA molecules into the micropore during the SALE process. Pristine MOF-808 possessed an absorption band in the UV region (250–310 nm) due to the π → π* transition of the BTC linker19. Solid-state UV–vis spectra of MOF-808-PBA exhibited an additional broad absorption band from 310 to 500 nm with a maximum at 350 nm, along with a shoulder band at 420 nm, which is utterly different from vibronic monomeric spectral features of PBA (Fig. 2d and Supplementary Fig 10)38. Moreover, the solid-state UV–vis spectrum of PBA showed a broad absorption band from 250 to 700 nm (Fig. 2d). This result indicated ground-state aggregation of PBA molecules in the confined MOF pore, presumably through aromatic π–π stacking interactions. Afterward, photoluminescence (PL) measurements were carried out to obtain further insight into the optical properties of MOF-808-PBA. The PL spectra of MOF-808-PBA showed a red-shifted, broad, structureless excimer emission band at 480 nm (λex = 370 nm), which can be attributed to the close proximity of PBA molecules in the confined nanospace of MOF, hence enabling excimer formation (Fig. 2e, Supplementary Figs. 11, 12)38,39. An exceptionally long lifetime of the PBA-confined MOF-808 (τav = 23.2 ns) over PBA monomer (τav = 3.01 ns) further corroborates the formation of PBA excimer. (Fig. 2f, Supplementary Fig. 13 and Supplementary Table 1)40,41. Moreover, the confinement effect of MOF pore contributes to the restriction of the nonradiative pathway, which resulted in an unexpectedly higher quantum yield of 23.88% (solid-state) and a long lifetime of 23.2 ns compared to solid PBA ligand (QY% = 9.3%, τav = 14.51 ns) (Fig. 2f, Supplementary Fig. 14 and Supplementary Table 1)42.
Methyl viologen (MV2+) molecules were introduced as an acceptor in MOF-808-PBA to form a donor–acceptor supramolecular CT complex inside the confined nanospace. The UV–vis spectrum of the resulting dark brown color MOF-808-PBA-MV compound showed a distinct CT band from 450 to 700 nm with an absorption maximum at 540 nm (Fig. 2d and Supplementary Fig. 5). MOF-808-PBA-MV was digested with KOH/D2O/DMSO-d6 and subjected to 1H NMR measurement which suggested the precise ratio of PBA and MV2+ is of 4:3 per formula unit of MOF (Supplementary Fig. 15). Time-dependent density functional theory (TDDFT) calculation suggested the CT band between pyrenebutyric acid (PBA) and methyl viologen (MV) appears at 596.01 nm, which is in agreement with the experimental one (Fig. 3a). Detailed analysis of the results obtained from TDDFT calculation revealed that the band at 596.01 nm could be attributed to the transition from HOMO to LUMO + 1 (49.5% contribution), which is found to be the charge transfer transition from PBA to MV as obtained from the molecular orbital analysis (Supplementary Table 2). Next, the PL and the excited-state lifetime of the PBA in MOF-808-PBA-MV were studied to understand the charge transfer properties. As shown in Fig. 2e and Supplementary Fig. 16, the excimer emission of MOF-808-PBA was significantly quenched, reflecting the exciton quenching due to CT complex formation43. Quenching of the emission intensity of PBA excimer in MOF-808-PBA-MV was also observed under UV illumination (Inset of Fig. 2e). The generation of CT complex displayed a drastic decline of PBA lifetime from 23.2 to 1.2 ns (λex = 370 nm, λcol = 480 nm) (Fig. 2f, Supplementary Table 1). Further, EPR studies showed a single resonance peak at g = 1.98 of viologen radical, confirming the formation of charge-separated species (PBA+•-MV+•) inside the MOF pore (Supplementary Figs. 17, 18)44. Importantly, to understand the nature of the CT complex we also prepared the MOF-808-PBA-MV under completely dark condition designated as MOF-808-PBA-MV_d, which showed no recognizable EPR signal in that state, which implies daylight act as a stimulus to form the charge-separated species (details have been provided in the Supplementary Information; Supplementary Fig. 19). Apart from that, the redox signature of MV2+ from the cyclic voltammetry exhibited two reversible one-electron waves at \({{E}}_{1/2}^{1}=-0.38\,{{{{{\rm{V}}}}}}\) and \({{E}}_{1/2}^{2}=-0.78\,{{{{{\rm{V}}}}}}\) vs. Ag/AgCl corresponding to the MV2+/MV+•, and MV+•/MV redox transitions, respectively (Supplementary Fig. 20)45. The cyclic voltammetry of MOF-808-PBA-MV exhibited both the cathodic and anodic peaks corresponding to the generation of MV2+ ⇌ MV•+ and MV•+ ⇌ MV (Supplementary Fig. 21). To further clarify the decrease of the anodic peak corresponding to MV•+\(\to\) MV2+ formation, three consecutive CV cycles were scanned, where we noticed after 1st segment, the peak current corresponding to MV2+ ⇌ MV•+ was significantly decreased due to the formation of stable PBA•+-MV•+ CT complex inside the MOF pore. The PXRD and HRTEM analysis of MOF-808-PBA-MV confirmed that the structural integrity and morphology of pristine MOF remained intact after PSM (Supplementary Figs. 1 and 22–24). In addition, MOF-808-PBA-MV possessed BET surface area of 357 m2 g−1 with a significant contraction of micropore volume to 0.09 cm3 g−1, whereas mesopore volume remained almost constant with Vmicro/Vmeso ratio of 0.125 (Fig. 2c and Supplementary Fig. 4). All these experiments suggest the formation of supramolecular donor–acceptor CT complex in the confined nanospace of MOF. Next, CO2 adsorption measurement of MOF-808-PBA-MV was carried out at 298 K and showed an uptake of 12 cm3 g−1 at saturation, suggesting that CO2 can diffuse easily to the catalytic centre during photocatalytic CO2 reduction (Fig. 3b).

a Electron transfer feasibility from PBA-MV to Zr-cluster using DFT. b CO2 adsorption of MOF-808-PBA-MV at 298 K. c Mott–Schottky (MS) plot for MOF-808-PBA-MV in 0.2 M Na2SO4 aqueous solution. d HOMO–LUMO band position diagram for MOFs (MOF-808, MOF-808-PBA, MOF-808-PBA-MV) obtained from Mott–Schottky (MS) and DRS. e Tauc plot of MOF-808-PBA-MV evaluating the optical bandgap.
A low energy CT band appeared due to the formation of donor–acceptor CT complex inside the MOF pore, which can render a convenient path to regulate the electron flow based on visible light harvesting toward the Zr-oxo cluster, an active catalytic site for successive CO2 reduction. Apart from that, for efficient CO2 reduction, the photocatalyst is required to have suitable conduction and valence band. The band energy levels of MOF-808, MOF-808-PBA, and MOF-808-PBA-MV were examined using flat band potential from Mott-Schottky (MS) measurement (Fig. 3c and Supplementary Figs. 25a, 26a). All the MOFs exhibited positive slopes from C−2 values (vs. applied potential), which are consistent with the typical n-type nature of the semiconductor. Figure 3d summarized the band energy level of pristine MOF-808 as well as post-synthetically modified MOFs. From the MS measurement, LUMO of pristine MOF-808 was calculated, which was estimated to be −0.51 V vs. Ag/AgCl (i.e. −0.31 V vs. NHE at pH 7) (Supplementary Fig. 25a). Based on the Tauc-plot, the optical band gap of MOF-808 was calculated to be 4.06 eV (Supplementary Fig. 25b). The disfavored band alignment of pristine MOF clearly signifies the inability to reduce CO2. Upon successive grafting of PBA-MV into the MOF pore, the position of LUMO changed dramatically from −0.31 V (MOF-808) to −0.40 V (MOF-808-PBA) to −0.67 V (MOF-808-PBA-MV) vs. NHE at pH 7 (Fig. 3c and Supplementary Fig. 25a, 26a). Next, the optical band gap was calculated from the Tauc plot and found to be 2.75 and 1.93 eV for MOF-808-PBA, MOF-808-PBA-MV, respectively (Fig. 3e and Supplementary Fig. 26b). Accordingly, the valence band (HOMO) of MOF-808-PBA and MOF-808-PBA-MV was calculated to be 2.35 and 1.26 V (vs. NHE at pH 7), respectively. Hence, it can be concluded the introduction of supramolecular donor–acceptor CT complex in the confined nanospace of MOF-808 resulted in an energetically favorable LUMO to carry out CO2 reduction46. In addition, their energy level was further investigated by ultraviolet photoelectron spectroscopy (UPS) (Supplementary Fig. 27, Supplementary Table 3) which showed a high accordance with the MS measurement47,48. As aforementioned, all the optical and electronic features are hugely advantageous for visible-light-driven photocatalytic applications. To understand the importance of the homogeneous distribution of the CT complex in MOF-808-PBA-MV, a physical mixture of MOF-808, PBA, and MV was prepared (MOF-808(PBA + MV)-Phy mix.) (Supplementary Figs. 28, 29). This physical mixture can be considered a heterogeneous (supramolecular CT + MOF) system. It is worth mentioning that MOF-808(PBA + MV)-Phy mix. showed minimal CO2 reduction activity compared to MOF-808-PBA-MV (Supplementary Table 7). This poor activity can be attributed to the unstable CT complex in MOF-808(PBA + MV)-Phy mix. which disintegrates in the catalytic condition as realized in UV–vis spectra of the catalytic solution (Supplementary Fig. 29).
Photocatalytic CO2 reduction reaction

a The amount of CH4 and H2 evolution by MOF-808-PBA-MV as a function of time under visible light irradiation in water medium using BNAH and TEA as a sacrificial electron donor; error bars mean ± standard deviations calculated from three independent measurements. b Mass spectrum of produced 13CH4 (m/z = 17) via isotopic 13CO2 reduction under visible light over MOF-808-PBA-MV. c Comparison of photocatalytic product generation by using MOF-808-PBA-MV along with MOF-808-PBA and homogeneous PBA-MV in a water medium. d Control experiments of photocatalytic CO2 reduction reaction over MOF-808-PBA-MV under different conditions. e The amount of CH4 and H2 evolution by MOF-808-PBA-MV under sunlight irradiation in a water medium using BNAH and TEA as a sacrificial electron donor after 6 h.
The catalytic activity of MOF-808-PBA-MV was assessed in a CO2-saturated aqueous medium under visible light irradiation (300 W Xenon Arc lamp, λ > 400 nm) in the presence of 1-benzyl-1,4-dihydro-nicotinamide (BNAH) and triethylamine (TEA) as a sacrificial agent. Remarkably, MOF-808-PBA-MV showed impressive activity for CO2RR towards CH4 formation with >99% selectivity, with efficient suppression of H2 evolution in the aqueous medium. The gaseous products were monitored by gas chromatography and represented in Fig. 4a. The CH4 production was continuously increased with irradiation time, and a total of 7.3 mmol g−1 was produced in 16 h (Fig. 4a and Supplementary Figs. 30, 31). The maximum production rate was calculated to be 460 μmol g−1 h−1 with a maximum TON of 28 for CH4 formation, which is exceptionally high for photocatalytic CO2 reduction in the water medium (Supplementary Fig. 32, Supplementary Tables 4, 5). In this process, 0.369 mmol g−1 of H2 was also generated. Apparent quantum efficiency tests (AQE) were also performed with different monochromatic wavelengths. As illustrated in Supplementary Fig. 33, the highest AQE of 1.4% was achieved at a wavelength of 550 nm, which strongly suggested the charge transfer-assisted photocatalytic activity of MOF-808-PBA-MV (Supplementary Table 6, see Supplementary Information). To confirm the generated CH4 originated from the photocatalytic reduction of dissolved CO2, we performed the reaction using isotopic 13CO2. A peak at m/z = 17 clearly demonstrated that produced 13CH4 was generated from the photoreduction of dissolved CO2 in a water medium (Fig. 4b and Supplementary Fig. 34). To unveil the role of CT complex inside the MOF pore in prompting photocatalytic CO2 reduction, several control experiments were conducted under a similar reaction condition and discussed in the Supplementary information demonstrating the important role of electron flow in a confined nanospace (Fig. 4c, Supplementary Figs. 35, 36 and Supplementary Table 7). Furthermore, no detectable amount of product was obtained when the reaction was carried out without a catalyst or in the dark or in an Ar atmosphere (Fig. 4d, Supplementary Table 7). In CO2-saturated aqueous solution in the presence of only TEA, MOF-808-PBA-MV exhibited 1.2 mmol g−1 of CH4 along with 0.456 mmol g−1 of CO and 0.291 mmol g−1 of H2 in 16 h irradiation time (Fig. 4d, Supplementary Table 7). Similarly, the employment of BNAH as a sole sacrificial electron donor in water medium, MOF-808-PBA-MV produced 3.5 mmol g−1 of CH4 along with 0.230 mmol g−1 of CO and 0.265 mmol g−1 of H2 in 16 h irradiation time (Fig. 4d, Supplementary Table 7). Importantly, the combination of BNAH and TEA played a significant role in the process of CO2 reduction reaction to CH4 formation over MOF-808-PBA-MV49. Here, BNAH acts as an efficient hole scavenger to enhance the catalytic activity. However, TEA was required for efficient CO2RR as it suppressed back electron transfer from the reduced photosensitizer to BNAH•+50. Finally, the BNA radical (BNA•) produced by the deprotonation of BNAH•+ was further dimerized to produce BNA2, which was confirmed from the LCMS data (Supplementary Figs. 37, 38). Moreover, the reduction power of BNA2 (E0 = 0.26 V vs. SCE) is stronger than BNAH (E0 = 0.57 V vs. SCE) and, therefore, efficiently quenched the PBA+ photosensitizer51,52. Further, to understand the role of the defect in mesoporous MOF-808, we have also prepared a microporous analog of the catalyst (details have been provided in the Supplementary Information; Supplementary Figs. 39–43). A four-fold enhanced catalytic activity was observed in mesoporous MOF-808-PBA-MV (CH4: 7.3 mmol g−1) as compared to microporous MOF-808-PBA-MV (CH4: 1.8 mmol g−1), which can be attributed to the increased availability of coordinatively unsaturated ZrIV sites in the Zr-oxo cluster (Supplementary Fig. 44)31. Catalytic efficiency of MOF-808-PBA-MV was evaluated under direct sunlight irradiation on our laboratory’s rooftop from 5 to 11 October (2020). The catalytic efficiency was weather dependent as expected, and we achieved the highest CH4 production of 1.020 mmol g−1 on a sunny day, 11th Oct, after 6 h of sunlight irradiation (Fig. 4e, Supplementary Fig. 45). 1H NMR and high-performance liquid chromatography (HPLC) were carried out to analyze the liquid product. 1H NMR spectra suggested the presence of a trace amount of formate, which was formed as an intermediate of CH4 formation during the catalysis (Supplementary Fig. 46). No other liquid products were detected, further confirmed by HPLC analysis (Supplementary Fig. 47). In addition, MOF-808-PBA-MV was easily recovered from the reaction medium by centrifugation for post-catalytic analysis and again used for catalytic activity. Examining the stability of MOF-808-PBA-MV in the recycling experiment, it showed a negligible drop in CH4 production even after six consecutive cycles with a total of 66 h of light irradiation. (Supplementary Fig. 48). Additional experiment was performed using CO to verify the catalytic performance towards CO reduction. After the photocatalytic reaction, CH4 was found to be a major product with a production of 10.4 mmol g−1 in 16 h (Supplementary Fig. 49). In this case, the CH4 production is higher compared to photocatalytic CO2RR, which is obvious due to the conversion of CO2 to CO is a more thermodynamically uphill process. Post-catalytic analysis of MOF-808-PBA-MV manifested the structural integrity and retention of optoelectronic properties (Supplementary Figs. 50–54).
The enhanced photocatalytic CO2 reduction to CH4 over MOF-808-PBA-MV as compared to its donor module MOF-808-PBA can be justified by transient photocurrent measurement and impedance studies. The photo-responsive behavior was dramatically enhanced due to the formation of the CT complex (Fig. 5a). The measured photocurrent of MOF-808-PBA-MV was 11.24 μA cm−2 whereas; MOF-808-PBA exhibited a photocurrent of 1.18 μA cm−2, indicating a tenfold increase in photocurrent after the formation of the CT complex. Additionally, this experiment was also measured in the presence of a sacrificial electron donor and found a slight enhancement of current density (13.26 μA cm−2) was observed (Supplementary Fig. 55). The enhanced photo-induced current upholds the superior catalytic activity of MOF-808-PBA-MV. This assertion was also supported by the electrochemical impedance spectroscopy (Supplementary Fig. 56). MOF-808-PBA-MV showed a much smaller radius of semi-circle as compared to MOF-808-PBA, indicating a small charge transfer resistance. Moreover, after irradiation with visible light, the semicircle of MOF-808-PBA-MV got further decreased, suggesting a fast electron transfer kinetics.

a Transient photocurrent responses of MOF-808-PBA and MOF-808-PBA-MV in 0.2 M Na2SO4 aqueous solution under visible-light irradiation. b EPR spectra of MOF-808-PBA-MV in dark and under visible light irradiation. Magnified EPR spectra of MOF-808-PBA-MV showing the formation of ZrIII species. c Transient absorption spectra of MOF-808-PBA-MV at different time delays. d, e The transient decays at 440 and 650 nm. Solid lines in panels d and e are fitted data.
Exploration of the charge transfer pathway
To uncover the mechanism behind photocatalytic activity and gain deeper insight into the electron transfer process in MOF-808-PB-MV, EPR spectra were collected with light irradiation (Fig. 5b). As expected, the intensity of the signal at a g value of 1.98 increased significantly due to the increased concentration of charge-separated species [PBA•+-MV+•] into the MOF pore. Most importantly, a characteristic EPR signal at g = 2.01 was detected due to the formation of ZrIII species, which was revealed only upon light irradiation (Fig. 5b)53. Hence, the excited state electron generated from the CT complex is injected into the Zr-oxo cluster to reduce CO2 to CH422,54. To further support this statement, time-resolved photoluminescence (TR-PL) spectroscopy of MOF-808-PBA-MV was performed both in Ar and CO2 atmosphere (Supplementary Fig. 57 and Supplementary Table 8), which exhibited an average lifetime of 1.74 and 0.92 ns, respectively, suggesting that CO2 binding with the Zr-oxo cluster promoted fast excited state electron transfer kinetics from PBA-MV to the catalytic centre55.
The charge transfer interaction between PBA and MV was further investigated using femtosecond (fs) transient absorption (TA) techniques. The TA spectra (Fig. 5c) of MOF-808-PBA-MV showed a narrow excited state absorption (ESA) band at 440 nm due to the cationic pyrene moiety, PBA•+ and a very broad ESA band around 550–700 nm due to MV+•56. The instantaneous appearance of the PBA•+ and MV+• indicates very efficient electron transfer which takes place within the time resolution of our TA instrument (120 fs). The absence of the electron transfer process in a homogeneous solution of pyrene and MV2+ (Supplementary Fig. 58) suggests the importance of confinement in the electron transfer process. The decay kinetics of PBA•+ (440 nm) and MV+• (650 nm) was analyzed by multiexponential decay function, and the fitted parameters are 110 ps (49%) and 2460 ps (51%) for PBA•+ and 13.3 ps (11%), 73 ps (35%), 1250 ps (38%), 5 ns (17%) for MV+• (Fig. 5d, e). The 5 ns decay component at 650 nm is due to the pyrene excimer (see supplementary information)57. Distinct decay time constants for PBA•+ and MV+• suggested the absence of the energy-wasting back electron transfer process in MOF-808-PBA-MV. Significantly shorter lifetime of MV+• (average lifetime ~507 ps) in MOF-808-PBA-MV as compared to microsecond lifetime in other Zr-based MOF system58 also suggested enhanced transfer of electrons from MV+• to the catalytic center of MOF-808. Moreover, the frontier orbitals (FMOs) of PBA-MV and Zr-cluster of the MOF are aligned based on TDDFT analysis. LUMO of Zr-cluster (−2.532 eV) was found to be lower in energy than LUMO + 1 of PBA-MV (−2.286 eV). This indicates the feasibility of the photoexcited electron transfer from LUMO + 1 of PBA-MV photosensitizer to the LUMO of the Zr-cluster (Fig. 3a).
Insight into the CO2 reduction mechanism
Subsequently, in situ DRIFT measurement was performed to reveal the reaction intermediate during the CO2 reduction process (Fig. 6, Supplementary Fig. 59). The MOF-808-PBA-MV photocatalyst was first degassed at 120 °C to remove all the adsorbed gases. Before the introduction of CO2 and H2O vapor into the reactor, an FT-IR spectrum of the catalyst was recorded and selected as background to suppress catalyst peaks. A peak at 1377 cm−1 appeared after introducing CO2, and water vapor into the reactor could be assigned as monodentate carbonate (m-CO32−)59. The peak at 1436 cm−1 could be assigned to symmetric stretching of HCO3− 1. After light irradiation, several new peaks appeared. A peak that emerged at 1242 cm−1 can be assigned to the CO2− stretching59. Most importantly, a peak at 1619 cm−1 was detected, which can be assigned as COOH* species1. COOH* is a very crucial intermediate during CO2 reduction to CH4 via the formate pathway. In addition, a strong band at 1723 cm−1 appeared, which could be attributed to the bending vibration of C = O60. In the meantime, a small band appeared at 2101 cm−1, assigned to CO coordinated with metal61. Importantly, the absorption band at 1855, 1766, and 1111 cm−1 belongs to CHO*, CH2O*, and CH3O* species, respectively, which are pivotal intermediates for CH4 formation1,62,63. Besides, the band at 2872, 2933, 2970, 3035, and 1304 cm−1 were assigned to C–H stretching vibrations, which clearly suggests the formation of CH4 (see the supplementary information for more details)60. Furthermore, we have also performed an in-situ DRIFT experiment in the presence of BNAH and TEA, which followed a similar pathway as mentioned above (details have been provided in the Supplementary Information; Supplementary Fig. 60). In addition, the in-situ DRIFT study using isotopic CO2 (13CO2) provides an insightful conclusion, where we observed red shifted stretching bands corresponds to the crucial intermediate (Supplementary Fig. 61)64. The mechanistic aspect was further clarified by using CO as a feeding gas instead of CO2 (Supplementary Fig. 62).

Time-dependent in situ FT-IR spectra of photocatalytic CO2RR in a mixture of CO2 and H2O vapor over MOF-808-PBA-MV in the dark and under visible light irradiation. Irradiation times 10, 20, 30, 40, 50, 60, 70, 80 min, respectively.
Based on the DRIFTS study and DFT calculations, we have proposed a possible catalytic cycle for the photocatalytic CO2RR by MOF-808-PBA-MV (Fig. 7a, b, Supplementary Tables 9–36). For theoretical calculations, we have designated the MOF-808 catalyst, having the formula [Zr6(μ3-O)5(μ3-OH)3(HCOO)5(BTC)2(H2O)2] as f-[ZrIV(BTC)2(H2O)2] (f = Zr5(μ3-O)5(μ3-OH)3(HCOO)5, Zr-cluster fragment). Upon photoirradiation, the photogenerated hole of [PBA+-MV+]2+* will get reductively quenched by sacrificial electron donor BNAH (Supplementary Fig. 63), and the photoexcited electron will readily get transferred to the BTC ligand of Zr-cluster, and thus the catalytic CO2 reduction will be initiated on Zr-cluster as reported earlier22,23,24,54,65. As compared to microporous MOF-808, missing linker (formate) defect-regulated mesoporous MOF contains open metal sites (ZrIV) which are considered to be the active catalytic centers31. One electron reduction of f-[ZrIV(BTC)2(H2O)2] (1) generates f-[ZrIV(BTC.−)(BTC)(H2O)]− (2) with the removal of one water molecule (ΔG = −0.48 eV). The generation of transient state in f-[ZrIV(BTC.−)(BTC)(H2O)]− with BTC.− (2) is verified by the spin density distribution plot where the spin density of the extra electron is clearly seen on BTC ligand (Supplementary Table 12). Moreover, f-[ZrIV(BTC.−)(BTC)(H2O)]− (2) (Supplementary Table 12) is the active species for the catalytic cycle. Next, either reduction of f-[ZrIV(BTC.−)(BTC)(H2O)]− (2) followed by oxidative addition of CO2 to f-[ZrIII(BTC.−)(BTC)(H2O)]2− (3) (Fig. 7a and Supplementary Table 13) or first, oxidative addition of CO2 to f-[ZrIV(BTC.−)(BTC)(H2O)]− (2) followed by reduction of f-[ZrV(BTC)(BTC)(H2O)(CO22−)]− (4΄) (Supplementary Fig. 64, Supplementary Table 14) may occur. The binding of CO2 with f-[ZrIII(BTC.−)(BTC)(H2O)]2− (3) is an exergonic process (ΔG = −0.72 eV) with a lower activation barrier (ΔG⧧ = 0.55 eV) to generate CO2 bound species whereas the CO2 binding pathway with f-[ZrIV(BTC.-)(BTC)(H2O)]− (2) is an endergonic (ΔG = +0.36 eV) with a higher activation barrier (ΔG⧧ = 0.60 eV) (Fig. 7b and Supplementary Tables 16, 17). This inferred that the CO2 binding to f-[ZrIII(BTC.−)(BTC)(H2O)]2− (3) is the major pathway under the experimental conditions. This is quite expected because a lower oxidation state on Zr prior to the binding of CO2 will facilitate the oxidative addition of CO2. The next step in the photocatalytic cycle involves the protonation of CO2-bound intermediate f-[ZrIV(BTC)(BTC)(H2O)(CO22−)]2− (4) (Supplementary Table 15) leading to f-[ZrIV(BTC)(BTC)(H2O)(COOH−)]− (5) (ΔG = −2.07 eV) (Supplementary Table 18) which after subsequent protonation and water elimination will afford f-[ZrIV(BTC)(BTC)(H2O)(CO)] (6) (ΔG = −1.54 eV) (Supplementary Table 19). After that, one-electron reduction of f-[ZrIV(BTC)(BTC)(H2O)(CO)] (6) will lead to f-[ZrIV(BTC.−)(BTC)(H2O)(CO)]− (7) (ΔG = −0.77 eV) (Supplementary Table 20) which can either release CO molecule to regenerate f-[ZrIV(BTC.−)(BTC)(H2O)]− (2) or undergo proton-coupled reduction to form f-[ZrIV(BTC.−)(BTC)(H2O)(CHO)]− (8) (Supplementary Table 21) and enter the CH4 formation pathway. However, proton-coupled reduction of f-[ZrIV(BTC.−)(BTC)(H2O)(CO)]− (7) to f-[ZrIV(BTC.−)(BTC)(H2O)(CHO)]− (8) is highly exergonic (ΔG = −2.45 eV) compared to the release of CO from f-[ZrIV(BTC.−)(BTC)(H2O)(CO)]− (7) (ΔG = −0.34 eV) which indicates preferential higher- order reduction up to CH4 rather than the generation of CO. In the following steps of the photocatalytic cycle, the successive proton couple reduction of f-[ZrIV(BTC.−)(BTC)(H2O)(CHO)]− (8) will lead to f-[ZrIV(BTC.−)(BTC)(H2O)(CH3OH)]− (11), where all the intermediate steps are highly exergonic (Fig. 7a, b and Supplementary Tables 21–24). In the next step, proton-coupled reduction and subsequent water elimination from f-[ZrIV(BTC.−)(BTC)(H2O)(CH3OH)]− (11) intermediate will lead to the formation of f-[ZrIV(BTC.−)(BTC)(H2O)(CH3)]− (12) (Supplementary Table 25) which is found to be highly exergonic (ΔG = −2.47 eV). Finally, f-[ZrIV(BTC.−)(BTC)(H2O)(CH3)]− (12) intermediate will readily undergo proton-coupled reduction to result in the release of CH4 (ΔG = −2.71 eV), and thus the active species f-[ZrIV(BTC.−)(BTC)(H2O)]− (2) will get regenerated to re-enter into the catalytic cycle. The highly exergonic reduction pathway of f-[ZrIV(BTC.−)(BTC)(H2O)(CH3OH)]− (11) (∆G = −2.47 eV) suppressed the endergonic desorption of CH3OH from f-[ZrIV(BTC.-)(BTC)(H2O)(CH3OH)]− (11) (∆G = +0.28 eV) which explained the non-production of CH3OH (Fig. 7b). Furthermore, the role of proximal H2O was also investigated by DFT calculation, which revealed that the proximal H2O facilitates the desorption of hydrophobic CH4 (ΔG = −2.71 eV) (Supplementary Fig. 65). This result suggests the positive impact of the existing proximal H2O in releasing off the hydrophobic CH4 product to enhance desorption.

a Schematic illustration of the proposed reaction mechanism for the CO2-to-CH4 photo-conversion on MOF-808-PBA-MV in water (f = Zr-cluster fragment). b Free energy diagram for the photocatalytic CO2 reduction to CH4 promoted by MOF-808-PBA-MV.
Discussion
In summary, we have created a simple yet highly efficient integrated system by pore modulation of MOF-808 for visible-light-driven CO2 reduction to CH4 selectively and efficiently with excellent yield in an aqueous medium. The embedded CT complex comprising of PBA and MV was exploited as a backbone for the electron transfer process to provide photoexcited electrons to the ZrIV catalytic center to facilitate challenging multielectron reduction. The CT complex-induced electron delivery for the utilization of CO2RR is the first of its kind to the best of our knowledge. Encouragingly, this strategy is beneficial to achieve CH4 formation with >99% selectivity with excellent yield by suppressing H2 and other carbonaceous products. The mechanism was well explored through in situ DRIFTS by identifying reaction intermediates. Apart from that, EPR was utilized to identify ZrIII as an active catalytic site for CO2 photoreduction, and the rapid electron transfer kinetics was recognized from TA spectroscopy. Additionally, a catalytic cycle was constructed along with Gibbs’s free energy pathway to visualize detailed mechanistic insight and energetics of the reaction intermediates with the help of DFT calculation. This work provides the utilization of a D–A complex inside a confined nanospace of MOF that can lead to a design protocol to construct a highly efficient heterogeneous catalyst towards selective photoreduction of CO2 to a highly reduced product.
Methods
Reagents
All the reagents were commercially available and used as such without further purification. Zirconium tetrachloride (ZrCl4·8H2O, purity ≥ 98%), Zirconium oxychloride octahydrate (ZrOCl2·8H2O, purity ≥ 99.5%) 1,3,5-Benzenetricarboxylic acid (BTC), 1-Pyrenebutyric acid (PBA), and Methyl viologen (MV) were purchased from Sigma-Aldrich Chemical Co. Ltd. Dimethylformamide (DMF), formic acid (purity > 98%), anhydrous acetone and anhydrous methanol were purchased from Spectrochem Pvt. Ltd. All the photophysical studies were carried out using HPLC grade solvents. 13CO2 cylinder was purchased from Sigma-Aldrich Chemical Co. Ltd.
Preparation of mesoporous MOF-808
Mesoporous MOF-808 was synthesized by following a slightly modified reported procedure33. Benzene-1,3,5-tricarboxylic acid (H3BTC) (0.282 g, 1.3 mmol) and ZrCl4 (0.932 g, 3.9 mmol) were dissolved in 20 mL of formic acid and 40 mL of DMF and placed in a screw-capped glass vial and heated at 130 °C for one day. Afterward, the mixture was cooled to room temperature in the oven, and the precipitate was filtered by vacuum filtration and washed with DMF (2 × 40 mL), acetone (2 × 40 mL), and methanol (3 × 40 mL). The resulting precipitate was dried in the air followed by activating at 150 °C for 24 h under a vacuum to yield the desired compound. 1H solution NMR spectra of the digested MOF-808 (400 MHz, D2O, ppm) δ: 8.1 (s, BTC), 8.2 (s, HCOOH), peak area ratio (BTC: HCOOH) = 6.0:5.0. Isolated yield 80% based on Zr.
Preparation of MOF-808-PBA
In a 15 mL glass vial, 5 mL 0.1 M DMF/MeOH (1:2) solution of pyrenebutyric acid (PBA) molecule was added to 0.100 g of activated MOF-808. The reaction mixture was placed in a 60 °C oven for 15 days. Notably, during this period, the PBA molecule solution was replaced after 3 days. After 15 days, a pale yellow color MOF was collected by filtration and washed several times with methanol to remove the unreacted PBA molecule. The exchanged product was dried in an oven at 60 °C under a vacuum. 1H-NMR spectra of digested MOF-808-PBA (400 MHz, D2O, ppm) δ: 8.1 (s, BTC), 8.21 (s, HCOOH), peak area ratio (BTC:HCOOH) = 6.0:2.0.
Preparation of MOF-808-PBA-MV
PBA exchanged MOF, MOF-808-PBA (70 mg) was added to a solution containing Methyl viologen (5 mg, 23.8 mmol). A dark brown color compound was formed immediately after adding MV. The suspension was kept in a 60 °C oven for one day. The resulting powder sample was filtered and washed with water to remove any unreacted methyl viologen, affording MOF-808-PBA-MV.
Physical measurement
Elemental analysis was performed in Thermo Scientific Flash 2000 CHN analyzer. Bruker FT-IR spectrometer was used to record infrared spectra. Samples were prepared as pellets using KBr for IR measurement. 1H NMR spectra were recorded on Bruker AVANCE-400 spectrometer (at 400 MHz) with chemical shifts recorded as ppm. Mettler Toledo-TGA 850 instrument was used to measure the thermal stability in the N2 atmosphere within the temperature range 30–800 °C at a heating rate of 5 °C/min. Powder X-ray diffraction patterns for all the MOF samples were recorded in Bruker D8 discover instrument using Cu-Kα radiation. Morphological studies have been carried out using Bruker Leica-S440I field emission scanning electron microscope (FE-SEM) by placing samples on a silicon wafer under a high vacuum with an accelerating voltage of 100 kV. Transmission electron microscopy (TEM) analysis has been performed using JEOL JEM-3010 with an accelerating voltage at 300 kV. N2 adsorption measurements were carried out at 77 K in a QUANTACHROME QUADRASORD-SI analyzer. Samples were degassed at 150 °C and 1 × 10−1 Pa vacuum for 12 h before performing sorption isotherm measurement. UV–vis studies were performed in the solid state using Perkin Elmer Model Lambda 900 spectrophotometer instrument. The photoluminescence properties were performed in Fluorolog 3.21 spectrofluorimeter (Horiba Jobin-Yvon) instrument. The photoluminescence (PL) quantum yield was determined by an absolute method using the same fluorescence spectrometer equipped with a 120 mm integrating sphere with a BENFLEC-coated inner face (Horiba Instruments). Fluorescence decay profiles were recorded using a Horiba Delta Flex time-correlated single-photon-counting (TCSPC) instrument. A 405 nm laser diode with a pulse repetition rate of 1 MHz was used as the light source. The instrument response function (IRF) was collected using a scatterer (Ludox AS40 colloidal silica, Sigma-Aldrich).
Photocatalytic reaction
The stock solution was prepared by dispersing 7 mg of finely powdered sample in 30 mL water and sonicated for 30 min. In the photocatalytic reduction of CO2 for each experiment, 4 mL stock solution was taken, and 2 mL water was added. The experiments were carried out in a 30 mL stopper vessel with magnetic stirring containing 6 mL of catalyst solution along with 9.3 μM of BNAH. Highly pure CO2 (99.99%) gas was passed into the reactor containing a well-dispersed catalyst solution for 40 min. Then, 1 mL TEA was added to the reaction mixture as a base and sealed the setup making a total volume of 7 mL of the reaction mixture and 23 mL of void space. The reaction mixture was irradiated with a 300 W Xenon lamp (Newport) through a visible bandpass filter (400–800 nm) with constant stirring. We measured the intensity of the light source using a power meter (Newport-843-R). The measured intensity during the catalytic reaction (λ = 400–800) is 100 mW/cm2 (Note: The distance between the light source and the reactor is 8 cm.). The amount of CO and CH4 evolved was analyzed using gas chromatography–mass spectrometry (SHIMAZU GC-2010 PLUS). CO and CH4 were quantified by using RT® Molecular sieve 5 A column (45 m, 0.32 mm ID, 30 μm df) with a mass detector. The calibration was done by a standard gas mixture of H2, CO, and CH4 of different concentrations at ppm-level. Importantly, GC–MS has a detection limit of 1.0 ppm for H2, CO, and CH4. After the photocatalysis, the reaction mixture was filtered to remove the residual solid, and the solution was further analyzed to determine the amount of liquid product. The liquid product was detected using a RID detector in High-performance liquid chromatography (Agilent Infinity1260) with Hi-Plex H column (300 × 7.7 mm) and 5 mM H2SO4 aqueous solution as eluent. The liquid product was also verified with the NMR technique using the solvent suppression method. For 13CO2 and CO reduction reactions, we purged the reaction mixture with 13CO2 and CO, respectively. Ultraviolet photoelectron spectroscopy (UPS) was performed using a thermo-scientific K-alpha X-ray photoelectron spectrometer (Source He(I)−21.22 eV).
Computational details
The density functional theoretical (DFT) calculations are performed using the Gaussian 16 package of programs66. The plausible catalytic cycle and the corresponding free energy diagram for the photocatalytic CO2 reduction to CH4 by π-stacked PBA-MV moiety integrated inside the MOF pore as a photosensitizer and the MOF, f-[ZrIV(BTC)2(H2O)2] itself as catalyst (where f = Zr5(μ3-O)5(μ3-OH)3(HCOO)5) are established from the theoretical study. The optimizations are performed utilizing B3LYP67,68,69,70,71,72 exchange-correlation functional along with 6–31G(d) basis set for all atoms except Zr. LANL2DZ is used as a basis set as well as ECP for Zr73,74. The polarizable continuum model (PCM) is utilized to account for the solvation effect of water75. Grimme’s d3 dispersion is also used to tackle weak interactions76. The harmonic vibrational frequency analysis of the optimized geometries is performed to confirm the nature of stationary points. The absence of any imaginary vibrational mode in the frequency calculations of all the optimized intermediates suggest the optimized structures to be the minima on the potential energy surface (PES). The electronic absorption spectra are calculated using the time-dependent DFT (TDDFT) method. For this, B3LYP exchange-correlation functional along with 6-31+G (d,p) basis set is utilized, whereas the solvent effect (water) and dispersive interactions are tackled by PCM and Grimme’s d3 methods, respectively. Molecular orbital (MO) pictures and the spin density distribution plots are generated using GaussView 6.0.1677.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Li, X. et al. Selective visible-light-driven photocatalytic CO2 reduction to CH4 mediated by atomically thin CuIn5S8 layers. Nat. Energy 4, 690–699 (2019).
Rej, S. et al. Well-defined Cu2O photocatalysts for solar fuels and chemicals. J. Mater. Chem. A 9, 5915–5951 (2021).
Wu, J. et al. CO2 reduction: from the electrochemical to photochemical approach. Adv. Sci. 4, 1700194 (2017).
Song, H. et al. Solar-energy-mediated methane conversion. Joule 3, 1606–1636 (2019).
Ulmer, U. et al. Fundamentals and applications of photocatalytic CO2 methanation. Nat. Commun. 10, 3169 (2019).
Li, J. et al. Self-adaptive dual-metal-site pairs in metal-organic frameworks for selective CO2 photoreduction to CH4. Nat. Catal. 4, 719–729 (2021).
Neaţu, Ş. et al. Gold–copper nanoalloys supported on TiO2 as photocatalysts for CO2 reduction by water. J. Am. Chem. Soc. 136, 15969–15976 (2014).
Rao, H. et al. Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 548, 74–77 (2017).
Verma, P. et al. Charge-transfer regulated visible light driven photocatalytic H2 production and CO2 reduction in tetrathiafulvalene based coordination polymer gel. Nat. Commun. 12, 7313 (2021).
Leong, K. et al. Energy and charge transfer by donor–acceptor pairs confined in a metal-organic framework: a spectroscopic and computational investigation. J. Mater. Chem. A 2, 3389–3398 (2014).
Jeevan, A. K. et al. Self-assembly and photochemistry of a pyrene-methyl viologen supramolecular fiber system. J. Phys. Chem. B 125, 8539–8549 (2021).
Liu, Y. et al. Phase-enabled metal-organic framework homojunction for highly selective CO2 photoreduction. Nat. Commun. 12, 1231 (2021).
Jiang, Z. et al. Filling metal-organic framework mesopores with TiO2 for CO2 photoreduction. Nature 586, 549–554 (2020).
Xiao, J.-D. et al. Metal-organic frameworks for photocatalysis and photothermal catalysis. Acc. Chem. Res. 52, 356–366 (2019).
Trickett, C. A. et al. The chemistry of metal-organic frameworks for CO2 capture, regeneration and conversion. Nat. Rev. Mater. 2, 17045 (2017).
Benseghir, Y. et al. Co-immobilization of a Rh catalyst and a Keggin polyoxometalate in the UiO-67 Zr-based metal-organic framework: in depth structural characterization and photocatalytic properties for CO2 reduction. J. Am. Chem. Soc. 142, 9428–9438 (2020).
Wang, S. et al. Cobalt imidazolate metal-organic frameworks photosplit CO2 under mild reaction conditions. Angew. Chem. Int. Ed. 53, 1034–1038 (2014).
Qiu, X. et al. Applications of nanomaterials in asymmetric photocatalysis: recent progress, challenges, and opportunities. Adv. Mater. 33, 2001731 (2021).
Karmakar, S. et al. Covalent grafting of molecular photosensitizer and catalyst on MOF-808: effect of pore confinement toward visible light-driven CO2 reduction in water. Energy Environ. Sci. 14, 2429–2440 (2021).
Li, R. et al. Integration of an inorganic semiconductor with a metal-organic framework: a platform for enhanced gaseous photocatalytic reactions. Adv. Mater. 26, 4783–4788 (2014).
Fang, Z.-B. et al. Boosting interfacial charge-transfer kinetics for efficient overall CO2 photoreduction via rational design of coordination spheres on metal-organic frameworks. J. Am. Chem. Soc. 142, 12515–12523 (2020).
Xu, H.-Q. et al. Visible-light photoreduction of CO2 in a metal-organic framework: boosting electron–hole separation via electron trap states. J. Am. Chem. Soc. 137, 13440–13443 (2015).
Qin, J.-S. et al. Creating well-defined hexabenzocoronene in zirconium metal-organic framework by postsynthetic annulation. J. Am. Chem. Soc. 141, 2054–2060 (2019).
Elcheikh Mahmoud, M. et al. Metal-organic framework photocatalyst incorporating bis(4′-(4-carboxyphenyl)-terpyridine)ruthenium(II) for visible-light-driven carbon dioxide reduction. J. Am. Chem. Soc. 141, 7115–7121 (2019).
Song, Y. et al. Titanium hydroxide secondary building units in metal-organic frameworks catalyze hydrogen evolution under visible light. J. Am. Chem. Soc. 141, 12219–12223 (2019).
Lan, G. et al. Electron injection from photoexcited metal-organic framework ligands to Ru2 secondary building units for visible-light-driven hydrogen evolution. J. Am. Chem. Soc. 140, 5326–5329 (2018).
Ma, X. et al. Modulating coordination environment of single-atom catalysts and their proximity to photosensitive units for boosting MOF photocatalysis. J. Am. Chem. Soc. 143, 12220–12229 (2021).
Jiao, L. et al. Microenvironment modulation in metal-organic framework-based catalysis. Acc. Mater. Res. 2, 327–339 (2021).
Peng, Y. et al. A versatile MOF-based trap for heavy metal ion capture and dispersion. Nat. Commun. 9, 187 (2018).
Ardila-Suárez, C. et al. Synthesis, characterization, and post-synthetic modification of a micro/mesoporous zirconium–tricarboxylate metal-organic framework: towards the addition of acid active sites. CrystEngComm 21, 3014–3030 (2019).
Mautschke, H. H. et al. Catalytic properties of pristine and defect-engineered Zr-MOF-808 metal organic frameworks. Catal. Sci. Technol. 8, 3610–3616 (2018).
Wu, T.-S. et al. Enhancement of catalytic activity by UV-light irradiation in CeO2 nanocrystals. Sci. Rep. 9, 8018 (2019).
Liang, W. et al. Tuning pore size in a zirconium–tricarboxylate metal-organic framework. CrystEngComm 16, 6530–6533 (2014).
Sing, K. S. W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 57, 603–619 (1985).
Feng, L. et al. Creating hierarchical pores by controlled linker thermolysis in multivariate metal-organic frameworks. J. Am. Chem. Soc. 140, 2363–2372 (2018).
Singh, A. et al. Unraveling the effect on luminescent properties by postsynthetic covalent and noncovalent grafting of gfp chromophore analogues in nanoscale MOF-808. Inorg. Chem. 59, 8251–8258 (2020).
Mulas, G. et al. Anharmonic vibrational spectroscopy of polycyclic aromatic hydrocarbons (PAHs). Chem. Phys. 149, 144102 (2018).
Yu, C. et al. Glucose sensing via polyanion formation and induced pyrene excimer emission. Chem. Commun. 21, 1347–1349 (2009).
Majumdar, A. et al. Liquid–liquid phase separation is driven by large-scale conformational unwinding and fluctuations of intrinsically disordered protein molecules. J. Phys. Chem. Lett. 10, 3929–3936 (2019).
Dong, Y. et al. Synthesis and the aggregation induced enhanced emission effect of pyrene based polysiloxanes. Polym. Chem. 6, 7827–7832 (2015).
Prasad, K. et al. Rational design of a pyrene based luminescent porous supramolecular framework: excimer emission and energy transfer. RSC Adv. 5, 74986–74993 (2015).
Beyazkilic, P. et al. Nanoconfinement of pyrene in mesostructured silica nanoparticles for trace detection of TNT in the aqueous phase. Nanoscale 6, 15203–15209 (2014).
Mikhnenko, O. V. et al. Exciton diffusion in organic semiconductors. Energy Environ. Sci. 8, 1867–1888 (2015).
Wu, J. et al. Methyl viologen-templated zinc gallophosphate zeolitic material with dual photo-/thermochromism and tuneable photovoltaic activity. Chem. Sci. 6, 2922–2927 (2015).
Iehl, J. et al. π-Dimerization of viologen subunits around the core of C60 from twelve to six directions. Chem. Sci. 4, 1462–1469 (2013).
Lingampalli, S. R. et al. Recent progress in the photocatalytic reduction of carbon dioxide. ACS Omega 2, 2740–2748 (2017).
Debnath, B. et al. Visible light-driven highly selective CO2 reduction to CH4 using potassium-doped g-C3N5. Langmuir 38, 3139–3148 (2022).
Das, K. et al. Intrinsic charge polarization in Bi19S27Cl3 nanorods promotes selective C–C coupling reaction during photoreduction of CO2 to ethanol. Adv. Mater. 35, 2205994 (2023).
Barman, S. et al. Metal-free catalysis: a redox-active donor–acceptor conjugated microporous polymer for selective visible-light-driven CO2 reduction to CH4. J. Am. Chem. Soc. 143, 16284–16292 (2021).
Tamaki, Y. et al. Photocatalytic CO<sub>2</sub> reduction with high turnover frequency and selectivity of formic acid formation using Ru(II) multinuclear complexes. Proc. Natl Acad. Sci. USA 109, 15673 (2012).
Kuramochi, Y. et al. Reaction mechanisms of catalytic photochemical CO2 reduction using Re(I) and Ru(II) complexes. Coord. Chem. Rev. 373, 333–356 (2018).
Fukuzumi, S. et al. Energetic comparison between photoinduced electron-transfer reactions from NADH model compounds to organic and inorganic oxidants and hydride-transfer reactions from NADH model compounds to p-benzoquinone derivatives. J. Am. Chem. Soc. 109, 305–316 (1987).
Xu, M. et al. Interfacial microenvironment modulation boosting electron transfer between metal nanoparticles and MOFs for enhanced photocatalysis. Angew. Chem. Int. Ed. 60, 16372–16376 (2021).
Chen, D. et al. Highly efficient visible-light-driven CO2 reduction to formate by a new anthracene-based zirconium MOF via dual catalytic routes. J. Mater. Chem. A 4, 2657–2662 (2016).
Wang, S. et al. Intermolecular cascaded π-conjugation channels for electron delivery powering CO2 photoreduction. Nat. Commun. 11, 1149 (2020).
Gong, Z. et al. Generation dependent ultrafast charge separation and recombination in a pyrene-viologen family of dendrons. J. Phys. Chem. B 120, 4286–4295 (2016).
Yu, J. et al. Excited-state electronic properties in Zr-based metal-organic frameworks as a function of a topological network. J. Am. Chem. Soc. 140, 10488–10496 (2018).
Gutierrez, M. et al. Photochemistry of Zr-based MOFs: ligand-to-cluster charge transfer, energy transfer and excimer formation, what else is there? Phys. Chem. Chem. Phys. 18, 27761–27774 (2016).
Ma, Z. et al. Selectivity reversal of photocatalytic CO2 reduction by Pt loading. Catal. Sci. Technol. 8, 5129–5132 (2018).
Zhang, R. et al. Photocatalytic oxidative dehydrogenation of ethane using CO2 as a soft oxidant over Pd/TiO2 catalysts to C2H4 and syngas. ACS Catal. 8, 9280–9286 (2018).
Liu, L. et al. Understanding the reaction mechanism of photocatalytic reduction of CO2 with H2O on TiO2-based photocatalysts: a review. Aerosol Air Qual. Res. 14, 453–469 (2014).
Ewing, G. E. et al. Infrared detection of the formyl radical HCO. J. Chem. Phys. 32, 927–932 (1960).
Wang, W. et al. Hybrid carbon@TiO2 hollow spheres with enhanced photocatalytic CO2 reduction activity. J. Mater. Chem. A 5, 5020–5029 (2017).
Tan, T. H. et al. Unlocking the potential of the formate pathway in the photo-assisted Sabatier reaction. Nat. Catal. 3, 1034–1043 (2020).
SK, M. et al. An anthracene-based metal-organic framework for selective photo-reduction of carbon dioxide to formic acid coupled with water oxidation. Chem. Eur. J. 27, 4098–4107 (2021).
Frisch, M. J. et al. Gaussian 16, Revision C.01 (Gaussian, Inc., Wallingford, CT, USA, 2016).
Kim, K. et al. Comparison of density functional and MP2 calculations on the water monomer and dimer. J. Phys. Chem. 98, 10089–10094 (1994).
Stephens, P. J. et al. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 (1994).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988).
Lee, C. et al. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
Vosko, S. H. et al. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 59, 1200 (1980).
Becke, A. D. Density-functional thermochemistry. III. Role exact.Exch. J. Chem. Phys. 98, 5648 (1993).
Hay, P. et al. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 270 (1985).
Wadt, W. R. et al. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 82, 284 (1985).
Tomasi, J. et al. Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094 (2005).
Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Dennington, R. et al. GaussView, Version 6.0.16 (Semichem Inc., Shawnee Mission, KS, 2016).
Acknowledgements
The authors are grateful to Prof. S. Sampath, IISC, for UPS measurements. The authors are grateful for the support and the resources provided by the “PARAM Yukti Facility” under the National Supercomputing Mission, Government of India at the JNCASR, Bangalore. S.K. and F.A.R. thanks CSIR (Government of India) for the fellowship. S.B. thanks JNCASR for the fellowship. TKM acknowledges the Department of Science and Technology (DST, project no. CRG/2019/005951; SPR/2021/000592) RAKCAM (from UAE), SSL, ICMS, SAMat research facility, and JNCASR for financial support. All authors also acknowledge Life Sciences Research, Education, and Training at JNCASR (project no. DBT/JNCASR/D0004/2018/ 00159:− 4547)
Author information
Authors and Affiliations
Contributions
S.K. and T.K.M. designed the concept of this work. S.K. and S.B. performed major experiments. S.K., S.B., and T.K.M. analyzed the experimental data and wrote the manuscript. D.R. assisted during the synthesis. F.A.R. performed the computational study. S.N. performed the TA measurement. All the authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Karmakar, S., Barman, S., Rahimi, F.A. et al. Confining charge-transfer complex in a metal-organic framework for photocatalytic CO2 reduction in water. Nat Commun 14, 4508 (2023). https://doi.org/10.1038/s41467-023-40117-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40117-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
