
Abstract
Endonucleases are enzymes that cleave internal phosphodiester bonds within double-stranded DNA or RNA and are essential for biological functions. Herein, we use genetic code expansion to create an unnatural endonuclease that cleaves non-coding RNAs including short interfering RNA (siRNA) and microRNAs (miRNAs), a function that does not exist in nature. We introduce a metal-chelating unnatural amino acid, (2,2′-bipyridin-5-yl)alanine (BpyAla) to impart endonuclease activity to the viral suppressor of RNA silencing protein p19. Upon binding of copper, the mutant p19-T111BpyAla displays catalytic site-specific cleavage of siRNA and human miRNAs. Catalysis is confirmed using fluorescence polarization and fluorescence turn-on. Global miRNA profiling reveals that the engineered enzyme cleaves miRNAs in a human cell line. The therapeutic potential is demonstrated by targeting miR-122, a critical host factor for the hepatitis C virus (HCV). Unnatural endonuclease function is shown to deplete miR-122 levels with similar effects to an antagomir that reduces HCV levels therapeutically.
Similar content being viewed by others


Introduction
Endonucleases catalyze the cleavage of internal phosphodiester bonds in RNA and DNA leading to scissile rupture of genetic material or their transcription products. Endonuclease function in many organisms is involved in DNA damage repair mechanisms, restricting gene expression and antiviral responses to infection in bacteria1. Endonucleases that target RNA substrates are involved in mRNA splicing, RNA silencing pathways, maturation of t-RNAs, and other non-coding RNAs2,3,4,5. Adapting and engineering these natural enzyme catalysts has led to cornerstone biotechnologies from recombinant DNAs to gene editing technologies6,7,8.
Short non-coding RNA molecules (ncRNAs) such as microRNAs (miRNAs) have important regulatory roles9,10. miRNAs have been identified to play critical roles in gene expression regulation during transcriptional processes11, as well as different biological and biochemical processes ranging from cellular metabolism to immunity10,12,13. As products of the RNA silencing pathway, miRNAs are usually 21–25 nucleotides in length9,10,14. They exert their function by targeting specific messenger RNAs (mRNA) for degradation or translational repression10,13,15. Also, miRNA-based regulation has been linked to disease progression, which makes them interesting targets for protein-based therapeutics10,16. While there do exist natural suppressors of RNA silencing17,18,19,20, these typically bind and sequester RNA intermediates. No naturally occurring endonuclease exists that catalyzes the specific degradation of either siRNAs or miRNAs.
Designing endonuclease enzymes, and indeed any enzyme, based on substrate-binding scaffolds (e.g., proteins, aptamers, catalytic antibodies) have had limited success because the product closely resemble the bound transition state, causing product inhibition which prevents enzymatic turnover21,22,23,24,25. Thus, the product needs to be significantly different from the substrate and the transition state structures for the designed endonuclease enzyme to be successful. The binding protein needs to retain enough selectivity to its substrate, that once the preferential binding characteristics are lost, the product is released, and product inhibition is avoided25.
To engineer a new endonuclease capable of specifically targeting miRNAs, we started with the Tombusvirus p19 viral suppressor of RNA silencing (VSRS), which naturally binds and sequesters small RNA duplexes with picomolar affinity, thereby preventing the activation of the RNA silencing pathway17,18,26,27,28,29,30 (Fig. 1a, c). p19 binds RNA sequences with size selectivity26,27. Rational design and protein engineering of the p19 VSRS has allowed the development of biotechnological tools with diverse applications17,27,28,31,32,33,34,35,36,37. p19 has proved to be a potent inhibitor of small RNA function, which opens the door to further engineering the protein to exhibit “super suppressor” activity with added endonuclease functionality17,33,34,35,36. Previously, it has been shown that using the pioneering technologies of genetic code expansion in combination with unnatural amino acids (UAAs), different organisms can code for more than the 20 canonical amino acids allowing for the incorporation of unique chemical side chains into the desired location in a protein of interest38,39,40. It has been shown that the attachment of metal chelating molecules such as Ethylenediaminetetraacetic acid (EDTA) and phenanthroline derivatives to nucleic acid binding agents has led to oxidative and hydrolytic cleavage41.

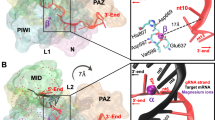
a Incorporation of BpyAla site-specifically into the VSRS protein p19 dimer introduces a metal binding site. Here we used copper to bind to BpyAla. If the metal can cleave the phosphodiester linkage within the p19 substrate, then it should induce site-specific strand cleavage of the bound siRNA or miRNA. Cleavage can occur on either strand. Created with BioRender.com. b Crystal structure of the p19 dimer with K67 (blue) and T111 (red) highlighted PDB: 1RPU. c Overall Schematic representation of the potential disruption of the RNA silencing pathway upon treatment with the engineered p19 endonuclease. Created with BioRender.com.
In this work, we explore the potential of introducing metals site-specifically into p19. The introduction of BpyAla chelated divalent ions is utilized, leading to site-specific RNA cleavage. Here we use p19, an RNA-binding protein with high selectivity to double-stranded small RNAs of 19–25 nucleotides in length but a very low affinity for single-stranded (ssRNA) and shorter double-stranded RNA (dsRNA). We successfully engineer it to cleave its native substrates. The designed endonuclease displays multiple turnover events since the protein no longer binds the cleaved products due to the loss of preferential binding characteristics. This strategy can provide a therapeutic tool targeting miRNAs involved in disease progression, as well as showing the potential of introducing catalytic activity into proteins of interest with affinities towards nucleic acids.
Results
Site-specific introduction of (2,2′-bipyridin-5-yl)alanine into the p19 dimer
We introduced (2,2′-bipyridin-5-yl)alanine (BpyAla)42,43, within the RNA-binding pocket of p19 to insert a catalytic site (Fig. 1b). The UAA bearing a bipyridyl moiety, provides an unnatural side chain capable of binding transition metal ions such as copper and iron that can catalyze the cleavage of the phosphodiester backbone of nucleic acids42,44.
We previously established the successful incorporation of Azidophenylalanine (AzF) into p19 at positions K67 and T111, in which full-length expression of the mutants and successful irreversible photo-cross-linking to their canonical substrates was achieved to produce cross-linked products with intermediates of the RNA silencing pathway37, with potential applications to CLIP assays. These two sites are found in the binding pocket of the p19 dimer, near the phosphate backbone of the RNA (Fig. 1b). To incorporate BpyAla, amber codons (TAG) were introduced in place of K67 and T111, to determine which position would allow for optimal cleavage of the bound RNA product. We expressed p19 mutants in the presence of the evolved tRNA/aaRS pair, and the presence of 1 mM BpyAla. (Supplementary Fig. 1). Bacterial cells (BL2l (DE3)) expressing the tRNA/BpyRS pair and the mutant p19 genes were grown in Lysogency Broth (LB). The full-length expression was detected using western blot analysis under the induction of Isopropyl β-d-1-thiogalactopyranoside (IPTG) (Supplementary Fig. 1)42.
We have confirmed the formation of p19-T111BpyAla enzyme-copper (II) complex using inductively coupled plasma mass spectrometry (ICP-MS). ICP-MS data shows that the associated copper (II) concentrations with p19-T111BpyAla are significantly higher than that associated with the control p19-WT samples, thus confirming copper ion chelation to T111BpyAla (Supplementary Fig. 2). In addition, the UV-Vis absorption spectra of p19-T111BpyAla showed two new absorption peaks at 319 nm and 304 nm upon the addition of CuSO4, and a decrease in absorption at 283 nm (Supplementary Fig. 3). Spectral changes are consistent with the red-shift of π–π* transition of the incorporated bipyridyl moiety upon chelation of Cu2+ ion, further confirming the formation of the artificial metal-bound enzyme. These data taken together confirm the specific binding of copper to the BpyAla residue within p19-T111BpyAla.
Small RNA cleavage using p19-T111BpyAla
Next, we sought to assess the binding affinity of the mutants to a 21nts siRNA targeting Firefly luciferase (GL2) and containing Cy3 at the 5′ end of the guide strand (Fig. 2a, Supplementary Fig. 4). We determined the binding affinities of the purified recombinant mutants using an electrophoretic mobility shift assay (EMSA). The binding affinity of p19-T111BpyAla was determined to be 2.38 ± 0.63 nmol/L, while p19-K67BpyAla is 8.41 ± 0.49 nmol/L (Fig. 2a, Supplementary Fig. 4). Overall, both mutants were shown to bind siRNA with high affinity, in which the incorporated UAA has minimal perturbation to the binding functions.

a Binding plots for p19-T111BpyAla and for p19-K67BpyAla using electrophoretic mobility shift assay. Binding plots were constructed by varying the concentrations of the protein (0–1 µM), while maintaining the concentration of Cy3-labeled GL2 at 2 nM. b An overall schematic representation of the experimental strategy, in which Cy3-GL2 siRNA cleavage was assessed using a denaturing urea-PAGE gel. Created with BioRender.com. c 20% urea-PAGE gel was used to visualize the degradation product of Cy3-Gl2 siRNA by the engineered p19-T111BpyAla mutant in the presence of reducing agent and CuSO4. Gel was independently reproduced 2 times, where different purified protein stocks were used.
To further assess the success of introducing the catalytic functionality into the selected sites in the p19 dimer, both mutants were tested for their ability to cleave Cy3 labeled 21 nucleotides long RNA duplex substrate (GL2 siRNA). The site-specific cleavage of RNA was investigated using a gel-retardation assay (Fig. 2b, c). The Cy3-tagged siRNA duplexes were incubated with p19-T111BpyAla, p19-K67BpyAla, or p19-WT, in the presence of Cu (II). The cleavage reactions were analyzed using denaturing urea-PAGE (Fig. 2b, c). The gel has revealed that only p19-T111BpyAla was able to catalyze the cleavage of the siRNA-tagged ligand leading to a lower-size band appearing on the gel. On the other hand, P19-K67BpyAla did not catalyze the cleavage of the substrate. No cleavage occurred with p19-WT under the same conditions (Fig. 2c).
Cleavage of siRNA by p19-T111BpyAla detected using fluorescence polarization
Next, we sought to validate the catalytic function using fluorescence polarization assays, in which p19-T111BpyAla was incubated with a fluorescently tagged siRNA. A decrease in polarization has been observed over time, which is expected as upon cleavage and release of the substrate. The fluorescently tagged RNA is released from the enzyme, leading to faster rotation and consequently, a lower polarization signal (Fig. 3). The fluorescence polarization assay measured the degree of polarization of the fluorophore-tagged RNA and the change in fluorescence polarization as measured by the ratio of parallel and perpendicular light45,46. We used a Cy3-tagged double-stranded siRNA, which when unbound exhibits a low signal, however, upon being bound to the target protein, the fluorophore exhibits a higher polarization signal due to the increase in molecular mass and resultant slower rotation of the fluorophore bound to the complex45. When p19-T111BpyAla is combined with Cy3-siRNA, a time-dependent decrease in fluorescence polarization signal is observed (Fig. 3b, c). The decrease in fluorescence polarization signal is attributed to the cleavage of the bound siRNA molecules, leading to the release of the Cy3-siRNA from the protein complex. The release of the fragmented RNA exhibits a faster rotation45,46. These results confirm the hydrolytic cleavage of small RNAs due to the site-specific introduction of BpyAla into p19.

a The overall hydrolytic RNA cleavage mechanism due to the incorporation of BpyAla into p19. b An overall schematic representation of the experimental strategy, in which Cy3-Gl2 siRNA cleavage was assessed using fluorescence polarization. Created with BioRender.com. c Fluorescence polarization assay was used to visualize the cleaving potential of p19-T111BpyAla. The assay was conducted using 5 µM p19-T111BpyAla or p19-WT and 200 nM RNA, n = 3, in which n values represent 3 independent replicates. Red dots represent p19-T111BpyAla, and black dots represent p19-WT.
Determination of the kinetic parameters of p19-T111BpyAla using a fluorescence turn-on assay
Next, we sought to utilize a fluorescence turn-on assay to determine the kinetic parameters of the engineered enzyme, p19-T111BpyAla. We designed a siRNA probe containing a 3′BHQ-2 quencher on the passenger strand, while the guide strand of the siRNA contains a 3′ Cy3 fluorophore. The distance between the quencher and fluorophore is 59.7 Å, which allows efficient quenching as it is within fluorescence resonance energy transfer (FRET) distance (1–100 Å). Upon incubation of p19-T111BpyAla and Cu2+ ions with various, excess concentrations of the siRNA probe, an increase of fluorescence over time is observed (Fig. 4a). The increase of fluorescence is only observed with p19-T111BpyAla and not with the p19-WT protein, indicating that cleavage and release of the siRNA take place, as an increase in the Cy3 fluorescence is observed over time (Supplemental Fig. 5). The kcat was determined to be 3.0 × 10−3 s−1 while KM was calculated to be 670 nM. The catalytic efficiency of the engineered enzyme was thus determined to be 4.47 × 103 M−1 s−1 (Fig. 4b).

a A schematic representation of the enzymatic cleavage assay using a fluorescence turn-on approach, where if cleavage occurs an increase of Cy3 fluorescence is observed. Created with BioRender.com. b Steady-state cleavage velocities (MTO rates) are plotted against the concentration of substrate. 100 nM p19-T111BpyAla or p19-WT was incubated with varying concentrations of Cy3-GL2-BHQ-2 substrate to calculate the kinetic parameters at 37 °C. siRNA cleavage was monitored using fluorescence, in which initial velocities were derived from the initial slopes. Error bars represent mean values ± SD for 3 independent replicates, representing 3 independent purified protein batches. Black circles represent p19-T111BpyAla and Black triangles represent p19-WT.
p19-T111BpyAla can cleave miRNAs isolated from hepatocellular carcinoma cell line
To further demonstrate the utility of p19-T111BpyAla as an artificial endonuclease for degrading small double-stranded RNA intermediates, we examined its ability to enzymatically cleave the small RNA fractions from Huh7 human hepatoma cells. The small RNA fractions from Huh7 cells were treated with either p19-WT treatment, or p19-T111BpyAla in the presence of CuSO4 for 4 h at 37 °C. The treated RNA was then further purified and concentrated to ensure the removal of the proteins from the samples (Fig. 5a). To uncover the identities of the substrates for the engineered endonuclease, we conducted a miRNA profiling experiment using Nanostring technology (Fig. 5b). We isolated the small RNA fractions from Huh7 cell line and treated with either p19-WT or p19-T111BpyAla. When comparing the profile of p19-T111BpyAla to that of the wild-type protein, we identified a list of statistically significant miRNAs that are differentially downregulated compared to the p19-WT (P ≤ 0.05) (Fig. 5b). These data suggest that the engineered protein degrades select miRNAs and gives rise to an amplified effect on the small RNA milieu as compared to that of its WT counterpart that can only sequester miRNAs but not degrade them (Supplementary Table 1). Interestingly, miR-122-5p was observed to be downregulated by 2.0 folds when treated with p19-T111BpyAla in comparison to treatment with WT. miR-122 is one of the miRNAs of highest abundance in the liver. Other miRNAs are identified in this screen, however, p19’s affinity to these identified has not been previously examined. It is possible that p19-T111BpyAla can potentially exhibit preferential binding to these miRNAs based on the positioning of the mismatched bulges in their secondary structure, which could give rise to enhanced binding, and thus preferential cleavage as previously examined with miR-12227. However, further structure-function analysis needs to be performed to validate the preferential cleavage pattern obtained from the Nanostring screen. p19 is known to exhibit a high degree of size selectivity, and some degree of sequence dependence when it comes to the binding of miRNAs27. miR-122 represents more than 70% of the miRNA pool in hepatoma cells47,48, and due to the instrumental role it exhibits in hepatitis C virus replication (HCV)48, we decided to proceed with examining the effects of T111BpyAla on miR-122 specifically.

a Schematic representation of the workflow for small RNA isolation for Nanostring profiling and RT-qPCR analysis. RNA used was isolated from Huh7 hepatocellular carcinoma cell line and treated in the presence of CuSO4. Created with BioRender.com. b A heat map depicting some of the significant downregulated fold changes in miRNAs upon treatment with p19-T111BpyAla relative to p19-WT treated miRNAs, n = 2, n represents 2 biological trials, where RNA is isolated from 2 independent cell passages. miRNAs with counts below 20 were eliminated from the heat map. Changes presented are more than 1.5-fold change. c qRT-PCR analysis of the relative miR-122 expression in p19-T111BpyAla treated samples in comparison to that treated with p19-WT in the presence of copper ions. Error bars represent the mean ± SD, n = 3, n values represent the treatment of RNA isolated from 3 different passages. d qRT-PCR analysis of the relative miR-122 expression in p19-T111BpyAla treated samples in the presence and absence of copper ions. Error bars represent the mean ± SD, n = 3, n values represent the treatment of RNA isolated from 3 different passages. e Derepression of miR-122’s downstream direct target STAT3 upon treatment with p19-T111BpyAla, Error bars represent the mean ± SD, n = 3. n values represent the transfection of cells from 3 different passages. Unpaired two-tailed t-test was used to evaluate statistical significance.
Next, we validated the Nanotring results by measuring the levels of miR-122, upon treatment with the mutant in the isolated samples using RT-qPCR. From Fig. 5c, it is evident that there is an observed decrease in the levels of miR-122 in p19-T111BpyAla samples, relative to treatment with p19-WT in the presence of copper, which aligns with the results from the Nanostring profiling (Fig. 5b). We attribute this decrease to the degradation of miR-122 by the mutant enzyme, as an enhanced degradation/decrease in miR-122 levels is only observed upon the introduction of copper ions (Fig. 5d). Derepression of a miR-122 direct target STAT3 was observed upon treatment with p19-T111BpyAla (Fig. 5e). These results together aid in demonstrating biocatalysis in a complex sample. In addition, there is no enhanced binding observed towards miR-122 by p19-T111BpyAla in comparison to the p19-WT when analyzed by EMSA (Supplemental Fig. 6).
For this study, we chose to next focus on miR-122 as it is a highly expressed miRNA in hepatic cells and is known to play a major role as a proviral host factor in the propagation of HCV, a positive-sense single-stranded RNA virus of the family Flaviviridae38,39,40. HCV is known to be a causative agent of hepatitis and hepatocellular carcinoma and lymphomas in humans. It is well established that HCV uniquely requires the liver-specific miR-122 for its replication. It exerts a positive effect on HCV RNA levels by binding directly to a site in the 5′-UTR of the viral RNA, and contributes to the stability, translation, and replication of the viral RNA49,50,51.
p19-T111BpyAla represses HCV replication
To investigate the effects of p19-T111BpyAla on the levels of miR-122, we sought to utilize a hepatocellular carcinoma cell line harboring the HCV subgenomic replicon pFK-I389neo/luc/NS3-3′/5.1 (Fig. 6a). This is a replicon system (our version is referred to as the E9 cell line) has been widely used to investigate the replication of HCV in cell culture52. Replication of HCV has been shown to be significantly diminished in miR-122 knockout cell line53. Thus, we wanted to validate the antiviral role that p19-T111BpyAla can potentially play. We have treated cells with p19-T111BpyAla by directly transfecting the enzyme-Cu2+ complex, pre-loaded with copper, into cells or transfecting miR-122 antagomir inhibitor as a control, to validate the effects of the mutant protein on HCV replication. The levels of miR-122 are directly correlated with HCV replication, which is linked to the relative luciferase signal in E9 cells. The effects of both miR-122 antagomir, and p19-T111BpyAla are investigated and correlated to the successful HCV subgenomic replication. Importantly, we were able to show that the designed protein led to almost the same level of HCV inhibition of replication as the levels achieved by the antagomir (Fig. 6b, c). In addition, upon transfecting the enzyme, without the addition of Cu2+, we were still able to observe a decrease in luciferase signal, and this can be attributed to the fact that BpyAla can still chelate other divalent metal ions in cells and cellular media used in the assay such as Zn2+ (Supplemental Fig. 7)

a Schematic representation of the subgenomic HCV replicon (pFK-I389neo/luc/NS3-3′/5.1) used in this study52 and HCV JFH1T strain. Created using BioRender.com. b Relative luciferase signal upon treatment of E9 cells with either miR-122 antagomir, c p19-T111BpyAla or p19-WT. The levels of miR-122 are directly correlated with HCV replication, which is linked to the relative luciferase signal in E9 cells. The effects of both miR-122 antagomir, and p19-T111BpyAla were investigated, with both treatments leading to a similar reduction in HCV subgenomic replication. For b and c, n = 3, and error bars represent mean values ± SD, p-values are presented. Unpaired two-tailed t-test was used to evaluate statistical significance. d Relative expression of HCV JFH1 IRES in Huh7.5 cells upon treatment with 1 µM p19-T111BpyAla, n = 3, error bars represent mean ± SD. P-values are presented in the figure. n values represent transfections of different cell passages. Control represents no treatment. Unpaired two-tailed t-test was used to evaluate statistical significance.
The basis for the antiviral effects of the antagomir arises from the fact that it binds to the guide strand of miR-122 more tightly than the competing genomic viral RNA. Thus, it prevents binding to and the sequestering of miR-122 by HCV. The p19-T111BpyAla enzyme (loaded with copper) can both sequester miR-122 away from HCV and reduce the overall amount of miRNA through biocatalysis. Thus, with the engineered enzyme, there is the advantage of cleaving the target miRNA reducing its overall levels in the infected cell through catalytic turnover, which was confirmed by quantification of miRNA-122 using Taqman qPCR (Fig. 5c).
In addition to validating the effects of p19-T111BpyAla on HCV subgenomic replication, we wanted to see the effects on Huh7.5 cells infected with JFH1T strain of HCV. Upon HCV infection and then direct protein transfection with 1 µM p19-T111BpyAla-Cu2+ (pre-loaded with copper), an inhibition of HCV replication of approximately 50% was achieved (Fig. 6d). Taken together these data support the conclusion that p19-T111BpyAla possesses unique anti-HCV properties by acting as an unnatural endonuclease with substrate selectivity demonstrated towards the proviral miRNA miR-122 (Fig. 6). Taken together, these experiments demonstrated that p19-T111BpyAla has a specific effect on HCV replication in a cellular model system by targeting and cleaving miR-122.
Discussion
Herein, we show the possibility of engineering endonucleases targeting small RNAs using expanded genetic code techniques to introduce the unnatural amino acid BpyAla site-specifically into p19 protein. We show that upon the addition of copper ions, the engineered p19 VSRS takes on artificial endonuclease activity not found in the wild-type protein or is naturally found in the eukaryotic kingdom54. Engineered endonucleases have been previously designed through protein fusions, which in nature can be bulky and could interfere with the functionality and proper folding of the designed enzyme55. Thus far, the engineering of RNA endonucleases has been achieved by combining different functional modules of proteins54. Similar designs have been applied to DNA binding proteins by introducing a zinc finger DNA binding domain along with an established DNA cleavage domain, with strict selectivity toward a specific DNA sequence54,55. However, for the first time, we showed that this approach can be successfully applied specifically to design endonucleases targeting miRNAs, to prevent their function.
An endonuclease based on the viral protein p19 is a good candidate for an RNA endonuclease design that specifically targets miRNAs due to the intrinsic size specificity associated with the protein26. We have confirmed that position T111 in the p19 dimer accommodates the introduction of unnatural amino acids as well as copper binding without drastically affecting substrate binding. Position T111 in the p19 dimer has been shown to accommodate different amino acids31,37.
As an artificial enzyme, p19-T111BpyAla, should have wide applications in catalytic suppression of the RNA silencing pathway. We demonstrated an application showing that p19-T111BpyAla catalytically degraded human miR-122, which resulted in the downregulation of HCV replication in a cell culture model. RNA therapy and usage of antagomirs, while effective, do have some disadvantages, mainly in costs associated with scale-up and challenges in terms of delivery. In contrast, protein therapeutics have been used and engineered to retain tissue specificity through the employment of protein fusions with tissue-specific peptides, thus holding an advantage over RNA therapeutics56,57,58.
For HCV, it is well established that the virus has evolved a dependence on human miR-122, and those oligonucleotide antagonists (antagomirs) for miR-122 can restrict and even eliminate HCV, even in clinical trials59,60. We demonstrated that p19-T111BpyAla was as effective as the corresponding antagomir at reducing miR-122 pools and limiting HCV replication while doing so catalytically and through enzyme-like function. This illustrates the potential for engineered enzymes like p19-T111BpyAla as antiviral strategies in the case where either the virus encodes its viral miRNA or interacts directly with the host’s miRNA as well as in other diseases where miRNAs are disease-causing10,48,61,62,63. Recently, the usage of catalytic enzymes to inhibit viral infections has been investigated, however, it is still a relatively recent concept27,64,65.
The p19-T111BpyAla endonuclease shows some selectivity and reasonable potency in different model systems. One challenge is the lack of sequence specificity17, which is overcome in the case of HCV replication by the high abundance of the target miRNA. In future iterations, this limitation can be mitigated by utilizing directed evolution techniques to identify other mutants with preferential binding to individual miRNAs of interest, while maintaining the characteristics of p19-T111BpyAla. The current system however can be expressed in specific cells with high expression of a miRNA of interest, in which the engineered endonuclease can preferentially cleave. In this study, miR-122 is the most highly expressed miRNA in liver cells, and thus preferentially targeted.
From the Nanostring profiling experiment, it is evident that other miRNAs exhibit a higher degree of cleavage, which can potentially be attributed to the enhanced binding of p19 mutant to these miRNAs. The differences in affinity could be attributed to the mismatches in the miRNA sequences, which create bulges in the secondary structure of the miRNA, thus affecting the potential binding affinity to p19, depending on the location of the bulge27,35,46.
Designing new enzymes based on adding catalytic residues/centers to high-affinity binding motifs has proved challenging mainly due to issues pertaining to product inhibition. Catalytic RNAs, antibodies, and other strategies have a common challenge of lack of substrate turnover due to product inhibition21,22,23,24. Designing endonucleases based on substrate-binding scaffolds faces the same challenges25. However, a key element of our system is the fact that recombinant p19 proteins retain their strong selectivity towards dsRNA of 20–25 nt in length17, and high selectivity towards the products of the dicer complex. p19-T111BpyAla at the same time displays little or no affinity towards the ssRNA or 10-11 nt dsRNA products of copper-mediated miRNA cleavage. The low affinity for cleaved products allows for the release of the cleaved product from the enzyme.
In conclusion, we have created the artificial enzyme, p19-T111BpyAla, capable of cleaving and eliminating the products of dicer, siRNAs and miRNAs. This enzyme should have wide applications in catalytic suppression of the RNA silencing pathway. The enzyme also has the potential to be deployed as a therapeutic. It is interesting to think about catalytic systems for targeting human viruses. Our work lays the groundwork for new versions with enhanced targeting.
Methods
Expression and purification of p19-WT, p19-K67BpyAla, and p19-T111BpyAla
The coding sequence of p19 retrieved from Carnation Italian Ringspot was cloned in pTriEX-Neo plasmid and subjected to site directed mutagenesis to incorporate amber mutations (TAG) in the positions of interest K67 and T111 using the following primers: forward K67 (5′-AGGAAAGCTGGGTTTCGGGTAGGTTGTCTTTAAGCGCTATC-3′), reverse K67 (5′-GATAGCGCTTAAAGACAACCTACCCGAAACCCAGCTTTCCT-3′), forward T111 (5′-AGCGAGATGCTGCATACTAAACGGAATCGCCGGTC-3′) and reverse T111 (5′-GACCGGCGATTCCGTTTAGTATGCAGCATCTCGCT-3′). Mutagenesis was performed using the Quick-change lighting Site-Directed Mutagenesis kit (Agilent) according to the manufacturer’s protocol. The plasmid was previously constructed in our lab as follows: the CIRV p19 sequence was synthesized by Genescript (Piscataway, NJ) in pUC57, followed by cloning by PCR in pTriEx 4-neo vector (EMD Biosciences, San Diego, CA). this was performed using the following primers: forward (5′-TAAGCCATGGAACGCGCTATCCAAGG-3′) and reverse (5′-CGACTCGAGCTCGCTTTCTTTCTTGAAGG-3′). The PCR product was subsequently digested using NcoI and XhoI (New England Biolabs) and inserted into multiple cloning site of pTriEx 4-neo with a C-terminal 8X histidine tag.
The plasmid encoding BpyAlaRS/tRNA (pEVOL-BpyAla) was a kind gift from Dr. Peter Schultz (The Scripps Research Institute). Transformation of the plasmid encoding BpyAlaRS/tRNA (pEvol-BpyAla) was transformed in OneShot BL21 (DE3) Chemically competent cells (ThermoFisher) and then selected under Chloramphenicol (25 µg/ml). Cells were then re-made competent and transformed with p19 plasmids bearing the appropriate amber mutations, where cells were then plated under the selection of Chloramphenicol and ampicillin (100 µg/ml). Colonies were then selected for protein expression. Cells were then grown in Lysogeny Broth in the presence of the appropriate antibiotics at 37 °C until the optical density of 0.6 was reached. Protein expression was induced using 1 mM IPTG in the presence of 1 mM BpyAla for 4 h at 25 °C. Cells were then harvested and purified using a two-step nickel affinity chromatography and size exclusion chromatography as previously described66. Briefly, cells were sonicated and the lysates were loaded onto a HisTrap FF column (GE Healthcare Life Sciences) and eluted using a buffer containing 50 mM Tris pH 8, 300 mM NaCl, 1 mM DTT and 250 mM imidazole. Fractions were further purified using size exclusion chromatography on an S200 column (GE Healthcare Life Sciences) in a buffer containing 20 mM Tris pH 8, 150 mM NaCl, and 1 mM DTT.
Immunoblotting
Cell pellets were harvested from 1 mL aliquots via centrifugation and lysed in 1x SDS Laemmli sample buffer. Cell lysates were resolved using 12% SDS gel electrophoresis (SDS-PAGE), and then transferred onto a TGX stain-free PVDF membrane (Bio-Rad). Membranes were blocked in 3% BSA in Tris-buffered saline with 0.05% tween (TBS-T). Blot was then incubated with 6xhistag HRP-conjugated antibody (1:5000 dilution; ThermoFisher, MA1-21315-HRP). Membranes were then washed and incubated for 5 min with the ECL Plus Western Blotting System (Bio-Rad). The blot was visualized using ChemiDoc MP imaging system (Bio-Rad). Uncropped blot is presented in Supplemental Fig. 8.
Substrates
GL2 siRNA cyanine 3 labeled guide strand (5′-Cy3-CGU ACG CGG AAU ACU UCG AUU-3′) and passenger strand (5′-UCG AAG UAU UCC GCG UAC GUU-3′; Sigma-Aldrich). Strands were annealed at 1:1.2 molar ratio in buffer containing 100 mM potassium acetate, 30 mM HEPES pH 7.5, and 2 mM magnesium acetate by heating to 95 °C for 2 min and cooling to 25 °C at a rate of 1 °C/min. The presence of duplexed siRNA was confirmed on a 6% non-denaturing agarose gels (ThermoFisher Scientific).
BHQ-2-GL2-Cy3 substrate: Cyanine 3 labeled guide strand (5′-CGU ACG CGG AAU ACU UCG AUU-Cy3-3′) and passenger strand (5′-UCG AAG UAU UCC GCG UAC GUU-BHQ-2-3′; Dharmacon). Strands were annealed at 1:1.2 molar ratio in buffer containing 100 mM potassium acetate, 30 mM HEPES pH 7.5, and 2 mM magnesium acetate by heating to 95 °C for 2 min and cooling to 25 °C at a rate of 1 °C/min. The presence of duplexed siRNA was confirmed on a 6% non-denaturing agarose gels (ThermoFisher Scientific).
Electrophoretic mobility shift assay (EMSA)
Varying concentrations of p19 up to 1 µM were incubated for 1 h with 2 nM GL2 siRNA or miR-122 duplexed substrate in 20 mM Tris pH 7, 100 mM NaCl, 1 mM EDTA, 0.02% v/v Tween-20, and 2 mM DTT. Following incubation, samples were resolved on a 6% non-denaturing agarose gels. Visualization of products was performed using a ChemiDoc MP imaging system (Bio-Rad). Data were analyzed using GraphPad Prism v 8.0 and fit to the Eq. (1):
Where KD is the dissociation constant, n is the number of equivalent sites on the p19 dimer, s is the concentration of labeled small RNA, Amax is the maximal change is fluorescence, and x is the concentration of the p19 dimer.
Denaturing urea-PAGE
For cleavage of the 21 bp GL2 double-stranded siRNA fragment, the reaction was initiated by adding 3-mercaptopropionic acid (final concentration 2.5 mM) to the reaction mixture containing 2 nM Cy3 labeled siRNA (21 bp), 100 nM p19-T111BpyAla, 200 nM CuSO4, in a buffer containing 20 mM HEPES, 100 mM NaCl and 0.02% Tween (pH 7.3) and incubated for 4 h. The reaction mixture RNA was then further purified and concentrated using RNA concentrator kit according to the manufacturer’s protocol (Zymo Research) and the RNA fragments were analyzed by 20% TBE gel containing 8 M urea.
Fluorescence polarization (FP)
The assay was performed in black 96-well plate to measure FP. Experiments were carried out using excess enzyme concentration [E] = 5000 nM, and Cy3-GL2 siRNA concentration of 200 nM. Reactions were performed in phosphate-buffered saline (PBS) with 10 μM CuSO4 and 2.5 mM β-mercaptopropionic acid.
Fluorescence polarization measurements were carried out using SpectraMax i3 (Molecular Devices). Polarization was monitored at 560 nm with excitation at 546 nm. Polarization is expressed as (2) where IV and IH are the vertically and horizontally polarized emission intensities, respectively.
Fluorescence turn-on assays
The assay was performed in black 96-well plate to measure fluorescence intensity at excitation of 520 nm, and emission of 570 nm. Experiments measuring multiple turnover reaction rates were carried out using enzyme concentration [E] = 100 nM, and varying excess concentrations BHQ2-GL2-Cy3 siRNA between 150 and 2000 nM. Reactions were performed in phosphate-buffered saline (PBS) with 10 mM DTT, 200 nM CuSO4, and 100 µM β-mercaptopropionic acid. Vo values are calculated using the initial slopes for the change in product formation over time under different substrate concentrations. Steady-state kinetic parameters were calculated using Graphpad Prism V8.
Cell culture, RNA Isolation, miRNA profiling, and RT-qPCR
Huh7 cells were seeded in a 6-well plate at 500,000 cells/well. Forty-eight hours post seeding, the cells were lysed using ML lysis buffer. Small RNA fractions were isolated using NucleoSpin miRNA (Macherey-Nagel). 900 ng RNA was then treated with 5 µM protein (p19-WT or T111BpyAla), 10 µM CuSO4, 2.5 mM β-mercaptopropionic acid and allowed to incubate for 4 h at 37 °C. 100 attomoles of Cel-miR248 was used as a spike-in oligo control (IDT technologies), serving as an internal monitor to account for any changes caused during RNA purification. RNA was then further purified and concentrated using RNA concentrator kit to remove any protein complexes, according to the manufacturer’s protocol (Zymo Research). Following treatment, relative miRNA levels (miR-122-5p) were quantified using the TaqMan miRNA Assay (Applied Biosystems) Assay ID: 002245, with 10 ng of total RNA used for reverse transcription using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). For miR-122 quantification, the 2−ΔΔCt method was used to calculate fold changes in expression relative to mock or control treated samples, with RNU6β levels being used for normalization, (Applied Biosystems) Assay ID: 001093.
To analyze miRNA expression on the Nanostring ncounter platform, the RNA treated with either p19-WT or p19-T111BpyAla was prepared as per manufacturer’s protocol for the ncounter human v3 miRNA expression kit. Briefly, 50 ng of RNA was hybridized to the Reporter and Capture probes for 16 h at 65 °C. The prepared samples were loaded onto the nCounter Prep Station to wash the unbound probes and immobilize the samples on the cartridge for analysis and data collection. The target probes were counted using the nCounter Digital Analyzer. The collected data were processed and analyzed using the nSolver analysis software version 3.0. The ratio of expression of miRNAs for p19-T111BpyAla treated samples against p19-WT treated samples were calculated, n = 2. Cel-miR248 was used as a spike-in oligo control (IDT technologies), serving as an internal monitor to account for any changes caused during RNA purification. Cel-miR248 levels were used to normalize the quantified RNA levels. Counts below 20 were eliminated from the analysis.
For quantification of STAT3 mRNA levels upon treatments with p19-T111BpyAla, cells were seeded at 100,000 cells in a 12-well plate. After 24 h, 1 µM p19-T111BpyAla-Cu2+ was combined transfection reagent ProteoJuice (Novagen) in opti-MEM media (Gibco) as per manufacturer’s protocol, then supplemented with full media. After another 24 h, cells were lysed for analysis and RNA isolation. RNA isolation from Huh7 cells was done using RNeasy kits (Qiagen) according to manufacturer’s protocol. 250 ng of total RNA was revese-transcribed using superscript II RT kit (life technologies). qPCR was performed using an iCycler (Bio-Rad), using SYBR green SSO Advanced supermix (Bio-Rad) following the manufacturer’s protocol. Primers targeting STAT3 were used (forward primer 5′-CAGCAGCTTGACACACACGGTA-3′; reverse primer 5′-AAACACCAAAGTGGCATGTGA-3′). Primers targeting the reference gene RNA18S5 were used (forward primer 5′-GCGATGCGGCGGCGTTATTC-3′; reverse primer 5′-CAATCTGTCAATCCTGTCCGTGTCC-3′).
E9 subgenomic replicon luciferase assay
1 µM p19-T111BpyAla pre-complexed with CuSO4 which the protein was pre-incubated with CuSO4 for 30 min on ice, and excess CuSO4 was removed by buffer exchange in PBS using 10KDa Amicon Ultra centrifugal filters (Millipore). Samples without CuSO4 were also prepared. HCV subgenomic replicon pFK-I389neo/luc/NS3-3′/5.1 (E9) cells were seeded at 4 × 104 cells/well in 24-well plates, then either treated with 1 µM p19-T111BpyAla combined with ProteoJuice transfection reagent, or 50 nM hsa-miR-122-5p mirVana miRNA inhibitor (Ambion) transfected using RNAiMax (Life Technologies). Transfections were performed according to manufacturer’s protocol. Cells were lysed after 24 h post transfection in 1× passive lysis buffer (Promega), and the Luciferase assay was carried out using a SpectraMax L luminometer (Molecular Devices).
JFH-1 infections
Huh7.5 Cells were seeded one day prior to infection in full media (DMEM, 10% FBS, NEAAs) at 20,000 cells/well in 24-well plates. After 24 h, the cells were incubated with the virus at MOI 0.1 in serum-free DMEM for 5 h. 1 µM of p19-T111BpyAla complexed with CuSO4 in which the protein was pre-incubated with CuSO4 for 30 min on ice, and excess CuSO4 was removed by buffer exchange in PBS using 10 kDa Amicon Ultra centrifugal filters (Millipore). p19-T111BpyAla was then combined transfection reagent ProteoJuice (Novagen) in opto-MEM media (Gibco) as per manufacturers protocol, then supplemented with full media and added to the cells. The cells were incubated for 48 h after which they were lysed in RL buffer and RNA was isolated using the Norgen Single Cell RNA purification kit (Norgen Biotek) as per manufacturer’s protocol. Reverse transcription and RT-qPCR were performed with primers targeting the HCV JFH1 IRES (forward primer 5′-GTCTGCGGAACCGGTGAGTA-3′; reverse primer 5′-GCCCAAATGGCCGGGATA-3′). 250 ng of total RNA was revese-transcribed using superscript II RT kit (Life Technologies). qPCR was performed using an iCycler (Bio-Rad), using SYBR green SSO Advanced supermix (Bio-Rad) following the manufacturer’s protocol.
Inductively coupled plasma mass spectrometry
20 µM p19-WT or p19-T111BpyAla was incubated with 40 µM CuSO4 in a total volume of 200 µL 1x phosphate-buffered saline (PBS) buffer and allowed to incubate for 30 min. Excess CuSO4 was removed by buffer exchange in PBS using 10 kDa Amicon Ultra centrifugal filters (Millipore). Washes were performed 3 times, to ensure removal of excess copper. All the above solutions were diluted gravimetrically by 10X with 1% HNO3, and were measured by Inductively Coupled Plasma Mass Spectrometry using an Agilent 8800QQQ triple quadrupole ICP-MS. Both 63Cu and 65Cu isotopes were monitored, which showed excellent correlation confirming the absence of any possible polyatomic ion interferences which potentially could have originated from the constituents in the buffer solutions. The measured Cu concentrations in all the measured solutions were well above the limit of detection at 0.09 parts per billion.
Statistical analysis
Data are presented as the mean of replicates, with error bars representing mean ± standard deviation. Statistical significance was evaluated using two-tailed Student’s t-test and p values of less than or equal 0.05 were deemed significant.
Synthesis of (2-pyridacyl)pyridium Iodide
(2-pyridacyl)pyridium Iodide was synthesized according to previously reported synthetic methods67. Iodine (2.0 g, 16 mmol, 0.6 eq.) was dissolved in warm pyridine (7.0 mL, 80 °C) under an argon atmosphere. 2-acetyl pyridine (3.0 mL, 26.7 mmol, 1.0 eq.) was slowly added and the mixture was stirred for 4 h. After cooling, the precipitated solid was collected by filtration and washed with cold pyridine (23 mL). The black solid was then added to boiling ethanol (100 mL) to which 2 spatulas of activated carbon was added. After 1 h, the mixture was filtered via hot filtration. The filtrate was cooled down in an ice bath and the precipitate was collected to yield the desired product as yellow crystals (5.0 g, 15.3 mmol, 57% yield). 1H-NMR data matched previously reported data67. 1H-NMR (300 MHz, DMSO-d6) δ, 9.00–9.01 (m, 2H), 8.87–8.89 (m, 1H), 8.70–8.76 (m, 1H), 8.25–8.32 (m, 2H), 8.12–8.17 (m, 1H), 8.06–8.09 (m, 1H), 7.81–7.86 (m, 1H), 6.50 (s, 2H). Raw spectra are presented in the Source data file.
Synthesis of 5-methyl-2,2′-bipyridine
5-methyl-2,2′-bipyridine was synthesized according to previously reported synthetic methods67. In a dry round-bottomed flask, (2-pyridacyl)pyridium iodide (5.0 g, 15.3 mmol, 1.0 eq.) was added with ammonium acetate (2.4 g, 30.6 mmol, 2 eq.). The solid mixture was dissolved in formamide (15 mL) and methacrolein (1.8 mL, 21.4 mmol, 1.4 eq.) was slowly added. The mixture was heated to 75 °C and stirred for 12 h. Once cooled to room temperature, the reaction mixture was poured into saturated NaHCO3 and extracted 6 times with 20 mL of EtOAc. The combined organic layers were washed with brine (100 mL), dried with Na2SO4, and concentrated under reduced pressure. The crude oil was passed through a short silica pad, eluting with 19:1 (DCM:MeOH). The desired product was obtained as a pale yellow oil (1.57 g, 9.2 mmol, 60% yield). 1H-NMR data matched previously reported data67. 1H-NMR (300 MHz, CDCl3) δ 8.66 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H), 8.50 (dt, J = 2.3, 0.8 Hz, 1H), 8.35 (dt, J = 8.0, 1.1 Hz, 1H), 8.28 (dd, J = 8.1, 0.8 Hz, 1H), 7.79 (td, J = 7.8, 1.8 Hz, 1H), 7.67–7.56 (m, 1H), 7.32–7.21 (m, 1H), 2.38 (s, 3H). Raw spectra are presented in the Source data file.
Synthesis of 5-bromomethyl-2,2′-bipyridine
5-bromomethyl-2,2′-bipyridine was synthesized according to modified previously reported synthetic methods67. 5-Methyl-2,2′-bipyridine (2.9 g, 17.3 mmol, 1.0 eq.) was dissolved in CCl4 (100 mL) and the solution was degassed via bubbling argon. N-bromosuccinimide (4.0 g, 22.5 mmol, 1.3 eq.) and AIBN (284 mg, 1.73 mmol, 0.1 eq.) were then added to the mixture. A reflux condenser was attached and the mixture was refluxed under inert atmosphere for 3 h. The solvent was then removed under reduced pressure and the crude product was dissolved in 100 mL of CH2Cl2. 100 mL saturated NaHCO3 was poured onto the mixture and it was extracted 3 times with 30 mL of CH2Cl2. The organic layers were combined, washed with 50 mL of saturated NaHCO3, dried with Na2SO4, and concentrated under reduced pressure. The obtained mixture of mono and bis-brominated products was then dissolved in THF (20 mL) under an argon atmosphere. Diethyl phosphite (0.8 mL, 6.2 mmol, 0.36 eq.) and N,N-diisopropylethylamine (1.1 mL, 6.2 mmol, 0.36 eq.) were added to the mixture and it was stirred for 18 h at room temperature. After full consumption of the dibromide (verified via 1H-NMR), the THF was evaporated under reduced pressure and 25 mL of saturated NaHCO3 was added to the residue. The mixture was extracted 3 times with 30 mL of EtOAc. The combined organic layers were dried over Na2SO4 and concentrated under vacuum. The crude product was loaded onto a silica gel column and eluted initially with 200 mL of CH2Cl2, followed by approx. 500 mL of 200:1 (CH2Cl2:MeOH). The mono-bromide was obtained as a white solid (2.25 g, 9.0 mmol, 52% yield). 1H-NMR data matched previously reported data67. 1H-NMR (300 MHz, CDCl3) δ 8.73–8.63 (m, 2H), 8.40 (dt, J = 8.0, 1.1 Hz, 2H), 7.91–7.74 (m, 2H), 7.32 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 4.54 (s, 2H). Raw spectra are presented in the Source data file.
Synthesis of 3-(2,2′-bipyridin-5-yl)-L-alanine
Under an inert atmosphere, 5-(bromomethyl)-2,2′-bipyridine (2.3 g, 9.0 mmol, 1.0 eq.), glycine t-butyl ester benzophenone imine (2.80 g, 9.5 mmol, 1.05 eq.) and Maruoka’s catalyst ((11bR)-(–)-4,4-Dibutyl-4,5-dihydro-2,6-bis(3,4,5-trifluorophenyl)-3H-dinaphth[2,1-c:1′,2′-e]azepinium bromide) (20.3 mg, 0.027 mmol, 0.03 eq.) were added to toluene (23 mL) and a 50% aqueous solution of KOH (4.5 mL) was subsequently added. The mixture was stirred for 40 h at −10 °C. The mixture was then diluted with brine (20 mL) and extracted 5 times with diethyl ether (20 mL); the organic layers were combined, dried with Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel, eluting with 50:1 (CH2Cl2:MeOH) to yield the target compound as a yellow oil (3.70 g, 8.0 mmol, 88% yield). 1H NMR (300 MHz, CDCl3) δ 8.71–8.61 (m, 1H), 8.42 (d, J = 2.2 Hz, 1H), 8.36 (d, J = 8.0 Hz, 1H), 8.24 (d, J = 8.1 Hz, 1H), 7.81 (d, J = 1.8 Hz, 1H), 7.65–7.51 (m, 3H), 7.42–7.27 (m, 7H), 6.75 (d, J = 7.1 Hz, 2H), 4.17 (d, J = 7.4 Hz, 1H), 3.27 (d, J = 5.3 Hz, 2H), 1.45 (s, 9H). Raw spectra are presented in the Source data file.
The intermediate product was then dissolved in 6 M HCl (8 mL) and stirred at 60 °C for 12 h. After heating, the solution was cooled down to room temperature and washed 3 times with diethyl ether to remove benzophenone. The aqueous phase was then concentrated and dried under reduced pressure to give the product as a pale white solid (Bpy-Ala•3 HCl) (2.81 g, 8.0 mmol, quant.). Spectral data matched previously reported data42. 1H NMR (300 MHz, MeOD-d4) δ 8.84–8.80 (m, 2H), 8.67 (dt, J = 8.2, 1.1 Hz, 1H), 8.55 (ddd, J = 8.2, 7.7, 1.6 Hz, 1H), 8.47 (dd, J = 8.3, 0.8 Hz, 1H), 8.21 (dd, J = 8.3, 2.2 Hz, 1H), 7.96 (ddd, J = 7.6, 5.6, 1.2 Hz, 1H), 4.40 (t, J = 6.8 Hz, 1H), 3.41 (qd, J = 14.6, 6.8 Hz, 2H). Raw spectra are presented in the Source data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the results and conclusions are available within this paper, the Supplementary Information and Source data files. Source data are provided as Source data file. The Nanostring profiling data generated in this paper have been deposited into NCBI GEO under the accession number: GSE229272. Figures 1b and 3 were made using IRPU https://doi.org/10.2210/pdb6RPU/pdb. Source data are provided with this paper.
Change history
21 November 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41467-023-43521-7
References
Jinek, M. et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Paroo, Z., Liu, Q. & Wang, X. Biochemical mechanisms of the RNA-induced silencing complex. Cell Res. 17, 187–194 (2007).
Song, M. S. & Rossi, J. J. Molecular mechanisms of Dicer: endonuclease and enzymatic activity. Biochem. J. 474, 1603–1618 (2017).
Kisiala, M. et al. Restriction endonucleases that cleave RNA/DNA heteroduplexes bind dsDNA in A-like conformation. Nucleic Acids Res. 48, 6954–6969 (2020).
Bentley, D. L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 15, 163–175 (2014).
Bondeson, D. P. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015).
Doudna, J. A. & Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096–1258096 (2014).
Nelles, D. A. et al. Programmable RNA tracking in live cells with CRISPR/Cas9. Cell 165, 488–496 (2016).
Li, Y. & Kowdley, K. V. MicroRNAs in common human diseases. Genomics Proteom. Bioinforma. 10, 246–253 (2012).
Peng, Y. & Croce, C. M. The role of MicroRNAs in human cancer. Signal Transduct. Target Ther. 1, 15004 (2016).
Wahid, F., Shehzad, A., Khan, T. & Young, Y. microRNAs: synthesis, mechanism, function, and recent clinical trials. BBA - Mol. Cell Res. 1803, 1231–1243 (2010).
Roberts, A. P. E., Lewis, A. P. & Jopling, C. L. in Cellular RNA Interference Mechanisms Vol. 102, 101–139 (Elsevier Inc., 2011).
Mendell, J. T. & Olson, E. N. Review microRNAs in stress signaling and human disease. Cell 148, 1172–1187 (2012).
Wahid, F., Shehzad, A., Khan, T. & Kim, Y. Y. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys. Acta Mol. Cell Res. 1803, 1231–1243 (2010).
Calin, G. A. & Croce, C. M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 6, 857–866 (2006).
Kuno, G., Chang, G. J., Tsuchiya, K. R., Karabatsos, N. & Cropp, C. B. Phylogeny of the genus Flavivirus. J. Virol. 72, 73–83 (1998).
Vargason, J. M., Szittya, G., Burgyán, J. & Hall, T. M. T. Size selective recognition of siRNA by an RNA silencing suppressor. Cell 115, 799–811 (2003).
Silhavy, D. et al. A viral protein suppresses RNA silencing and binds silencing-generated, 21- to 25-nucleotide double-stranded RNAs. EMBO J. 21, 3070–3080 (2002).
Calabrese, J. M. & Sharp, P. A. Characterization of the short RNAs bound by the P19 suppressor of RNA silencing in mouse embryonic stem cells. RNA 12, 2092–2102 (2006).
Kontra, L. et al. Distinct effects of p19 RNA silencing suppressor on small RNA mediated pathways in plants. PLoS Pathog. 12, 1–26 (2016).
Stewart, J. D. & Benkovic, S. J. Catalytic antibodies: mechanistic and practical considerations. Chem. Soc. Rev. 22, 213–219 (1993).
Palermo, G. et al. Catalytic metal ions and enzymatic processing of DNA and RNA. Acc. Chem. Res. 48, 220–228 (2015).
Miyashita, H., Karaki, Y., Kikuchi, M. & Fujii, I. Prodrug activation via catalytic antibodies. Proc. Natl Acad. Sci. USA 90, 5337–5340 (1993).
Hu, L., Arifuzzaman, M. D. & Zhao, Y. Controlling product inhibition through substrate-specific active sites in nanoparticle-based phosphodiesterase and esterase. ACS Catal. 9, 5019–5024 (2019).
Robinson, P. K. Enzymes: principles and biotechnological applications. Essays Biochem. 59, 1–41 (2015).
Danielson, D. C. & Pezacki, J. P. Studying the RNA silencing pathway with the p19 protein. FEBS Lett. 587, 1198–1205 (2013).
Cheng, J., Danielson, D. C., Nasheri, N., Singaravelu, R. & Pezacki, J. P. Enhanced specificity of the viral suppressor of RNA silencing protein p19 toward sequestering of human microRNA-122. Biochemistry 50, 7745–7755 (2011).
Ream, J. A., Lewis, L. K. & Lewis, K. A. Rapid agarose gel electrophoretic mobility shift assay for quantitating protein: RNA interactions. Anal. Biochem. 511, 36–41 (2016).
Campuzano, S. et al. Magnetobiosensors based on viral protein p19 for microRNA determination in cancer cells and tissues. Angew. Chem. Int. Ed. 53, 6168–6171 (2014).
Khan, N., Cheng, J., Pezacki, J. P. & Berezovski, M. V. Quantitative analysis of microRNA in blood serum with protein-facilitated affinity capillary electrophoresis. Anal. Chem. 83, 6196–6201 (2011).
Foss, D. V., Schirle, N. T., MacRae, I. J. & Pezacki, J. P. Structural insights into interactions between viral suppressor of RNA silencing protein p19 mutants and small RNAs. FEBS Open Bio. 9, 1042–1051 (2019).
Danielson, D. C., Sachrajda, N., Wang, W., Filip, R. & Pezacki, J. P. A novel p19 fusion protein as a delivery agent for short-interfering RNAs. Mol. Ther. Nucleic Acids 5, e303 (2016).
Rawlings, R. A., Krishnan, V. & Walter, N. G. Viral RNAi suppressor reversibly binds siRNA to outcompete dicer and RISC via multiple turnover. J. Mol. Biol. 408, 262–276 (2011).
Fagegaltier, D. et al. The endogenous siRNA pathway is involved in heterochromatin formation in Drosophila. Proc. Natl Acad. Sci. USA 106, 21258–21263 (2009).
Lecellier, C.-H. et al. A cellular microRNA mediates antiviral defense in human cells. Science 308, 557–560 (2005).
Nasheri, N. et al. An enzyme-linked assay for the rapid quantification of microRNAs based on the viral suppressor of RNA silencing protein p19. Anal. Biochem. 412, 165–172 (2011).
Ahmed, N., Foss, D. V., Powdrill, M. H. & Pezacki, J. P. Site-specific cross-linking of a p19 viral suppressor of RNA silencing protein and its RNA targets using an expanded genetic code. Biochemistry 58, 3520–3526 (2019).
Lang, K. & Chin, J. W. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev. 114, 4764–4806 (2014).
Liu, C. C. & Schultz, P. G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 79, 413–444 (2010).
Wang, Q., Parrish, A. R. & Wang, L. Review expanding the genetic code for biological studies. Chem. Biol. 16, 323–336 (2009).
Schultz, P. G. & Dervan, P. B. Sequence-specific double-strand cleavage of DNA by penta-N-methylpyrrolecarboxamide-EDTA X Fe(II). Proc. Natl Acad. Sci. USA 80, 6834–6837 (1983).
Hyun, S. L. & Schultz, P. G. Biosynthesis of a site-specific DNA cleaving protein. J. Am. Chem. Soc. 130, 13194–13195 (2008).
Drienovská, I., Rioz-Martínez, A., Draksharapu, A. & Roelfes, G. Novel artificial metalloenzymes by in vivo incorporation of metal-binding unnatural amino acids. Chem. Sci. 6, 770–776 (2015).
Lee, H. S., Spraggon, G., Schultz, P. G. & Wang, F. Genetic incorporation of a metal-ion chelating amino acid into proteins as a biophysical probe. J. Am. Chem. Soc. 131, 2481–2483 (2009).
He, Y.-C., Yin, B.-C., Jiang, L. & Ye, B.-C. The rapid detection of microRNA based on p19-enhanced fluorescence polarization. Chem. Commun. 50, 6236–6239 (2014).
Cheng, J., Sagan, S. M., Jakubek, Z. J. & Pezacki, J. P. Studies of the interaction of the viral suppressor of RNA silencing protein p19 with small RNAs using fluorescence polarization. Biochemistry 47, 8130–8138 (2008).
Jopling, C. L. Liver-specific microRNA-122: biogenesis and function. RNA Biol. 9, 137–142 (2012).
Jopling, C. L., Yi, M. K., Lancaster, A. M., Lemon, S. M. & Sarnow, P. Molecular biology: modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309, 1577–1581 (2005).
Luna, J. M. et al. Argonaute CLIP defines a deregulated miR-122-bound transcriptome that correlates with patient survival in human liver cancer. Mol. Cell 67, 400–410 (2017).
Luna, J. M. et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell 160, 1099–1110 (2015).
Schult, P. et al. MicroRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat. Commun. 9, 2613 (2018).
Lohmann, V. et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113 (1999).
Ono, C. et al. Characterization of miR-122-independent propagation of HCV. PLoS Pathog. 13, e1006374 (2017).
Shotwell, C. R., Cleary, J. D. & Berglund, J. A. The potential of engineered eukaryotic RNA-binding proteins as molecular tools and therapeutics. Wiley Interdiscip. Rev. RNA 11, 1–21 (2020).
Choudhury, R., Tsai, Y. S., Dominguez, D., Wang, Y. & Wang, Z. Engineering RNA endonucleases with customized sequence specificities. Nat. Commun. 3, 1147 (2012).
Jung, E. et al. Identification of tissue-specific targeting peptide. J. Comput. Aided Mol. Des. 26, 1267–1275 (2012).
Zahid, M. & Robbins, P. D. Cell-type specific penetrating peptides: therapeutic promises and challenges. Molecules 20, 13055–13070 (2015).
Svensen, N., Walton, J. G. A. & Bradley, M. Peptides for cell-selective drug delivery. Trends Pharm. Sci. 33, 186–192 (2012).
van der Ree, M. H. et al. Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment Pharm. Ther. 43, 102–113 (2016).
van der Ree, M. H. et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. Lancet 389, 709–717 (2017).
Sullivan, C. S., Grundhoff, A. T., Tevethia, S., Pipas, J. M. & Ganem, D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435, 682–686 (2005).
Gupta, A., Gartner, J. J., Sethupathy, P., Hatzigeorgiou, A. G. & Fraser, N. W. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature 442, 82–85 (2006).
Bruscella, P. et al. Viruses and miRNAs: more friends than foes. Front. Microbiol. 8, 1–11 (2017).
Lopez-Tejedor, D., Claveria-Gimeno, R., Velazquez-Campoy, A., Abian, O. & Palomo, J. M. In vitro antiviral activity of tyrosinase from mushroom Agaricus bisporus against hepatitis C virus. Pharmaceuticals 14, 759 (2021).
Li, J. et al. Antiviral activity of a purine synthesis enzyme reveals a key role of deamidation in regulating protein nuclear import. Sci. Adv. 5, eaaw7373 (2019).
Ahmed, N. et al. Visualization of the delivery and release of small RNAs using genetic code expansion and unnatural RNA-binding proteins. Bioconjug. Chem. 29, 3982–3986 (2018).
Ballardini, R. et al. Photoinduced electron transfer in a triad that can be assembled/disassembled by two different external inputs. Toward molecular-level electrical extension cables. J. Am. Chem. Soc. 124, 12786–12795 (2002).
Acknowledgements
J.P.P. thanks the Natural Sciences and Engineering Council of Canada (NSERC) for funding for this work. This work was supported by Natural Sciences and Engineering Research Council Grant 298496 and Canadian Institutes of Health Research Grant 136807. No.A. and Na.A. thank NSERC for funding in the form of post-graduate scholarships. We thank Dr. Peter G. Schultz for providing the plasmid encoding BpyAlaRS/tRNA (pEVOL-BpyAla). The JFH1T strain of the hepatitis C virus was a kind gift from the laboratory of Dr. R.S. Russell (Memorial University, NL, Canada).
Author information
Authors and Affiliations
Contributions
J.P.P. and No.A. conceptualized the study. No.A., Na.A., and D.A.B. performed experiments. No.A. and J.P.P. wrote the manuscript. All authors edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks George Calin, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmed, N., Ahmed, N., Bilodeau, D.A. et al. An unnatural enzyme with endonuclease activity towards small non-coding RNAs. Nat Commun 14, 3777 (2023). https://doi.org/10.1038/s41467-023-39105-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-39105-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
