
Abstract
Solid electrolyte is vital to ensure all-solid-state batteries with improved safety, long cyclability, and feasibility at different temperatures. Herein, we report a new family of amorphous solid electrolytes, xLi2O-MCly (M = Ta or Hf, 0.8 ≤ x ≤ 2, y = 5 or 4). xLi2O-MCly amorphous solid electrolytes can achieve desirable ionic conductivities up to 6.6 × 10−3 S cm−1 at 25 °C, which is one of the highest values among all the reported amorphous solid electrolytes and comparable to those of the popular crystalline ones. The mixed-anion structural models of xLi2O-MCly amorphous SEs are well established and correlated to the ionic conductivities. It is found that the oxygen-jointed anion networks with abundant terminal chlorines in xLi2O-MCly amorphous solid electrolytes play an important role for the fast Li-ion conduction. More importantly, all-solid-state batteries using the amorphous solid electrolytes show excellent electrochemical performance at both 25 °C and −10 °C. Long cycle life (more than 2400 times of charging and discharging) can be achieved for all-solid-state batteries using the xLi2O-TaCl5 amorphous solid electrolyte at 400 mA g−1, demonstrating vast application prospects of the oxychloride amorphous solid electrolytes.
Similar content being viewed by others


Introduction
Along with the fast growing market of rechargeable electric vehicles (REVs), the development of all-solid-state batteries (ASSBs) is of high expectation due to their promises of safety, reliability, and high energy density1,2. A key component for ASSBs is solid electrolyte (SE) which can potentially enable the use of high-voltage cathodes and Li metal anode to boost the energy density3,4. One of the essential requirements for a favorable SE is high ionic conductivity. Crystalline SEs with long-range ordered structures have shown continuous and fast Li-ion conduction. For example, representative sulfide-based SEs, such as Li Argyodites5,6 and Li10GeP2S12 (LGPS)-type7,8, exhibit attractive ionic conductivities in the order of 10−2 S cm−1. Other types of crystalline SEs including oxide-based SEs (e.g., perovskite-type9, sodium superionic conductor (NASICON)-type10, and garnet-type11,12) and halide-based SEs (e.g., Li-M-Cl system, M = Y, In, Sc13,14,15,16,17) also demonstrate good conductivities of 10−4–10−3 S cm−1. While the ion conduction mechanisms of crystalline SEs have been widely studied to provide guidance for the search of new superionic conductors, some amorphous SEs also show good potentials but are less studied18.
Amorphous SEs present the primary advantages of softness, easy fabrication, low grain boundaries, wider compositional variations, and isotropic ionic conduction19, which are expected to compensate the drawbacks of some crystalline SEs with high grain boundary resistance, poor processibility, and high cost. Despite the diligent efforts, the research for amorphous SEs has been proceeding slowly. One major challenge is that the ionic conductivities of amorphous SEs are generally lower than those of the typical crystalline SEs. Another challenge is lacking long-range periodicity that makes it difficult to understand the ion conduction mechanism in amorphous materials. There are very limited established universal theories for structure modeling and ionic diffusivity prediction for amorphous materials20. Different types of reported amorphous SEs have their own advantages and disadvantages. Early researches for amorphous SEs have been reported since the 1960s19. The sulfide-based amorphous SEs, such as Li2S-P2S521 and Li2S-SiS222, show decent ionic conductivities around 10−4 S cm−1, but their narrow electrochemical stability window (1.5–2.5 V vs. Li+/Li)23,24 and poor electrode compatibility25 significantly limit their application in ASSBs. Oxide-based amorphous SEs (such as Li2O-MOx, M = Si, B, P, Ge, etc.26,27,28,29, and lithium phosphorus oxynitride30) exhibit improved (electro)chemical stability compared to the sulfide compounds, but their poor ionic conductivities of 10−9–10−6 S cm−1 at room temperature (RT, 25 °C) are far away from the benchmark (10−3 S/cm) for bulk-type ASSBs31. Nevertheless, if amorphous SEs were to be competitive with the crystalline SEs, both high conductivity and good compatibility with favorable layered oxide cathodes are required.
Herein, we report a family of lithium-based oxychloride amorphous SEs (xLi2O-MCly, M = Ta or Hf, 0.8 ≤ x ≤ 2, y = 5 or 4). The newly developed SEs display several desirable features compared to the existing SEs. (1) Facile synthesis method. One-step ball-milling method can easily yield the desired products in amorphous state. (2) High ionic conductivity. The optimized 1.6Li2O-TaCl5 amorphous SE possesses a high ionic conductivity of 6.6 × 10−3 S cm−1 at 25 °C, surpassing most of the other amorphous SEs. Similarly high ionic conductivities can be maintained within an amorphous formation region (x = 1.1‒1.8) for xLi2O-TaCl5. X-ray absorption spectroscopy (XAS) and other advanced techniques are combined to clarify that Ta-centered trigonal bipyramids with rich terminal chlorines are predominant for fast Li-ion diffusion. The optimized 1.5Li2O-HfCl4 amorphous SE also exhibits a good ionic conductivity of 1.97 × 10−3 S cm−1 at 25 °C. (3) Outstanding electrochemical performance. The xLi2O-MCly amorphous SEs have good compatibility with different favorable oxide cathodes without any additional cathode coatings. ASSBs using xLi2O-MCly amorphous SEs showed promising long-life cycling performance at both 25 °C and −10 °C.
Results
Preparation of the xLi2O-MCly materials
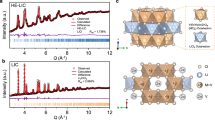
The xLi2O-TaCl5 amorphous SEs were prepared by simply ball-milling Li2O and TaCl5 at various stoichiometric ratios. Lab-based X-ray diffraction (XRD) patterns of the as-prepared xLi2O-TaCl5 (1 ≤ x ≤ 2) are shown in Fig. 1a. When x = 1, the pattern was generally amorphous with a few unknown impurities. Slightly increasing the molar ratio of Li2O/TaCl5 to 1.1 led to a completely amorphous feature (see zoom-in figures in Supplementary Fig. 1). Interestingly, further increase of the Li2O content (x ≤ 1.8) did not change the amorphous nature of xLi2O-TaCl5 SEs. As proved by synchrotron-based 2D diffraction patterns in Fig. 1b, the similar vague halos were recorded for the selected 1.1Li2O-TaCl5, 1.6Li2O-TaCl5, and 1.8Li2O-TaCl5 samples, indicating amorphous states for the xLi2O-TaCl5 SEs. Therefore, the amorphous formation region for the xLi2O-TaCl5 system was identified as 1.1 ≤ x ≤ 1.8. For a higher feeding ratio of Li2O (x ≥ 1.9), LiCl impurity appeared. SEM showed the continuous and compact surface morphology of one compound (1.6Li2O-TaCl5 pellet) in Supplementary Fig. 2, which was sharply contrast to other crystalline SE under the similar ball-milling conditions. In addition to the xLi2O-TaCl5, lithium-based oxychloride amorphous SEs can be extended to the other systems involving high-valence transition metal chlorides, for example, using Hf4+ instead of Ta5+. Figure 1c depicts the lab-based XRD patterns for a series of xLi2O-HfCl4 (0.8 ≤ x ≤ 2) samples. In comparison with the xLi2O-TaCl5 amorphous SEs, the formation of amorphous xLi2O-HfCl4 solids was more difficult. Only 1.5Li2O-HfCl4 exhibited relatively highest amorphous content among all the prepared xLi2O-HfCl4 (Fig. 1d). For other xLi2O-HfCl4 compositions (x ≠ 1.5), LiCl or unknown crystalline impurities could be easily observed.

a, c, Lab-based XRD patterns for the as-prepared xLi2O-TaCl5 (1 ≤ x ≤ 2) (a) and xLi2O-HfCl4 (0.8 ≤ x ≤ 2) (c). The broaden peaks before 25o are the diffraction peaks of the Kapton films used to seal the tested powders to avoid any air exposure. b, d Synchrotron-based 2D diffraction images of xLi2O-TaCl5 (x = 1.8, 1.6, and 1.1) (b) and xLi2O-HfCl4 (x = 2, 1.5, and 1.4) (d). Source data are provided as a Source Data file.
Ionic conductivity and Li-ion conduction analyses of the xLi2O-MCly materials
The ionic conductivities of the new amorphous SEs were determined by measuring the electrochemical impedance spectroscopy (EIS). The xLi2O-TaCl5 (x = 1.0, 1.1, 1.2, 1.4, 1.6, 1.8, 1.9, and 2.0) powders were cold-pressed into pellets for measurements. Their temperature-dependent EIS plots are shown in Supplementary Fig. 3. Figure 2a shows the extracted conductivity values at 25 °C. When the molar ratio of Li2O increased from 1 to 1.1, a surge of ionic conductivity (from 0.41 × 10−3 to 5.3 × 10−3 S cm−1) could be observed. Remarkably, the xLi2O-TaCl5 (1.1 ≤ x ≤ 1.8) pellets retained similarly high ionic conductivities of around 6 × 10−3 S cm−1, which is at the top level among all other reported SEs (Supplementary Table 1). The highest ionic conductivity was 6.6 × 10−3 S cm−1 for an optimized composition of 1.6Li2O-TaCl5. The activation energies determined from Arrhenius plots of xLi2O-TaCl5 amorphous SEs showed low values from 0.241 eV to 0.277 eV (Fig. 2b and Supplementary Fig. 4). Meanwhile, Fig. 2c compares the ionic conductivities for xLi2O-HfCl4 system (x = 0.8, 1, 1.2, 1.4, 1.5, 1.6, 1.8, and 2) at 25 °C (see corresponding Nyquist plots in Supplementary Fig. 5). The 1.5Li2O-HfCl4 SE in mostly amorphous state showed the highest ionic conductivity (1.97 × 10−3 S cm−1) and a low activation energy (0.328 eV) among the xLi2O-HfCl4 series (Fig. 2d and Supplementary Fig. 6). Direct current (DC) measurements for the representative 1.6Li2O-TaCl5 and 1.5Li2O-HfCl4 amorphous SEs under ion-blocking and electron-blocking conditions13,32 were also conducted as shown in Supplementary Fig. 7. The determined electronic conductivities were negligible (at 10−10 S cm−1 order). The Li-ion conductivities calculated from the DC measurements agree well with the values we derived from the EIS measurements, confirming the xLi2O-MCly amorphous SEs as excellent Li-ion conductors.

a, c The RT (25 °C) ionic conductivities for xLi2O-TaCl5 (a) and xLi2O-HfCl4 (c). b, d The corresponding activation energy values for xLi2O-TaCl5 (b) and xLi2O-HfCl4 (d) compounds. e The static 7Li motional narrowing spectra of 1.6Li2O-TaCl5 and 1.5Li2O-HfCl4. f, g Temperature-dependent 7Li SLR NMR rates for 1.6Li2O-TaCl5 (f) and 1.5Li2O-HfCl4 (g) measured in the laboratory frame of the reference. Source data are provided as a Source Data file.
In order to analyze the Li-ion mobility in xLi2O-MCly amorphous SEs, solid-state nuclear magnetic resonance (SSNMR) spectroscopy was adopted to provide the nuclide-specific information on structure and dynamics. First, qualitative information on Li-ion mobility33,34 of the xLi2O-TaCl5 and xLi2O-HfCl4 systems was provided via 7Li SSNMR temperature-dependent line-shape analyses. The selected 7Li SSNMR spectra of the 1.6Li2O-TaCl5 and 1.5Li2O-HfCl4 samples in a temperature range of 293–443 K are displayed in Supplementary Fig. 8. For both samples, a strong resonance corresponding to the central transition (CT) of 7Li (I = 3/2) was observed in each spectrum. The peaks due to the CT were narrow. The linewidths (Fig. 2e) were less than 1 kHz (i.e. around 450 Hz for 1.6Li2O-TaCl5 and between 520–610 Hz for 1.5Li2O-HfCl4) without significant variations in the temperature range of 293–443 K, implying that both systems were in the extreme narrowing regime. Such observation seemed to suggest a narrow distribution of slightly different Li-ion jumping rates and, by extension, a distribution of Li diffusion pathways, which is consistent with the amorphous nature of the materials.
Second, 7Li NMR spin-lattice relaxation (SLR) time (T1) allowed us to quantitatively determine the Li-ion jumping rates and activation energies corresponding to short-range as well as long-range ion diffusions in bulk electrolytes35,36,37. Fig. 2f, g show the plots of ln (1/T1) versus reciprocal temperature (1/kT) for 1.6Li2O-TaCl5 and1.5Li2O-HfCl4, respectively. A 1/T1 maximum was reached for each system, corresponding to 358 and 378 K for 1.6Li2O-TaCl5 and for 1.5Li2O-HfCl4, respectively. The fact that the peak maximum of 1.6 Li2O-TaCl5 appeared at a lower temperature than 1.5Li2O-HfCl4 suggested higher Li-ion mobility in 1.6Li2O-TaCl538. Li-ion jump frequency can be deduced from the maximum condition (τ·ω0 ≈ 1) at the relaxation rate peak34,36,39,40. Since the Larmor frequency of 7Li at 9.4 T was 155.2 MHz, the Li-ion jump frequency (1/τ) of 9.8 × 108 s−1 occurred at 358 and 378 K for the 1.6Li2O-TaCl5 and the 1.5Li2O-HfCl4, respectively. The activation energies, EaHT and EaLT, derived from the slopes of high-temperature (HT) and low-temperature (LT) flanks were not equal. Specifically, for the 1.6Li2O-TaCl5 sample, the activation energies were EaHT = 0.138 eV and EaLT = 0.047 eV, whereas the EaHT = 0.119 eV and EaLT = 0.081 eV for 1.5Li2O-HfCl4. Generally, the EaHT and EaLT are related to by a relationship of EaLT = (β − 1) EaHT where 1 < β ≤ 2. For uncorrelated isotropic diffusion described by the BPP (Bloembergen, Purcell and Pound) model, EaHT and EaLT are equal, corresponding to β = 216. In the present case, β values were determined to be 1.34 and 1.68 for the 1.6Li2O-TaCl5 and the 1.5Li2O-HfCl4 samples, respectively, indicating structurally complex Li-ion conductions40,41,42. Generally, correlation effects (e.g., Coulomb interactions, correlated ion dynamics, structural disorders, etc.) are considered closely associated with the impacted Li-ion conduction43,44. In our studies of 7Li SLR NMR for the amorphous SEs, the native structural disorder of the two amorphous samples was regarded as the major contributor towards the deviation of β value off 2 (1 <β < 2), leading to smaller EaLT values compared to those of EaHT33,42. Elaboration on the relevant correlation effects of locally disordered structure on the Li-ion migration is proposed as an interesting research direction that appeals to further attention.
Local structure exploration of xLi2O-MCly amorphous SEs
The Li-ion transport environment and structural information of representative xLi2O-TaCl5 were investigated by using Raman spectroscopy, X-ray Photoelectron Spectroscopy (XPS) and XAS. Figure 3a depicts the Raman spectra of xLi2O-TaCl5 (x = 1.2, 1.4, 1.6, 1.8, and 2). It was interesting to find that the bands at 180 cm−1 and 406 cm−1 for xLi2O-TaCl5 (x = 1.2, 1.4, 1.6 and 1.8) corresponded to Ta–Cl vibrations in trigonal bipyramidal TaCl545,46, which implied a dissociation of Ta2Cl10 bi-octahedra to TaCl5 trigonal bipyramid when introducing a moderate amount of Li2O into TaCl5 under high-energy ball-milling conditions. In comparison with noticeable Ta–Cl features, Ta–O fingerprints in xLi2O-TaCl5 were broader to show double/triple-coordinated oxygen stretching (O-3Ta/O-2Ta) vibrations47,48. This was a key signal that O atoms mainly act as bridges to connect Ta-centered trigonal bipyramids in xLi2O-TaCl5 amorphous SEs. Such an observation could also be verified in O 1 s XPS spectra in Fig. 3b, in which bridging oxygens with a binding energy of 532.1 eV49,50 showed increased fraction along with the growth of the feeding Li2O. This was consistent with the previous reports for oxysulfide amorphous SEs, which indicated oxygens showed strong amorphous formation ability to preferentially become bridging oxygens to connect polyhedra in a short range51.

a Raman spectra of the xLi2O-TaCl5 compounds, Ta2O5, and TaCl5. b–d O 1 s XPS (b), Cl 2p XPS (c), and Cl K-edge XAS (d) for xLi2O-TaCl5 amorphous SEs. Source data are provided as a Source Data file.
Since oxygens mainly contributed to form the structure of amorphous SEs, it was highly possible that chlorines took the responsibility to conduct Li ions. The chloride anion chemistry has been proved being beneficial for Li-ion migration because of a relatively large anion radius, large anion polarizability, and weak interaction with Li-ion52. Because of that, unsaturated Ta–Cl···Li bonds were proposed in xLi2O-TaCl5 amorphous SEs (Fig. 3c). The interactions between lithium and chlorine could be proved by Cl K-edge XAS. As shown in Fig. 3d, despite a pre-edge feature P1 (2822.3 eV) was indistinctly found in xLi2O-TaCl5 amorphous SEs because a mixing of Cl p-orbitals and Ta d-orbitals increased covalency53, the near-edge spectra of xLi2O-TaCl5 amorphous SEs (P2, P3, P4 and P5) were very similar to that of the LiCl. These peaks reflected the Cl 1 s electron transition process to unoccupied states and multiple scatterings, giving direct proof that Li atom was the nearest neighbor around Cl atom. Besides, it was interesting to find the oscillation in the extended range (from 2843 to 2920 eV) of each xLi2O-TaCl5 spectrum was identical with that of the LiCl, while the xLi2O-TaCl5 curves even showed a stronger amplitude than the LiCl one. This meant the Li-ion mobility environment in xLi2O-TaCl5 amorphous SEs was similar to that in LiCl but with rich Li···Cl or Cl···Li···Cl interactions.
Then, the Ta L3-edge extended X-ray absorption fine structure (EXAFS) spectra were employed to determine the atomic-scale chemical environment of amorphous xLi2O-TaCl5 SEs. In the Ta L3-edge XAS spectra, since the white line (WL) at Ta L3-edge corresponds to the dipolar transition from 2p3/2 core levels to unoccupied Ta 5d states, the WL intensity and peak position increase when the oxidation state of Ta increases, and vice versa54. As depicted in Fig. 4a, the XANES spectra of xLi2O-TaCl5 (x = 1.2, 1.4, 1.6, 1.8) possessed a similar feature (9888 to 9898 eV) with that of the TaCl5. The absorption edge energy (E0) at Ta L3-edge for xLi2O-TaCl5 was between 9883 eV (Ta2O5) and 9882 eV (TaCl5), ascribing to the oxidation of TaCl5 when Li2O was added. Figure 4b shows Ta L3-edge EXAFS of xLi2O-TaCl5 in k-space. The weakened amplitude in the range of 4 to 8 Å−1 and a low-k phase shift could be observed especially for 1.6Li2O-TaCl5 and 1.8Li2O-TaCl5. This was a signal for elongation and disorder of Ta–Cl bonds caused by Ta–O bonding, which could also be reflected in the XPS results (Fig. 3c). To further resolve the coordination of Ta in xLi2O-TaCl5, phase-uncorrected radial distribution functions (RDF) after Fourier Transformed (FT) EXAFS and wavelet transformed (WT) EXAFS were conducted55,56. Based on the Ta–O and Ta–Cl scattering paths in referential TaCl5 and Ta2O5 (Supplementary Fig. 9), Ta in each xLi2O-TaCl5 amorphous SE could be recognized to be coordinated by O and Cl (Fig. 4c–e, j, h). Intensive Ta-O signals could be observed for 1.6Li2O-TaCl5 and 1.8Li2O-TaCl5. EXAFS fitting (Supplementary Fig. 10) provided a semi-quantitative explanation about these differences in xLi2O-TaCl5 amorphous SEs, and the results were listed in Supplementary Table 2. The coordination number (CN) nearest to Ta could be estimated to be 5 in each xLi2O-TaCl5 compound, consistent with the coordination situations for Ta-centered trigonal bipyramids. As a result, we could determine that the local structure in superionic 1.2Li2O-TaCl5 and 1.4Li2O-TaCl5 amorphous SEs was mainly [TaCl4O]- trigonal bipyramid (Fig. 4f). Feeding more Li2O induced mixed short-order structures as [TaCl4O]- and O-rich [TaCl5-aOa]a- (2 ≤ a < 5) in 1.6Li2O-TaCl5 and 1.8Li2O-TaCl5 amorphous SEs (Fig. 4i). Besides, the connection of local structures in xLi2O-TaCl5 amorphous SEs could also be found as noticeable Ta–O–Ta (also write as Ta–Ta) resonances (12 Å−1) in Supplementary Fig. 11, which was of highly similarity with the Ta–O–Ta resonance in Ta2O5. Therefore, [TaCl4O]- and [TaCl5-aOa]a- shared most of the oxygens to form the networks of amorphous SEs were further confirmed.

a–c XANES (a), EXAFS (b), and FT-EXAFS (c) for xLi2O-TaCl5 amorphous SEs, as well as Ta2O5 and TaCl5 at Ta L3-edge. d, e, g, h WT spectra of 1.2Li2O-TaCl5 (d), 1.4Li2O-TaCl5 (e), 1.6Li2O-TaCl5 (g), and 1.8Li2O-TaCl5 (h) at Ta L3-edge with a k2 weighting. f, i The schematics of the local structures in superionic xLi2O-TaCl5 amorphous SEs (x = 1.2 and 1.4 in (f); x = 1.6 and 1.8 in (i)). (The local structures mainly display the coordination and possible geometry of xLi2O-TaCl5 amorphous SEs. Bond angles and Li-ion numbers are not accurate.) Source data are provided as a Source Data file.
Based on above structural information, explanations for high conductivity of xLi2O-TaCl5 amorphous SEs could be proposed as following. First, disordered and irregular [TaCl5-aOa]a- (1 ≤ a < 5) arrangements in amorphous xLi2O-TaCl5 SEs made it possible to form rich Li–Cl interactions and distorted Li–Cl sublattices. Second, corner-shared oxygen (O-2Ta) networks could induce a much wider range of distortions in Li sites57. The distorted lithium sites in O-2Ta networks were the predominance to realize a Li-ion energy landscape with low migration energy. At the same time, oxygens at the bridging positions would enlarge the doorway radius for easy access of Li ions58. Third, the unsaturated Ta–Cl···Li bonds in [TaCl5-aOa]a- (1 ≤ a < 5) showed weak Coulombic forces between lithium and chlorine, making it easier for Li ions escape from one site and jump to another. In short, oxygen incorporation is beneficial to the amorphization of xLi2O-TaCl5. The induced disordered structures in amorphous xLi2O-TaCl5 lead to a sharply increased ionic conductivity and decreased activation energy compared to the single-anion Li-Ta-Cl sample (see comparison data in Supplementary Fig. 12).
The local structures of amorphous 1.5Li2O-HfCl4 were also explored by Hf L3-edge XANES and EXAFS. Similar to the behaviors in xLi2O-TaCl5, the E0 of Hf L3-edge XANES in 1.5Li2O-HfCl4 was between those of the in HfCl4 and HfO2 (Supplementary Fig. 13). Combing with the WT-EXAFS spectra (Supplementary Fig. 14), it was shown that in 1.5Li2O-HfCl4, Hf was also nearest coordinated by O and Cl (Supplementary Fig. 15 and Supplementary Table 3). The XAS of superionic conductive 1.5Li2O-HfCl4 amorphous SE and poor ionic conductor 2Li2O-HfCl4 were compared, which showed 1.5Li2O-HfCl4 amorphous SE with more Hf–Cl bonds while 2Li2O-HfCl4 with obvious Hf–O interactions (Supplementary Fig. 13c). Based on the structural information in xLi2O-TaCl5, it was reasonable to know that the formation the abundant terminal chlorines and moderate bridging oxygens in 1.5Li2O-HfCl4 amorphous SEs was predominant to the fast Li-ion conduction.
Electrochemical performance of ASSBs
As promising SEs for bulk-type all-solid-state batteries (ASSBs), the electrochemical stability windows of 1.6Li2O-TaCl5 and 1.5Li2O-HfCl4 amorphous SEs were measured (Supplementary Fig. 16). Although the amorphous SEs were thermodynamically unstable against metal anodes (such as Li and Li-In) due to the reduction of Ta5+ or Hf4+, they showed good oxidative stability and are promising to tolerate cathode active materials beyond 4 V. The electrochemical performances of 1.6Li2O-TaCl5 and 1.5Li2O-HfCl4 amorphous SEs were then evaluated with LiNi0.83Co0.11Mn0.06O2 (NCM83) and LiCoO2 (LCO). The NCM83 ASSB with 1.6Li2O-TaCl5 SE exhibited good rate performance as shown in Fig. 5a. The charge-discharge curves depict high reversible capacities of 189.5 mAh g−1, 177.1 mAh g−1, 161.7 mAh g−1, 138 mAh g−1, 105.7 mAh g−1 and 83 mAh g−1 at 0.1 C, 0.2 C, 0.5 C, 1 C, 2 C, and 3 C, respectively (1 C = 200 mA g−1). As for the NCM83 ASSB with 1.5Li2O-HfCl4 SE, the rate performance was comparable with the 1.6Li2O-TaCl5 cell at lower rates (191.1 mAh g−1 @ 0.1 C, 171.9 mAh g−1 @ 0.2 C, 149.2 mAh g−1 @ 0.5 C) (Fig. 5b). NCM83 ASSBs integrated with either amorphous SE showed superior long cycle life at designated specific currents. As shown in Fig. 5c, the ASSB with 1.6Li2O-TaCl5 SE demonstrated highly stable cycling performance for over 300 cycles with a capacity retention of 92.9% at 1 C. The 1.5Li2O-HfCl4 ASSB also retained 89.6% of its initial reversible capacity after 300 cycles at 0.5 C (Fig. 5d). In particular, the impressive cycling durability at 2 C was demonstrated for the solid cell after evaluation of its rate capability (Fig. 5a). As depicted in Fig. 5e, there was a capacity retention of 90.7% after 2400 cycles. The charge-discharge curves suggested effective electrochemical reversibility during the long-cycling measurement (Supplementary Fig. 17). Due to the high ionic conductivity of 1.6Li2O-TaCl5, we further examined the ASSBs at a low temperature of −10 °C. The LCO ASSB exhibited an initial reversible capacity of 100.6 mAh g−1 with a Coulombic efficiency (CE) of 94.9%. The cell maintained a high capacity of 93.7 mAh g−1 after 100 cycles and stably operated for over 300 cycles with a capacity retention of 79.2% (average CE: 99.94%) (Fig. 5f). Meanwhile, despite that the 1.5Li2O-HfCl4 SE possessed a lower ionic conductivity of 3.4 × 10−4 S cm−1 at −10 °C, the LCO solid cell with 1.5Li2O-HfCl4 could still stably cycle over 300 cycles at 0.2 C (1 C = 140 mA g−1) (Fig. 5g). The rate performance of LCO ASSBs at −10 °C was also decent with either Ta-based or Hf-based amorphous SE (Supplementary Fig. 18). Finally, in order to demonstrate the practical prospect of amorphous SEs, 1.6Li2O-TaCl5 was chosen to be applied in pouch cell, which exhibited decent cycling performance in Supplementary Fig. 19.

a, b Charge–discharge curves extracted from rate performance measurements of NCM83 ASSBs at room temperature (RT, 25 °C) with 1.6Li2O-TaCl5 (a) and1.5Li2O-HfCl4 (b). c–e RT cycling performances of NCM83 ASSBs with 1.6Li2O-TaCl5 (c) and 1.5Li2O-HfCl4 (d), and long-life cycling of NCM83 ASSB using 1.6Li2O-TaCl5 at 2 C (e). f, g −10 °C cycling performances of LCO ASSBs using 1.6Li2O-TaCl5 (f) and 1.5Li2O-HfCl4 (g). Note: The specific currents of 1 C for NCM83 and LCO cathode materials are corresponding to 200 mA g−1 and 140 mA g−1, respectively. Source data are provided as a Source Data file.
In summary, we report a series of new amorphous superionic conductors, xLi2O-TaCl5 (x = 1.1‒1.8) and xLi2O-HfCl5 (x = 1.5), which can be prepared via a one-step ball-milling method. Among them, the optimized 1.6Li2O-TaCl5 and 1.5Li2O-HfCl4 amorphous SEs possess high ionic conductivities of 6.6 × 10−3 S cm−1 and 1.97 × 10−3 S cm−1, respectively, at 25 °C. The local environment in the representative superionic Ta-based amorphous SEs is identified as [TaCl5-aOa]a- (1 ≤ a < 5) trigonal bipyramids in which abundant terminal chlorines directly interact with Li ions weakly. Bridging oxygens that serve as joints in the networks of amorphous SEs can induce a wide range of distortions in Li sites. Fast Li-ion conduction in xLi2O-TaCl5 amorphous SEs benefits from such a mixed anion chemistry. The 1.6Li2O-TaCl5 and 1.5Li2O-HfCl4 amorphous SEs also show good cathode compatibility with conventional layered oxide cathode materials (NCM83 and LCO), performing outstanding electrochemical performances at both 25 °C and −10 °C. This study shall provide insights into the Li-ion dynamics and design principles of amorphous SEs, leading to a key advancement for ASSBs.
Methods
Synthesis of xLi2O-MCly (M = Ta or Hf) solid electrolytes
TaCl5 (Sigma Aldrich, 99.99%), HfCl4 (Sigma Aldrich, 98%), LiCl (Sigma Aldrich, reagent grade), and Li2O (Alfa Aesar, 99.5%) were used as the raw materials. The starting materials for each compound were mixed in an argon-filled glovebox (H2O < 0.1 ppm, O2 < 0.1 ppm). The resulting mixture (1 g) was then placed in a zirconia ball milling pot along with 40 g zirconia balls. Low-speed ball milling (100 rpm for 2 h) was first run to ensure all the precursors mixed well, followed by a high-speed ball milling process of 500 rpm for 10 h. Next, the ball-milled products were transferred into the glovebox for further use. Similarly, the Li3YCl6 electrolyte was prepared by ball milling YCl3 and LiCl in mole ratio of 1: 3. The starting materials (1 g) were weighted and pre-mixed in an argon-filled glovebox, which then placed in a zirconia ball milling pot along with 40 g zirconia balls. Low-speed ball milling (100 rpm for 2 h) was first run to ensure all the precursors mixed well, followed by a high-speed ball milling process of 500 rpm for 10 h.
Measurements of ionic and electronic conductivities
Ionic conductivity of as-prepared SEs was evaluated using electrochemical impedance spectroscopy (EIS) with two stainless steel rods as blocking electrodes. The SE powders (100~150 mg) were cold-pressed into pellets under ~300 MPa. The thickness of the pellets was between 0.04 cm and 0.06 cm. EIS measurements were performed using a multichannel potentiostat 3/Z (German VMP3). The applied frequency range was 1 Hz‒7 MHz and the voltage amplitude was 20 mV. The temperature control was realized in an ESPEC Environmental Test Chamber. The cell assembly process for DC measurements was similar with that for EIS tests. To determine the electronic conductivity, the current responses of the cell was measured at a range of constant voltages for 60 min each. The applied voltage ranged from 0.1 to 0.5 V with a step size of 0.1 V. The DC Li-ion conductivity was evaluated with a symmetric cell configuration of Li|Li6PS5Cl | xLi2O-MCly | Li6PS5Cl | Li under a bias voltage for 30 min. The bias voltage was applied at 5, 10, 15, 20, and 25 mV consecutively. The Li6PS5Cl SE (provided by China Automotive Battery Research Institute Co, Ltd) was used to prevent direct contact between Li metal and xLi2O-MCly SE.
Linear sweep voltammetry (LSV) test
Approximately 80 mg of the 1.6Li2O-TaCl5 or 1.5Li2O-HfCl4 amorphous SE powder was cold-pressed into a pellet. A 10-mg mixture of amorphous SE and carbon black (CB) (8:2 wt./wt.) was uniformly covered on one side of the pellet as working electrode. Li foil was attached on the other side of the pellet as both counter and reference electrode. A Li6PS5Cl interlayer (~40 mg) was adopted to avoid the incompatibility between amorphous SE and metallic Li. The LSV measurements were conducted using a versatile multichannel potentiostat 3/Z (VMP3) with a positive scan range from open-circuit voltage (OCV) to 6 V and a negative scan range from OCV to 0 V. The scan rate was 0.1 mV s−1.
Assembly and electrochemical characterizations of ASSBs
60 mg of 1.6Li2O-TaCl5 and 1.5Li2O-HfCl4 amorphous SEs were pressed at ~300 MPa to form a solid electrolyte layer (10 mm diameter), respectively. 10 mg of amorphous SE/NCM83 composite (3:7 mass ratio) was uniformly spread onto the surface of the one side of electrolyte layer and pressed with ~360 MPa for 5 min. NCM83 (LiNi0.83Co0.11Mn0.06O2) cathode material (polycrystalline particle size: ~3 μm) was provided by China Automotive Battery Research Institute Co, Ltd. Subsequently, Li-In alloy was placed on the other side of the electrolyte layer and pressed by ~120 MPa for 3 min. The Li-In alloy was prepared by pressing a piece of In foil (ϕ 10 mm, thickness 0.1 mm) and a piece of Li foil (ϕ 10 mm, thickness 20 μm) together under ~60 MPa for 5 min. To prevent the direct contact between amorphous SE and Li-In. 40 mg of Li3YCl6 powder pressed into pellet was served as the interlayer at the anode side. The obtained internal pellet cell was sandwiched between two stainless-steel rods as current collectors. Finally, a stack pressure of ~80 MPa was applied to the solid cell for various electrochemical tests. All cell fabrication processes were carried out in an Ar-filled glove box (H2O, O2 < 0.1 ppm). For the −10 °C full-cell test, the assemble process was similar as above. However, we changed the anode electrolyte to Li10GeP2S12 (LGPS) to provide a high ionic conductivity at −10 °C. LGPS was purchased from MSE Supplies LLC, showing a high ionic conductivity around 6 × 10−3 S cm−1 at 25 °C with cold-pressed pellet. LCO was used as cathode material. The specific currents of 1 C for NCM83 and LCO cathode materials are corresponding to 200 mA g−1 and 140 mA g−1, respectively. The electrochemical performances were evaluated using the Neware and Land battery testing system. The temperature of 25 °C (RT) for the battery testing was realized in a designated battery testing lab equipped with a temperature control system. The battery tests under the temperature of −10 °C were realized by a freezer manufactured by Thermo Fisher Scientific. Prior to the tests, all cells were rest and equilibrated for 12 h to reach target temperatures.
Fabrication of all-solid-state pouch cell
The all-solid-state pouch cell was fabricated by stacking layers of NCM83/1.6Li2O-TaCl5 cathode, SE separators (Li3YCl6 and 1.6Li2O-TaCl5), and Li-In alloy anode. The membranes of cathode composites and SEs were made by dry-film processing method59, where 0.5 wt% Polytetrafluoroethylene (PTFE) were added to induce the formation of doughs and followed by calendaring to the target thickness (~80 um). The loading of the cathode was 13.125 mg cm−2. Stacking each layers was completed in the Ar-filled glovebox, which was then sealed in plastic vacuum bag for transferring to a dry room for further packing in the aluminum-plastic bag. A pressure of ~10 MPa was applied on the pouch cell during the cycling performance test using Neware battery testing system.
Characterization methods
Lab-based XRD measurements were performed on Bruker AXS D8 Advance with Cu Kα radiation (λ = 1.5406 Å). Kapton tape was used to cover the sample holder to prevent from the air exposure.
Raman spectra were measured with a HORIBA Scientific LabRAM HR Raman spectrometer operated under laser beam at 532 nm. Electrolyte powders were attached on a carbon tape and covered by a transparent cover glass for the test.
7Li SSNMR SLR measurements were performed on a Varian Infinity Plus wide-bore NMR spectrometer equipped with an Oxford wide-bore magnet (B0 = 9.4 T). The 7Li Larmor frequency was 155.248 MHz. The π/2 and π pulse length were determined to be 2.3 and 4.5 μs, respectively. Chemical shifts were referenced with respect to a 1.0 M LiCl solution. The electrolyte sample was sealed in custom-made Teflon tubes (ɸ = 4.7 mm) in an argon-filled glovebox (H2O < 0.1 ppm, O2 < 0.1 ppm). The 7Li spin-lattice relaxation times (T1) at different temperatures were determined using an inversion-recovery NMR experiment. The testing temperature ranges from 293–443 K.
Ta L3-edge and Hf L3-edge XAS data were collected at the 44 A beamline of Taiwan Photon Source (TPS) of the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. The spectra were recorded in transmission mode. Cl K-edge XAS (FY mode) were collected at the SXRMB beamline at Canadian Light Source (CLS). The above data were processed with Athena and Artemis softwares. Synchrotron-based 2D XRD images were collected at VESPERS beamline at CLS. The 2D diffraction data were recorded on a Pilatus 1 M detector with a photon energy of 13 keV (λ = 0.9537 Å). Profex and ALBULA softwares were used to process the data.
XPS were collected at Surface Science Western (SSW) in Canada by using Krotos AXIS Ultra Spectrometer system. Monochromatic Al K(alpha) source was adopted. There was a specially designed inert transfer vessel allowing for SEs samples in a glove box, and transferring to the instrument without air exposure. High resolution analyses were carried out with an analysis area of 100 microns and a pass energy of 40 eV.
Data availability
All data that support the findings of this study are provided within the paper and its Supplementary Information. All additional information is available from the corresponding authors upon request. Source data are provided with this paper.
References
Janek, J. & Zeier, W. G. A solid future for battery development. Nat. Energy 1, 1–4 (2016).
Schmuch, R., Wagner, R., Horpel, G., Placke, T. & Winter, M. Performance and cost of materials for lithium-based rechargeable automotive batteries. Nat. Energy 3, 267–278 (2018).
Manthiram, A., Yu, X. W. & Wang, S. F. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2, 1–16 (2017).
Zhao, W. J., Yi, J., He, P. & Zhou, H. S. Solid-state electrolytes for lithium-ion batteries: fundamentals, challenges and perspectives. Electrochem. Energy Rev. 5, 574–605 (2022).
Adeli, P. et al. Boosting solid-state diffusivity and conductivity in lithium superionic argyrodites by halide substitution. Angew. Chem. Int. Ed. 58, 8681–8686 (2019).
Zhou, L. D., Assoud, A., Zhang, Q., Wu, X. H. & Nazar, L. F. New family of argyrodite thioantimonate lithium superionic conductors. J. Am. Chem. Soc. 141, 19002–19013 (2019).
Kamaya, N. et al. A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011).
Kato, Y. et al. High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 1, 1–7 (2016).
Inaguma, Y. et al. High ionic-conductivity in lithium lanthanum titanate. Solid State Commun. 86, 689–693 (1993).
Luo, J. Y. & Xia, Y. Y. Aqueous lithium-ion battery LiTi2(PO4)3/LiMn2O4 with high power and energy densities as well as superior cycling stability. Adv. Funct. Mater. 17, 3877–3884 (2007).
Wu, J. F. et al. Gallium-doped Li7La3Zr2O12 garnet-type electrolytes with high lithium-ion conductivity. ACS Appl. Mater. Interfaces 9, 1542–1552 (2017).
Bernuy-Lopez, C. et al. Atmosphere controlled processing of Ga-substituted garnets for high Li-ion conductivity ceramics. Chem. Mater. 26, 3610–3617 (2014).
Asano, T. et al. Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 30, 1803075 (2018).
Li, X. N. et al. Water-mediated synthesis of a superionic halide solid electrolyte. Angew. Chem. Int. Ed. 58, 16427–16432 (2019).
Zhou, L. D. et al. A new halospinel superionic conductor for high-voltage all solid state lithium batteries. Energy Environ. Sci. 13, 2056–2063 (2020).
Zhou, L. D. et al. High areal capacity, long cycle life 4 V ceramic all-solid-state Li-ion batteries enabled by chloride solid electrolytes. Nat. Energy 7, 83–93 (2022).
Zhang, S. M. et al. Advanced high-voltage all-solid-state Li-ion batteries enabled by a dual-halogen solid electrolyte. Adv. Energy Mater. 11, 2100826 (2021).
Grady, Z. A., Wilkinson, C. J., Randall, C. A. & Mauro, J. C. Emerging role of non-crystalline electrolytes in solid-state battery research. Front. Energy Res. 8, 218 (2020).
Chandra, A., Bhatt, A. & Chandra, A. Ion conduction in superionic glassy electrolytes: an overview. J. Mater. Sci. Technol. 29, 193–208 (2013).
Bunde, A., Funke, K. & Ingram, M. D. Ionic glasses: history and challenges. Solid State Ion-. 105, 1–13 (1998).
Mizuno, F., Hayashi, A., Tadanaga, K. & Tatsumisago, M. New, highly ion-conductive crystals precipitated from Li2S-P2S5 glasses. Adv. Mater. 17, 918–921 (2005).
Kondo, S., Takada, K. & Yamamura, Y. New lithium ion conductors based on Li2S-SiS2 system. Solid State Ion. 53, 1183–1186 (1992).
Zhu, Y. Z., He, X. F. & Mo, Y. F. Origin of outstanding stability in the lithium solid electrolyte materials: insights from thermodynamic analyses based on first-principles calculations. ACS Appl. Mater. Interfaces 7, 23685–23693 (2015).
Nolan, A. M., Zhu, Y. Z., He, X. F., Bai, Q. & Mo, Y. F. Computation-accelerated design of materials and interfaces for all-solid-state lithium-ion batteries. Joule 2, 2016–2046 (2018).
Lau, J. et al. Sulfide solid electrolytes for lithium battery applications. Adv. Energy Mater. 8, 1800933 (2018).
Charles, R. J. Some structural and electrical properties of lithium silicate glasses. J. Am. Ceram. Soc. 46, 235–243 (1963).
Ehrt, D. Structure, properties and applications of borate glasses. Glass Technol. 41, 182–185 (2000).
Martin, S. W. Ionic-conduction in phosphate-glasses. J. Am. Ceram. Soc. 74, 1767–1784 (1991).
Murthy, M. K. & Ip, J. Studies in germanium oide sstems .1. I, phase equilibria in the system Li2O—GeO2. J. Am. Ceram. Soc. 47, 328–331 (1964).
Kozen, A. C., Pearse, A. J., Lin, C. F., Noked, M. & Rubloff, G. W. Atomic layer deposition of the solid electrolyte LiPON. Chem. Mater. 27, 5324–5331 (2015).
Kaup, K. et al. A lithium oxythioborosilicate solid electrolyte glass with superionic conductivity. Adv. Energy Mater. 10, 1902783 (2020).
Kwok, C. Y., Xu, S. Q., Kochetkov, I., Zhou, L. D. & Nazar, L. F. High-performance all-solid-state Li2S batteries using an interfacial redox mediator. Energy Environ. Sci. 16, 610–618 (2023).
Wilkening, M. & Heitjans, P. From micro to macro: access to long-range Li+ diffusion parameters in solids via microscopic 6,7Li spin-alignment echo NMR spectroscopy. Chemphyschem 13, 53–65 (2012).
Uitz, M., Epp, V., Bottke, P. & Wilkening, M. Ion dynamics in solid electrolytes for lithium batteries. J. Electroceram. 38, 142–156 (2017).
Grey, C. P. & Greenbaum, S. G. Nuclear magnetic resonance studies of lithium-ion battery materials. MRS Bull. 27, 613–618 (2002).
Heitjans, P. & Wilkening, M. Ion dynamics at interfaces: nuclear magnetic resonance studies. MRS Bull. 34, 915–922 (2009).
Yu, C. et al. Unravelling Li-ion transport from picoseconds to seconds: bulk versus Interfaces in an Argyrodite Li6PS5Cl-Li2S all-solid-state Li-ion battery. J. Am. Chem. Soc. 138, 11192–11201 (2016).
Yu, C. et al. Superionic conductivity in lithium argyrodite solid-state electrolyte by controlled Cl-doping. Nano Energy 69, 104396 (2020).
Yu, C. et al. Revealing the relation between the structure, Li-ion conductivity and solid-state battery performance of the argyrodite Li6PS5Br solid electrolyte. J. Mater. Chem. A 5, 21178–21188 (2017).
Yu, C. et al. Facile synthesis toward the optimal structure-conductivity characteristics of the Argyrodite Li6PS5Cl solid-state electrolyte. ACS Appl. Mater. Interfaces 10, 33296–33306 (2018).
Epp, V., Gun, O., Deiseroth, H. J. & Wilkening, M. Highly mobile ions: low-temperature NMR directly probes extremely fast Li+ hopping in Argyrodite-type Li6PS5Br. J. Phys. Chem. Lett. 4, 2118–2123 (2013).
Hanghofer, I. et al. Substitutional disorder: structure and ion dynamics of the argyrodites Li6PS5Cl, Li6PS5Br and Li6PS5I. Phys. Chem. Chem. Phys. 21, 8489–8507 (2019).
Kuhn, A. et al. Li self-diffusion in garnet-type Li7La3Zr2O12 as probed directly by diffusion-induced 7Li spin-lattice relaxation NMR spectroscopy. Phys. Rev. B 83, 094302 (2011).
Xu, Z. M., Chen, X., Zhu, H. & Li, X. Anharmonic cation-anion coupling dynamics assisted lithium-ion diffusion in sulfide solid electrolytes. Adv. Mater. 34, 2207411 (2022).
Huglen, R., Poulsen, F. W., Mamantov, G. & Begun, G. M. Characterization of tantalum pentachloride containing melts by Raman-spectroscopy. Inorg. Chem. 18, 2551–2555 (1979).
Beattie, I. R. & Ozin, G. A. Gas-phase raman spectroscopy of trigonal bipyramidal pentachlorides and pentabromides. J. Chem. Soc. A, 1691–1693 (1969).
Joseph, C., Bourson, P. & Fontana, M. D. Amorphous to crystalline transformation in Ta2O5 studied by Raman spectroscopy. J. Raman Spectrosc. 43, 1146–1150 (2012).
Stavrou, E., Zaug, J. M., Bastea, S. & Kunz, M. A study of tantalum pentoxide Ta2O5 structures up to 28GPa. J. Appl. Phys. 121, 175901 (2017).
Minami, T., Hayashi, A. & Tatsumisago, M. Preparation and characterization of lithium ion-conducting oxysulfide glasses. Solid State Ion. 136, 1015–1023 (2000).
Nesbitt, H. W. et al. Bridging, non-bridging and free (O2-) oxygen in Na2O-SiO2 glasses: an X-ray Photoelectron Spectroscopic (XPS) and nuclear magnetic resonance (NMR) study. J. Non-Cryst. Solids 357, 170–180 (2011).
Christensen, R., Olson, G. & Martin, S. W. Ionic conductivity of mixed glass former 0.35Na2O+0.65[xB2O3 + (1-x)P2O5] glasses. J. Phys. Chem. B 117, 16577–16586 (2013).
Liu, Y. S., Wang, S., Nolan, A. M., Ling, C. & Mo, Y. F. Tailoring the cation lattice for chloride lithium-ion conductors. Adv. Energy Mater. 10, 2002356 (2020).
Li, W. H. et al. Unraveling the origin of moisture stability of halide solid-state electrolytes by in situ and operando synchrotron X-ray analytical techniques. Chem. Mater. 32, 7019–7027 (2020).
Tsuchiya, T. et al. X-Ray absorption, photoemission spectroscopy, and Raman scattering analysis of amorphous tantalum oxide with a large extent of oxygen nonstoichiometry. Phys. Chem. Chem. Phys. 13, 17013–17018 (2011).
Xia, Z. M., Zhang, H., Shen, K. C., Qu, Y. Q. & Jiang, Z. Wavelet analysis of extended X-ray absorption fine structure data: theory, application. Phys. B Condens 542, 12–19 (2018).
Munoz, M., Argoul, P. & Farges, F. Continuous cauchy wavelet transform analyses of EXAFS spectra: a qualitative approach. Am. Mineral. 88, 694–700 (2003).
Jun, K. et al. Lithium superionic conductors with corner-sharing frameworks. Nat. Mater. 21, 1–8 (2022).
Kim, Y., Saienga, J. & Martin, S. W. Anomalous ionic conductivity increase in Li2S+GeS2+GeO2 glasses. J. Phys. Chem. B 110, 16318–16325 (2006).
Wang, C. H. et al. Solvent-free approach for interweaving freestanding and ultrathin inorganic solid electrolyte membranes. ACS Energy Lett. 7, 410–416 (2022).
Acknowledgements
This research was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Research Chair Program (CRC), the Canada Foundation for Innovation (CFI), Ontario Research Foundation (ORF), and the University of Western Ontario (UWO). The synchrotron research was performed at the Canadian Light Source, a national research facility of the University of Saskatchewan, which was supported by the CFI, NSERC, the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.
Author information
Authors and Affiliations
Contributions
S.Z., F.Z., and X.S. conceived and designed the experiments. S.Z. prepared samples and carried out the main experiments. S.Z. and F.Z. examined the electrochemical performance. S.Z. analyzed the XAS data and discussed with J.C. and T.S. S.A. and Y.H. contributed to the NMR characterizations and data analysis. J.F., J.L., L.C., R.F., and M.S. carried out the synchrotron-related tests. J. Liang and X.L. helped with the Raman measurements and analysis. Y.Z helped with the SEM measurements. S.Z. and F.Z. wrote and revised the paper. All the authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, S., Zhao, F., Chen, J. et al. A family of oxychloride amorphous solid electrolytes for long-cycling all-solid-state lithium batteries. Nat Commun 14, 3780 (2023). https://doi.org/10.1038/s41467-023-39197-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-39197-8
This article is cited by
-
A solid-state lithium-ion battery with micron-sized silicon anode operating free from external pressure
Nature Communications (2024)
-
Inorganic lithium-ion conductors for fast-charging lithium batteries: a review
Journal of Solid State Electrochemistry (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
