
Abstract
Self-adaptability is highly envisioned for artificial devices such as robots with chemical noses. For this goal, seeking catalysts with multiple and modulable reaction pathways is promising but generally hampered by inconsistent reaction conditions and negative internal interferences. Herein, we report an adaptable graphitic C6N6-based copper single-atom catalyst. It drives the basic oxidation of peroxidase substrates by a bound copper-oxo pathway, and undertakes a second gain reaction triggered by light via a free hydroxyl radical pathway. Such multiformity of reactive oxygen-related intermediates for the same oxidation reaction makes the reaction conditions capable to be the same. Moreover, the unique topological structure of CuSAC6N6 along with the specialized donor-π-acceptor linker promotes intramolecular charge separation and migration, thus inhibiting negative interferences of the above two reaction pathways. As a result, a sound basic activity and a superb gain of up to 3.6 times under household lights are observed, superior to that of the controls, including peroxidase-like catalysts, photocatalysts, or their mixtures. CuSAC6N6 is further applied to a glucose biosensor, which can intelligently switch sensitivity and linear detection range in vitro.
Similar content being viewed by others


Introduction
The establishment of indispensable adaptability plays a crucial role in living systems under a wide variety of environmental stimuli. For example, under normal conditions, thyroid hormones are primarily responsible for the regulation of metabolism at the basal level in humans. Nonetheless, when suddenly exposed to severe cold stimuli, the sympathetic nervous system releases high concentrations of norepinephrine. It significantly accelerates metabolism as the second pathway, which produces more heat to compensate for the cold. It features primary transduction in the normal state and a large gain in exceptional circumstances. For the same reason, mimicking such self-adaptability is highly envisioned in artificial devices1,2,3, such as robots, brain-machine interfaces, and the Internet of Things, to freely perceive weak and strong external signals, but remains a grand challenge. From a chemical point of view, to realize these imperative basic activities and prominent gain effect, seeking catalysts with multiple and modulable reaction pathways is the crux of the matter4,5,6,7,8,9. However, the negative internal interferences and inconsistency of reaction conditions, such as temperature, pH, and solvents, generally exist in different reaction pathways, hampering the effective coordination of multiple catalytic pathways simultaneously10.
As a metal-free semiconductor, polymeric carbon nitride (PCN) has been intensively explored not only as a metal-free photocatalyst11,12,13,14,15,16,17,18,19,20 but also as a solid ligand to anchor single metal atom21,22,23, owing to its engineerable conjugated repetitive units, rich lone pair electrons in the framework, and high physicochemical stability. For example, carbon nitrides with different topological structures (e.g., C3N4, C3N2, C5N2, and C2N)24,25,26,27,28,29,30 and metal dopants (e.g., K and Cu)31,32,33 have been developed for a wide range of photocatalytic oxidation reactions34,35,36,37 (e.g., clean water and sanitation) and oxidase38,39,40,41/peroxidase-like42,43,44 activities. Interestingly, these oxidation reactions are catalyzed by carbon nitrides via different reactive oxygen-related intermediates under similar conditions, except for light irradiation45,46,47,48,49. As such, we reason that engineered multiformity of carbon nitrides in oxidation processes would offer an intriguing way to solve the inconsistency of reaction conditions and negative internal interference for multiple reaction pathways in realizing self-adaptability; however, to our knowledge, this has rarely been reported.
Herein, we report an adaptable copper single-atom catalyst supported on C6N6 with a specialized donor-π-acceptor linker (CuSAC6N6). CuSAC6N6 could not only drive basic oxidation of peroxidase substrates through bound high-valent copper-oxo pathway, but also initiate a second gain reaction under light irradiation via free hydroxyl radical pathway under the same conditions. Moreover, the unique topological structure of CuSAC6N6, along with the specified donor-π-acceptor linker, promoted intramolecular charge separation and migration, thus successfully inhibiting the negative interference of electron transfers between the above two pathways. As a result, a sound basic activity and a superb gain up to 3.6 times under household light were obtained, significantly higher than that of its control systems. CuSAC6N6 was further successfully applied to a single glucose sensor with intelligent switching of sensitivity and linear detection range in vitro.
Results
Synthesis and molecular structure of CuSAC6N6
As illustrated in Fig. 1a, copper acetate, as the Cu source, was first complexed with dicyandiamide (DCDA) in ethylene glycol (EG) at 60 °C for 3 h to produce a reddish-brown DCDA-Cu complex. Subsequently, microwave-assisted condensation36 using EG as the solvent was utilized to synthesize the pale-yellow intermediate, denoted as Cu-CNint., with a yield of 85%. Owing to the pre-coordinated Cu-DCDA complex in EG, the possible formation of a metal or metallic oxide in Cu-CNint. was avoided. As displayed in Supplementary Fig. 1, the X-ray diffraction (XRD) pattern of Cu-CNint. showed the only peak at 26.7°, assigning to interlayer stacking. In contrast, the microwave-assisted polymerization of the blue Cu2+ and DCDA mixture resulted in a product with additional XRD peaks for CuO, indicating the significance of pre-complexation in the preservation of the single-atom state. The final product, CuSAC6N6, was obtained via thermal polymerization at 550 °C. As a control, CNmw was fabricated by the same microwave-assisted polymerization of DCDA in EG without copper and subsequent thermal condensation.

a Brief synthesis procedure for CuSAC6N6. b FTIR spectra of CuSAC6N6, CNmw and PCN. Solid-state c 13C and d 1H NMR spectra of CuSAC6N6. e C 1 s XPS spectrum of CuSAC6N6. f Normalized XRD patterns of CuSAC6N6, CNmw, and PCN. g LDI-TOF mass spectrum of CuSAC6N6. h Proposed condensation processes and molecular structure of CuSAC6N6.
The combustion elemental analysis in Supplementary Table 1 showed the transformation of the molar C/N value from Cu-CNint. of 0.73 to CuSAC6N6 of 0.95, which is ~1. FTIR spectroscopy was used to determine the chemical structure of the new carbon nitride (Fig. 1b). The FTIR spectrum of CuSAC6N6 showed strong vibration peaks around 800 and 1200–1700 cm−1, assigning to typical triazine rings (CN heterocycles). A characteristic peak at approximately 2900 cm−1, which was ascribed to C–H stretching50, was also observed. Solid-state NMR spectroscopy provided further insights into the nature of the building blocks of CuSAC6N6. As shown in Fig. 1c, the carbon atoms in the triazine units corresponded to the peak at ~163 (1) ppm, confirming the existence of conjugated triazine rings in CuSAC6N6. The adjacent chemical shift at ca. 156 (2) ppm was assigned to the carbon atom indirectly connected to the triazine ring51. Those two types of carbon atoms and FTIR spectrum demonstrated the existence of triazine rings and non-cyclization groups of -N=CH-. The 1H magic angle spinning (MAS) NMR spectrum was also employed to determine the local environment of the H atom. As shown in Fig. 1d, the 1H NMR spectrum of CuSAC6N6 exhibited a main characteristic peak around 8.3 ppm, ascribing to the -N=CH- group. Notably, this chemical shift of the 1H NMR spectrum was often assigned to the aromatic carbon environment52, indicating the existence of a triazine ring and a second conjugated carbon atom.
The high-resolution X-ray photoelectron spectroscopy (XPS) in Fig. 1e provided additional bonding information. The C 1s XPS spectra exhibited two main peaks at 284.6 eV (C1) and 286.8 eV (C2), which were attributed to the C-C peak and C species in triazine rings (C−N=C), respectively53. Interestingly, the carbon peak (ca. 284.6 eV) in PCN and CNmw was assigned to random adventitious carbon, while C-(N)3 shifted to high binding energy at 288.1 eV (Supplementary Fig. 2). These results demonstrated that the non-cyclization group of -N=CH- was coupled, forming -N=CH-CH=N- moieties in the thermal polymerization process. The crystalline texture was further explored using XRD spectroscopy (Fig. 1f). Compared to PCN and CNmw, the diffraction peak of CuSAC6N6 at ca. 26.0° (002) corresponding to interlayer stacking reflection (002), was broadened and down-shifted, indicating a slightly enlarged interlayer spacing which may arise from the insertion of Cu atoms between C6N6 layers.
To verify the precise molecular structure of CuSAC6N6, matrix-free laser desorption/ionization time-of-flight (LDI-TOF) mass spectra were measured. The m/z peaks resulted from the ablation products of the repetitive C6N6 units in CuSAC6N6. Figure 1g illustrates a series of m/z peaks, including [M + H+] of 127.11, assigning to C3N6H6 (M1, melamine, calc.: 126), m/z [M + H+] of 139.15 attributable to C4N6H6 (M2, calc.: 138), and m/z [M + H+] of 242.15 attributable to C9N9H7 (M3, calc.: 241). The other m/z peaks shown in Supplementary Fig. 3 also supported the ablation unit’s information. Therefore, these structural explorations demonstrated that CuSAC6N6 featured a repetitive basic triazine core and a -N=CH-CH=N- linker. The possible condensation processes and the molecular structure of CuSAC6N6 are shown in Fig. 1h.
Cu single-atom structure of CuSAC6N6
The scanning electron microscopy (SEM) images in Supplementary Fig. 4a shows the disordered and porous structure of CuSAC6N6, which distinguishes it from the blocky structures of PCN and CNmw (Supplementary Fig. 4b, c). The high-resolution TEM images in Supplementary Fig. 5 revealed the ultrathin nanosheet-like morphology of CuSAC6N6 and no obvious Cu/CuO nanoparticles existed on the surface of the C6N6 framework, which were in agreement with the XRD results. The large-area high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image (Fig. 2a and Supplementary Fig. 6) corroborated the existence of uniformly dispersed single-atom Cu on the C6N6 matrix, evident by abundant isolated bright spots highlighted by a white circle. The high Cu content in CuSAC6N6 was quantitatively measured by inductively coupled plasma mass spectrometry (ICP-MS) as ∼2.36 wt%. The corresponding high-resolution STEM-EDS elemental mapping images showed that the C, N, and Cu species were atomically and homogeneously dispersed across the entire C6N6 nanosheet (Supplementary Fig. 7 and Fig. 8).

a HAADF-STEM image of CuSAC6N6. b Cu k-edge XANES profiles of Cu foil, Cu2O, CuO, CuSAC6N6, and CuPc. c Cu k-edge EXAFS spectra in the R-space of the CuSAC6N6, CuPc, CuO, and Cu foil samples. d EXAFS fitting curve for CuSAC6N6 in R-space.
To further investigate the chemical state and local coordination environment of Cu species in CuSAC6N6, X-ray absorption fine structure (XAFS) measurements of the Cu K-edge were performed. From the normalized Cu K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. 2b), the absorption edge of CuSAC6N6 was located between the control samples of Cu foil and CuO, indicating that the coexisting Cu+ (which acted as a predominant oxidation state)54 and Cu2+ carried a partial positive charge between 0 and +2. Meanwhile, the related FT k3-weighted Extended X-ray absorption fine structure (EXAFS) spectrum in the R space for CuSAC6N6 was also measured (Fig. 2c). The presence of a characteristic peak located at 1.51 Å indicated the first coordination shell of Cu-N and there were no obvious metallic Cu–Cu and Cu–O–Cu interaction at around 2.2 and 2.7 Å, respectively55, demonstrating that Cu atoms were atomically dispersed in the CuSAC6N6 matrix. This result was consistent with the HAADF-STEM images. To further explore the atomic coordination of Cu, the related FT k3-weighted EXAFS fitting in R-spaces (Fig. 2d) was performed to reveal the detailed structural information56. The fitting analysis result (Supplementary Table 2) showed that the average Cu-N coordination number in the first coordination shell was 2.0 and the Cu-N bond length was 1.93 Å, indicating that one Cu atom was coordinated by two N atoms, forming a Cu-N2 moiety.
The existence of Cu-N chemical bonding information was also verified by the N 1s and Cu 2p XPS spectra. As shown in Supplementary Fig. 9, the N 1s peak could be deconvoluted into four peaks, in which the dominant N species around 398.0 eV was mainly ascribed to the formation of pyridinic nitrogen (C═N-C) in triazine rings; The peak at 399.7 and 400.7 eV were corresponding to pyrrolic N and graphitic N. Interestingly, a new bond at 398.7 eV was observed, demonstrating the formation of a Cu-N bond, which was also in agreement with the EXAFS spectrum in the R-space measurements (Fig. 2c). The Cu 2p spectrum (Supplementary Fig. 10) exhibited two main peaks with a binding energy of 932.9 and 952.9 eV, which were assigned to Cu 2p3/2 and Cu 2p1/2, respectively. Furthermore, a weak satellite peak was recorded at 944.8 eV, indicating the presence of Cu2+ species in CuSAC6N6. The spectrum for Cu 2p3/2 could be deconvoluted into two peaks at 932.6 and 934.8 eV, corresponding to Cu1+ and Cu2+, respectively55,57. This result further proved the coexistence of Cu+ and Cu2+ in CuSAC6N6, consisting of the XANES spectroscopy (Fig. 2b).
It should be noted that CuSAC6N6 in this work was practically a transition metal complex of a conjugated polymer. But unlike conventional polymers that demonstrate molecular behaviors, polymeric carbon nitrides are almost not dissolvable like graphite; thus, it is often used to support single-atom metals in catalysis58,59. The control experiments and comprehensive characterizations, such as XRD, high-resolution TEM, HAADF-STEM, EXAFS, and XPS, collaboratively demonstrated that Cu emerged as a single-atom state in the C6N6 matrix, rather than Cu/CuO nanoparticles or clusters. To keep consistency with previous reports22,32,54,58,59,60,61, the term of graphitic C6N6-based copper single-atom catalyst (CuSAC6N6) is used in this study.
Basic catalytic activity and Gain effect of CuSAC6N6
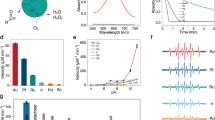
The filling of Cu-N coordination into the C6N6 framework endowed it with basic peroxidase-like catalytic activity. As shown in Fig. 3a and Supplementary Fig. 11, taking catalytic oxidation of 2, 2’-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) in the presence of H2O2 as the model reaction, an evident color change from transparency to green was observed62. Moreover, owing to the π-conjugated hybridization of energy levels along the polymer linker and π-stacking between linkers, CuSAC6N6 demonstrated a narrow optical gap (1.30 eV, Supplementary Fig. 12 and Fig. 13). Thus, CuSAC6N6 was able to effectively utilize the lower excitation energy of light to realize a gain. As shown in Fig. 3b, when irradiated by a household white LED lamp (400–900 nm, 50 mW/cm2, Supplementary Fig. 14), the color of ABTSox turned dark green, indicating a considerable enhancement in the oxidation of ABTS. The quantitative absorbance of ABTSox at 417 nm under light irradiation (0.72) was 3.6 times that in the dark (0.20). This result was equivalent to an improved total peroxidase-like rate constant of 3.4 times (Supplementary Fig. 15).

a Equation and photographs of standard ABTS catalytic oxidation using different catalysts as shown in (b) and (c). b Absorbance of ABTSox catalyzed by CuSAC6N6, PCN, CNmw, Cu2+-PCN, Cu-N-C, and Cu-N-C/CNmw without (basic) and with (gain) light irradiation. c Absorbance of ABTSox catalyzed by CuSAC6N6 at different temperatures. ABTS: 2, 2’-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid). Error bars represent the standard error derived from three independent measurements.
In contrast to CuSAC6N6, the basic catalytic activities of control PCN and CNmw were negligible (Fig. 3b). Under light irradiation, their catalytic activities were enhanced, indicative of typical photocatalysts, but were still much lower than that of CuSAC6N6. This result demonstrated that the basic activity of photocatalysts without external light irradiation is essentially lacking, which is indispensable for maintaining imperative activity in normal mode. To further understand the excellent basic activity and gain the effect of CuSAC6N6, two well-known copper-containing nanozymes, i.e., Cu2+-PCN and Cu-N-C with similar Cu-N coordination were also prepared. Interestingly, although higher basic activities of Cu2+-PCN and Cu-N-C were observed (Fig. 3b), none of them demonstrated a similar large gain effect as CuSAC6N6 under light irradiation, indicating that the second reaction pathway was not effectively initiated. For the former, although charge separation would occur in the C3N4 framework under light irradiation, the Cu ions significantly quenched it as recombination sites. For the latter, the graphitic carbon framework has intrinsically poor charge-separation ability. These results indicated that conventional carbon nitride-based (photo)catalysts were only applicable for effectively driving a single enzyme-like or photocatalytic reaction because of the negative internal interference or inconsistent reaction conditions in the basic and gain reactions. It was supposed that the unique conjugated linkers (-N=CH-CH=N-) not only elongated the distance of D-A to reduce recombination at Cu sites but also simultaneously compensated for the charge transfer between D-A in CuSAC6N6, thus coordinating the basic and gain reactions.
As additional controls, two highly efficient enzyme-like catalysts and photocatalysts, Cu-N-C and CNmw, were mixed into a nanocomposite (Cu-N-C/CNmw, Supplementary Figs. 16, 17). As shown in Fig. 3b, Cu-N-C/CNmw exhibited a gain effect under light irradiation; however, it reached only approximately one-third of that of CuSAC6N6. A series of characterizations, including electrochemical impedance spectra (EIS, Supplementary Fig. 18), photoluminescence (PL) spectra (Supplementary Fig. 19), and photoelectrochemical measurements (Supplementary Fig. 20) of PCN, Cu-N-C/CNmw, and CuSAC6N6 demonstrated a faster velocity of intramolecular charge migration in the C6N6 matrix than in PCN and Cu-N-C/CNmw played a crucial role in the boosted gain effect.
Considering that temperature induction is often used to obtain the gain effect, particularly in the photothermal manner in tumor therapies63, the influence of temperature on ABTS oxidation was also explored. As shown in Fig. 3c and Supplementary Fig. 21, the peroxidase-like activity of CuSAC6N6 showed elevated velocity with the increase of temperature. When the temperature was increased from room temperature (25 °C) to 55 °C, the maximum limit for most lives that could endure, the catalytic activity reached 2 times of the original one, but was much smaller than that by mild light irradiation (3.6 times). In this sense, the gain reaction driven by the photocatalytic method was more efficient by a factor of 80% than that driven by the thermal stimuli.
The intrinsic mechanism of enhancement of the peroxidase-like activity of CuSAC6N6 under light irradiation was further investigated. In the first set of experiments, the temperature of the reactor after irradiation using the LED lamp was measured to exclude the photothermal effect. It was found that the irradiation for 10 min made the temperature only improve by 3 oC (Supplementary Fig. 22), whereas the practical reaction in this study only took 3 min, indicating an even smaller temperature fluctuation. The enhancement of peroxidase-like activity with such a minor increase in temperature was further measured (Supplementary Figs. 23, 24), confirming that the photothermal-induced gain effect here was marginal. A series of control experiments, including examination of the oxidase-like activity of CuSAC6N6, direct photocatalytic oxidation of ABTS, and decomposition of H2O2, were also performed, which excluded the potential interferences for the profound gain of peroxidase-like activity (Supplementary Fig. 25). Moreover, the basic catalytic activity and photocatalytic processes of CuSAC6N6 can be easily modulated by tuning the Cu content and irradiation power density (Supplementary Figs. 26, 27).
Coordinated basic and gain reactions mechanism of CuSAC6N6
To understand the coordinated basic and gain reaction mechanisms of CuSAC6N6 in peroxidase-like activity under light irradiation, the possible intermediate reactive species were first studied using scavenger trapping experiments. As illustrated in Supplementary Fig. 28, both superoxide dismutase (SOD) and isopropanol, corresponding to the superoxide (O2·–) and hydroxyl radical (·OH) scavengers, respectively, had negligible influence on the oxidation of ABTS catalyzed by CuSAC6N6, indicating that these two radicals were not formed during the activation of H2O2. Nevertheless, the catalytic activity was notably reduced when isopropanol was added to the reaction solution under light irradiation (Supplementary Fig. 29), indicating that the redox reaction of H2O2 generated ·OH as a major step in CuSAC6N6 catalyzed ABTS oxidation. Other trapping experiments using radical probes such as nitrotetrazolium blue chloride (NBT) and coumarin (Supplementary Figs. 30, 31), and electron spin resonance (ESR) spectra (Fig. 4a, b) also supported this speculation. For instance, there was no ESR signal for any ROS-trapping agent adduct, reminiscent of the case catalyzed by HRP. Interestingly, under light irradiation, a typical characteristic peak of the DMPO-·OH spin adduct with a typical signal intensity of 1:2:2:1 was observed (Fig. 4b), indicating the existence of ·OH, in agreement with the scavenger trapping experiments in Supplementary Fig. 28.

ESR spectra of the spin adduct of ·OH, O2·–, and 1O2 generated during the activation of H2O2 by CuSAC6N6 in 0.2 M HAc-NaAc (pH 5.0) under the a basic and b gain reactions. c EPR spectra of CuSAC6N6 in air and after degassing at 200 °C in vacuum for 12 h. d XPS spectra of dynamic changes in the valence state of CuSAC6N6 in the light-on and light-off states. e Proposed mechanism for dual peroxidase-like and photocatalytic pathways mimicking the basic activity and gain effect using CuSAC6N6.
The solid-state ESR signal of CuSAC6N6 (Fig. 4c) further demonstrated the probability of the formation of Cu=O species during the peroxidase-like reaction. Given that Cu had a much stronger ESR intensity in the air than in degassed conditions at g = 2.08, it was speculated that CuSAC6N6 might have a similar catalytic mechanism to HRP via a bound ROS pathway45. Meanwhile, to investigate the charge transfer as well as the chemical-bond evolution of CuSAC6N6 under light irradiation, the synchronous illumination X-ray photoelectron spectroscopy technique was employed to clarify the dynamic changes of Cu-N60. As shown in Fig. 4d, the spectrum for Cu 2p3/2 was deconvoluted into two peaks at 932.6 and 934.8 eV, which were assigned to Cu1+ and Cu2+, respectively. Interestingly, the percentage of Cu2+ decreased from 52 to 40% after light irradiation, while the percentage of Cu1+ and Cu0 increased from 48 to 60%, supporting the acceptance of electrons for Cu atoms in the gain reaction64. Besides, as a significant amount of Cu2+ was retained under light irradiation, the basic reaction pathway should also be activated during the gain reaction, that was much more efficient.
Taking all these experimental evidences into consideration, the mechanism for the light-gained peroxidase-like catalytic activity of CuSAC6N6 was proposed (Fig. 4e). Briefly, in the basic reaction, H2O2 was first bound to the N-coordinated metal site of CuI in CuSAC6N6 to form CuI-superoxo species. Then, O-O cleavage occurred to decompose the adsorbed H2O2 into H2O upon the oxidation of CuI to CuII=O. Next, the CuII=O was reduced by the ABTS substrate owing to the affinity of ABTS for CuII=O and the electron-donating ability of ABTS. In contrast, when the light was turned on, the photocatalytic processes were activated, providing a new pathway for enhancing the catalytic activity. The terminal mixed-valence Cu species emerged as collectors of photogenerated electrons, and these photoelectrons subsequently reduced Cu+ to Cu0. Subsequently, Cu0 contributed to the in situ decomposition of H2O2 to produce ·OH via a Fenton-like process61. Finally, H2O2 oxidized Cu0 to promote the formation of Cu+ species, completing the photocatalytic cycle and maintaining the mixed-valence states. Therefore, for CuSAC6N6, the multiformity of reactive oxygen-related intermediates for the same oxidation reaction made the reaction conditions capable of being the same.
The aforementioned results further demonstrated that CuSAC6N6 not only had a peroxidase-like Cu-N coordination active center but also owned an unusual donor-π-acceptor (D-π-A) unit (Fig. 1h)65,66,67, where the single Cu atom acted as an electron acceptor, the triazine rings emerged as electron donors (photovoltaic center), and the -N=CH-CH=N- linkers offered π-conjugated charge transfer channels for D-A couples. Notably, as aforementioned in Fig. 3b, these π-interconnected D-A couples played a crucial role in addressing the negative internal interferences of basic and gain reactions by not only reducing recombination (control sample: Cu2+-PCN) and promoting charge separation (control sample: Cu-N-C), but also accelerating intramolecular charge transfer (control sample: Cu-N-C/CNmw).
These experimental results were further supported by density functional theory (DFT) calculations. As controls, two more samples were computed. One was Cu-g-C3N4, which was made from CuSAC6N6, but the -N=CH-CH=N- linkers were substituted with N atoms. The other was the most studied Cu-PCN, for which, the triazine ring in Cu-g-C3N4 was further altered into the heptazine ring. The first 50 excited states of these three systems were calculated by using the time-dependent DFT (TD-DFT) method, and the absorption spectra were simulated (Supplementary Fig. 32). The three highest intensity of delocalization excitations were selected for comparison for each molecule. The electron-charge center-of-mass distance (D) of CuSAC6N6, Cu-g-C3N4, and Cu-PCN demonstrate the crucial role of π-interconnected D-A couples (Fig. 5a and Supplementary Fig. 33). The hole-electron analysis was performed using Multiwfn68,69. Based on the hole-electron theory, the D value was applied to evaluate the hole-electron separation, and a larger D value was indicative of a more evident hole-electron separation. For Cu-g-C3N4 and Cu-PCN, the D value were 0.24 Å/2.08 Å/2.18 Å and 0.72 Å/0.72 Å/3.08 Å, respectively. Such short electron-charge center-of-mass distance leading to rapid electron-hole recombination without outside assistance. Interestingly, after inserting the -N=CH-CH=N- linkers in CuSAC6N6, the attraction of metal atoms and electrons was reinforced: the electron center had a strong tendency to approach the Cu atom, while the hole center remained concentrated in the excitation triazine ring, resulting in a further increasing the centroid distance of the electrons and holes to 3.08 Å/3.25 Å/3.53 Å. Such sound spatial separation would not only reduce the recombination of electrons and holes, but also promote intramolecular charge separation and migration, which well addressed the negative internal interference of basic and gain reactions. Furthermore, the most possible D-π-A electron transfer was qualitatively evaluated by analyzing the transitions form from the occupied molecular orbitals to the unoccupied molecular orbitals (Supplementary Figs. 34–36). It was observed that the electron distribution of the occupied molecular orbitals mainly resided on the triazine/heptazine rings in CuSAC6N6, Cu-g-C3N4, and Cu-PCN. While for the unoccupied molecular orbitals, delocalization electrons in CuSAC6N6 were transferred from three edge triazine rings to both -N=CH-CH=N- linkers and Cu atom, whereas electrons in Cu-g-C3N4 and Cu-PCN transitioned to other triazine/heptazine ring, resulting in few electrons delocalized on the Cu atom moieties.

a Isosurfaces of hole and electron distribution of the highest intensity of delocalization excitation for CuSAC6N6, Cu-g-C3N4, and Cu-PCN. The corresponding excitation and electron-charge center-of-mass distance (D) were marked below the picture. Calculated smooth description of electron (yellow isosurface) and hole (pink isosurface) spatial population distributions (isovalue = 0.001 au), respectively. b Ball-and-stick model structure of CuSAC6N6. Dashed areas indicate donor (D), π-conjugated charge transfer channels (π), and acceptor (A). c Simulated absorption spectrum and CTS of CuSAC6N6. Electron excitations were calculated with M06-2X/def2-TZVP level based on optimized ground-state geometries. Carbon atoms (black), Nitrogen atoms (blue), Hydrogen atoms (white), Cu atoms (orange).
To further understand the inter-fragmental charge transfer during the first 50 excited states in CuSAC6N6, the absorption spectra were deconvoluted into charge-transfer spectra (CTS), which were used to visually observe the contribution of each fragment in the model molecules70. Dashed areas in the ball-and-stick model structure of CuSAC6N6 (Fig. 5b) indicated donor (D), π-conjugated charge transfer channels (π), and acceptor (A). For simplicity, the D-π-A electron transfer was divided into D-π and D-A transitions. As shown in Fig. 5c, the local excitations in CuSAC6N6 were mainly concentrated in the D (23.5%) and π (14.6%), and the strongest charge transfer transitions were D-π (38.9%) and D-A (9.6%), in contrast, the electronic transitions from A and π to D were negligible. It was suggested that except for the local transition of the D and π parts, the excitation of the charge separation mainly consisted of D-π and D-A. A series of computational descriptions of the CuSAC6N6 systems, including conformational symmetries and the involvement of the transition metal (Supplementary Fig. 37), delocalization (Supplementary Fig. 38), conformational flexibility (Supplementary Fig. 39), and hybrid functionals (Supplementary Fig. 40) were also considered, which supported the above calculation. Therefore, owing to the existence of π-conjugated linkers, CuSAC6N6 essentially underwent intramolecular charge transfer from the triazine ring unit to the Cu atom (i.e., D-π-A) upon light irradiation.
Adaptable sensing of glucose
Improving quality of life is an enduring topic in modern society. Therefore, the intelligent response of glucose has received increasing attention as a powerful tool in the field of human health. In recent years, many biosensing methods for detecting glucose based on nanozymes have found broad utility owing to their simplicity, sensitivity, and high selectivity71,72,73. Despite enormous advances, in vitro recording of the narrow linear concentration range of glucose cannot directly provide physiological information for better comprehension of the dynamic fluctuation of glucose in the brain74. An intelligent response to glucose with a random concentration range at a single sensing interface in vivo is highly envisioned to understand the pathological process, but it still faces great challenges.
As proof of this concept, we propose an intelligent response sensor for monitoring glucose in vitro (Fig. 6a). As shown in Fig. 6b, for the conventional biosensors (0 W/cm2), at a low concentration range, the absorbance of ABTSox at 417 nm was proportional to the first order with glucose. In the high concentration range, the absorbance reached a plateau, leading to a limited detection range. In this sense, relating the response with full-scale analyte concentrations at a logarithmic scale primarily considering a fitting coefficient of determination (R²) close to 1 is widely used, but is questioned in uncertainty in theory75. In principle, a reliable fitting should follow the signal transformation mechanism rather than merely the data statistics76. Nonetheless, although the strict nonlinear rate equations (Fig. 6b, inset) could match the experimental data well (see the fitted calibration curve in Fig. 6b), it was noted that at the plateau region, the slopes were essentially zero, indicating an extremely poor sensitivity.

a Principle of adaptable sensing of glucose based on CuSAC6N6. Inset: rate equations of basic and gain reactions under tuned light intensity (Iλ). kB and kG are the intrinsic rate constants of the basic and gain reactions, respectively. c1 and c2 are constants. b Absorbance of ABTSox at 417 nm catalyzed by CuSAC6N6 with different concentrations of glucose in the presence of glucose oxidase under tuned light intensity (0, 30, and 50 W/cm2). Inset: nonlinear rate equations of basic and gain reactions with different concentrations of glucose. Bold lines are linear relationship between the absorbance of ABTSox at 417 nm and concentrations of glucose under tuned light intensity. Error bars represent the standard error derived from three independent measurements.
Developing adaptive sensitivity and linear detection concentration ranges is an ideal solution to obtain a strict full-scale range of reliable detection. As shown in Fig. 6b, glucose was normally detected in a linear range of 800–6000 μM. The linear range can be extended from 50–1000 μM at 30 mW/cm2 to 5–80 μM at 50 mW/cm2. In the same manner, the limit of detection (LOD) for glucose was tuned from 195.02 μM in the normal mode, to 15.07 and 1.20 μM under irradiation at 30 and 50 mW/cm2, respectively. The rate equations in Fig. 6a inset (see the detailed elementary reactions and derivations in Supplementary Information) well supported these facts that the concentration of ABTSox was in linear with that of glucose, and the slope (i.e., sensing sensitivity) was positively correlated with light intensity. Therefore, a full-scale range of reliable detection could be successfully realized by intelligent switching among different ranges of a single sensor in the measuring progress, enabled by the high adaptability of CuSAC6N6 using basic and gain reactions.
It should be noted that although several pioneering papers regarding C3N4-based biosensors have been reported40,77, to our knowledge, intelligent biosensors with self-adaptability have been rarely reported so far. Unlike most switchable systems that have no activities in the off-state, CuSAC6N6 had a considerable basic activity, reminiscent of the living system in maintaining the necessary activity under normal conditions. Indeed, due to limited room, intelligent artificial devices are preferred to equip biosensors as few as possible and mechanically switching of them would lead to a low operation efficiency. In this sense, one single biosensor with an adaptable linear detection range and sensitivity via an automatic light switch would be helpful. It is foreseeable that further considering the intrinsic outstanding temporal and spatial resolution of light irradiation, this intelligent biosensor would supply a prospective candidate for dynamic chemical noses for intelligent artificial devices, such as robots, brain-machine interface, and internet-to-things in a high level of integration.
Discussion
In summary, we proposed a CuSAC6N6 single-atom catalyst to address the inconsistency of reaction conditions and negative internal interference for basic and gain reactions in mimicking self-adaptability from nature. CuSAC6N6 had single Cu atoms, repetitive triazine cores, and -N=CH-CH=N- D-π-A linkers, which successfully coordinated the peroxidase-like Cu-N coordination center and light responsive center. CuSAC6N6 could not only drive the basic ABTS oxidation through bound high-valent copper-oxo pathway, but also undertake a second gain reaction triggered by light via a free hydroxyl radical pathway under the same conditions. The multiformity of reactive oxygen-related intermediates for the same oxidation reaction made the reaction conditions capable of being the same. The comprehensive experiments and DFT calculations further verified the unique topological structure of CuSAC6N6 along with the specified D-π-A linker promoted intramolecular charge separation and migration, thus successfully inhibiting the negative interference of electron transfers between peroxidase-like Cu-N coordination center and photo-responsive center. As a result, CuSAC6N6 demonstrated a sound basic catalytic oxidation activity and a superb gain up to 3.6 times under household light (50 mW/cm2). This performance was significantly higher than that of the control systems, including peroxidase-like catalysts, photocatalysts, or their mixtures, and even CuSAC6N6 that under thermal stimuli (by a factor of 80% rising from room temperature to 55 oC, the maximum temperature for most lives that can endure). As a proof-of-concept application, CuSAC6N6 was successfully applied in a biosensor of glucose, which intelligently switched sensitivity and linear detection range in vitro, simply by tuning the light intensity. It is highly envisioned that an adaptable graphitic C6N6-based copper single-atom catalyst, along with the further intrinsic temporal and spatial resolution of light, would supply a prospective candidate for adaptable chemical noses for artificial devices, such as robots, brain-machine interface, and internet-to-things in a higher level of integration.
Data availability
The data supporting the conclusions of this study are present in the paper and the Supplementary Information. The raw data sets used for the presented analysis within the current study are available from the corresponding authors upon request.
References
Wang, X. Q. et al. Somatosensory, light‐driven, thin‐film robots capable of integrated perception and motility. Adv. Mater. 32, 2000351 (2020).
Liao, F. et al. Bioinspired in-sensor visual adaptation for accurate perception. Nat. Electron. 5, 84–91 (2022).
Potyrailo, R. A. Multivariable sensors for ubiquitous monitoring of gases in the era of internet of things and industrial internet. Chem. Rev. 116, 11877–11923 (2016).
Zhang, X. et al. Enhanced photoresponsive ultrathin graphitic-phase C3N4 nanosheets for bioimaging. J. Am. Chem. Soc. 135, 18–21 (2012).
Wang, F., Zhang, Y., Du, Z., Ren, J. & Qu, X. Designed heterogeneous palladium catalysts for reversible light-controlled bioorthogonal catalysis in living cells. Nat. Commun. 9, 1209 (2018).
Neri, S., Garcia Martin, S., Pezzato, C. & Prins, L. J. Photoswitchable catalysis by a nanozyme mediated by a light-sensitive cofactor. J. Am. Chem. Soc. 139, 1794–1797 (2017).
Zhang, J., Wu, S., Lu, X., Wu, P. & Liu, J. Lanthanide-boosted singlet oxygen from diverse photosensitizers along with potent photocatalytic oxidation. ACS Nano 13, 14152–14161 (2019).
Liu, Y. et al. Structurally engineered light-responsive nanozymes for enhanced substrate specificity. Anal. Chem. 93, 15150–15158 (2021).
Li, S. et al. A nanozyme with photo‐enhanced dual enzyme‐like activities for deep pancreatic cancer therapy. Angew. Chem. Int. Ed. 58, 12624–12631 (2019).
Rudroff, F. et al. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 1, 12–22 (2018).
Wang, X. et al. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 8, 76–80 (2008).
Ghosh, I. et al. Organic semiconductor photocatalyst can bifunctionalize arenes and heteroarenes. Science 365, 360–366 (2019).
Zhao, D. et al. Boron-doped nitrogen-deficient carbon nitride-based Z-scheme heterostructures for photocatalytic overall water splitting. Nat. Energy 6, 388–397 (2021).
Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374–384 (2021).
Volokh, M., Peng, G., Barrio, J. & Shalom, M. Carbon nitride materials for water splitting photoelectrochemical cells. Angew. Chem. Int. Ed. 58, 6138–6151 (2019).
Kuriki, R., Sekizawa, K., Ishitani, O. & Maeda, K. Visible-light-driven CO2 reduction with carbon nitride: enhancing the activity of ruthenium catalysts. Angew. Chem. Int. Ed. 54, 2406–2409 (2015).
Ong, W.-J., Tan, L.-L., Ng, Y. H., Yong, S.-T. & Chai, S.-P. Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability? Chem. Rev. 116, 7159–7329 (2016).
Lakhi, K. S. et al. Mesoporous carbon nitrides: synthesis, functionalization, and applications. Chem. Soc. Rev. 46, 72–101 (2017).
Schlomberg, H. et al. Structural insights into poly (heptazine imides): a light-storing carbon nitride material for dark photocatalysis. Chem. Mater. 31, 7478–7486 (2019).
Liu, M. et al. Fully condensed poly (triazine imide) crystals: extended π‐conjugation and structural defects for overall water splitting. Angew. Chem. Int. Ed. 61, e202113389 (2021).
Wu, W., Huang, L., Wang, E. & Dong, S. Atomic engineering of single-atom nanozymes for enzyme-like catalysis. Chem. Sci. 11, 9741–9756 (2020).
Chen, Z. et al. Stabilization of single metal atoms on graphitic carbon nitride. Adv. Funct. Mater. 27, 1605785 (2017).
Wang, Y. et al. Catalysis with two-dimensional materials confining single atoms: concept, design, and applications. Chem. Rev. 119, 1806–1854 (2018).
Yang, H. et al. Carbon nitride of five-membered rings with low optical bandgap for photoelectrochemical biosensing. Chem 7, 2708–2721 (2021).
Ma, J. et al. Extended conjugation tuning carbon nitride for non‐sacrificial H2O2 photosynthesis and hypoxic tumor therapy. Angew. Chem. Int. Ed. 61, e202210856 (2022).
Mahmood, J. et al. Nitrogenated holey two-dimensional structures. Nat. Commun. 6, 6486 (2015).
Zhou, G. et al. Half-metallic carbon nitride nanosheets with micro grid mode resonance structure for efficient photocatalytic hydrogen evolution. Nat. Commun. 9, 3366 (2018).
Kumar, P. et al. C3N5: a low bandgap semiconductor containing an azo-linked carbon nitride framework for photocatalytic, photovoltaic and adsorbent applications. J. Am. Chem. Soc. 141, 5415–5436 (2019).
Kim, I. Y. et al. Ordered mesoporous C3N5 with a combined triazole and triazine framework and its graphene hybrids for the oxygen reduction reaction (ORR). Angew. Chem. Int. Ed. 57, 17135–17140 (2018).
Mahmood, J. et al. Two-dimensional polyaniline (C3N) from carbonized organic single crystals in solid state. Proc. Natl Acad. Sci. USA 113, 7414–7419 (2016).
Han, D. et al. Photoelectron storages in functionalized carbon nitrides for colorimetric sensing of oxygen. ACS Sens. 7, 2328–2337 (2022).
Zhang, T. et al. Preassembly strategy to fabricate porous hollow carbonitride spheres inlaid with single Cu–N3 sites for selective oxidation of benzene to phenol. J. Am. Chem. Soc. 140, 16936–16940 (2018).
Wang, W. et al. Photocatalytic C–C coupling from carbon dioxide reduction on copper oxide with mixed-valence copper(I)/copper(II). J. Am. Chem. Soc. 143, 2984–2993 (2021).
Cai, J. et al. Crafting mussel‐inspired metal nanoparticle‐decorated ultrathin graphitic carbon nitride for the degradation of chemical pollutants and production of chemical resources. Adv. Mater. 31, 1806314 (2019).
Liang, Y. et al. Highly dispersed bismuth oxide quantum dots/graphite carbon nitride nanosheets heterojunctions for visible light photocatalytic redox degradation of environmental pollutants. Appl. Catal. B Environ. 295, 120279–120291 (2021).
Huang, C. et al. Unraveling fundamental active units in carbon nitride for photocatalytic oxidation reactions. Nat. Commun. 12, 320 (2021).
Teng, Z. et al. Edge-functionalized g-C3N4 nanosheets as a highly efficient metal-free photocatalyst for safe drinking water. Chem 5, 664–680 (2019).
Fan, K. et al. In vivo guiding nitrogen-doped carbon nanozyme for tumor catalytic therapy. Nat. Commun. 9, 1440 (2018).
Zhou, Q. et al. Cascaded nanozyme system with high reaction selectivity by substrate screening and channeling in a microfluidic device. Angew. Chem. Int. Ed. 61, e202112453 (2021).
Zhang, P. et al. Modified carbon nitride nanozyme as bifunctional glucose oxidase-peroxidase for metal-free bioinspired cascade photocatalysis. Nat. Commun. 10, 940 (2019).
Huang, Y., Ren, J. & Qu, X. Nanozymes: classification, catalytic mechanisms, activity regulation, and applications. Chem. Rev. 119, 4357–4412 (2019).
Vázquez-González, M. et al. Mimicking horseradish peroxidase functions using Cu2+-modified carbon nitride nanoparticles or Cu2+-modified carbon dots as heterogeneous catalysts. ACS Nano 11, 3247–3253 (2017).
Ju, E. et al. Copper(II)-graphitic carbon nitride triggered synergy: improved ROS generation and reduced glutathione levels for enhanced photodynamic therapy. Angew. Chem. Int. Ed. 55, 11467–11471 (2016).
Wu, J. et al. Nanomaterials with enzyme-like characteristics (nanozymes): next-generation artificial enzymes (II). Chem. Soc. Rev. 48, 1004–1076 (2019).
Chen, X. et al. Bound oxygen-atom transfer endows peroxidase-mimic M–N–C with high substrate selectivity. Chem. Sci. 12, 8865–8871 (2021).
Ji, S. et al. Matching the kinetics of natural enzymes with a single-atom iron nanozyme. Nat. Catal. 4, 407–417 (2021).
Zhang, R. et al. Edge‐site engineering of defective Fe–N4 nanozymes with boosted catalase‐like performance for retinal vasculopathies. Adv. Mater. 34, 202205324 (2022).
Jiao, L. et al. Densely isolated FeN4 sites for peroxidase mimicking. ACS Catal. 10, 6422–6429 (2020).
Wan, K., Jiang, B., Tan, T., Wang, H. & Liang, M. Surface‐mediated production of complexed •OH radicals and Fe=O species as a mechanism for iron oxide peroxidase‐like nanozymes. Small 18, 2204372 (2022).
Wang, H. et al. Covalent triazine frameworks for carbon dioxide capture. J. Mater. Chem. A 7, 22848–22870 (2019).
Xie, J. et al. Efficient visible light-driven water oxidation and proton reduction by an ordered covalent triazine-based framework. Environ. Eng. Sci. 11, 1617–1624 (2018).
Devautour-Vinot, S. et al. Structure and dynamics of the functionalized MOF type UiO-66(Zr): NMR and dielectric relaxation spectroscopies coupled with DFT calculations. Chem. Mater. 24, 2168–2177 (2012).
Zhu, X. et al. A superacid-catalyzed synthesis of porous membranes based on triazine frameworks for CO2 separation. J. Am. Chem. Soc. 134, 10478–10484 (2012).
Xiao, X. et al. A promoted charge separation/transfer system from Cu single atoms and C3N4 layers for efficient photocatalysis. Adv. Mater. 32, 2003082 (2020).
Yang, J. et al. Dynamic behavior of single-atom catalysts in electrocatalysis: identification of Cu-N3 as an active site for the oxygen reduction reaction. J. Am. Chem. Soc. 143, 14530–14539 (2021).
Liu, H. et al. Atomically dispersed Cu catalyst for efficient chemoselective hydrogenation reaction. Nano Lett. 21, 10284–10291 (2021).
Sun, T. et al. Engineering of coordination environment and multiscale structure in single-site copper catalyst for superior electrocatalytic oxygen reduction. Nano Lett. 20, 6206–6214 (2020).
Tian, S. et al. Temperature-controlled selectivity of hydrogenation and hydrodeoxygenation in the conversion of biomass molecule by the Ru1/mpg-C3N4 catalyst. J. Am. Chem. Soc. 140, 11161–11164 (2018).
Huang, P. et al. Selective CO2 reduction catalyzed by single cobalt sites on carbon nitride under visible-light irradiation. J. Am. Chem. Soc. 140, 16042–16047 (2018).
Zhang, L. et al. Direct observation of dynamic bond evolution in single‐atom Pt/C3N4 catalysts. Angew. Chem. Int. Ed. 59, 6224–6229 (2020).
Zhou, Y., Zhang, L. & Wang, W. Direct functionalization of methane into ethanol over copper modified polymeric carbon nitride via photocatalysis. Nat. Commun. 10, 506 (2019).
Rahim, M. A. et al. Low-temperature liquid platinum catalyst. Nat. Chem. 14, 935–941 (2022).
Dong, S. et al. GSH‐depleted nanozymes with hyperthermia‐enhanced dual enzyme‐mimic activities for tumor nanocatalytic therapy. Adv. Mater. 32, 2002439 (2020).
Luo, L. et al. Binary Au-Cu reaction sites decorated ZnO for selective methane oxidation to C1 oxygenates with nearly 100% selectivity at room temperature. J. Am. Chem. Soc. 144, 740–750 (2021).
Shiraishi, Y. et al. Resorcinol–formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion. Nat. Mater. 18, 985–993 (2019).
Zhang, L., Morshedi, M., Kodikara, M. S. & Humphrey, M. G. Giant multi‐photon absorption by heptazine organometalation. Angew. Chem. Int. Ed. 61, e202208168 (2022).
Fan, X. et al. Construction of graphitic C3N4-based intramolecular donor–acceptor conjugated copolymers for photocatalytic hydrogen evolution. ACS Catal. 5, 5008–5015 (2015).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Liu, Z., Lu, T. & Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: electronic structure, electronic spectrum, and optical nonlinearity. Carbon 165, 461–467 (2020).
Liu, Z., Wang, X., Lu, T., Yuan, A. & Yan, X. Potential optical molecular switch: Lithium@cyclo[18]carbon complex transforming between two stable configurations. Carbon 187, 78–85 (2022).
Wei, H. & Wang, E. Fe3O4 magnetic nanoparticles as peroxidase mimetics and their applications in H2O2 and glucose detection. Anal. Chem. 80, 2250–2254 (2008).
Wang, L. et al. A glucose/O2 fuel cell-based self-powered biosensor for probing a drug delivery model with self-diagnosis and self-evaluation. Chem. Sci. 9, 8482–8491 (2018).
Wang, Q., Zhang, X., Huang, L., Zhang, Z. & Dong, S. GOx@ZIF-8(NiPd) nanoflower: an artificial enzyme system for tandem catalysis. Angew. Chem. Int. Ed. 56, 16082–16085 (2017).
Ye, X. et al. FRET modulated signaling: a versatile strategy to construct photoelectrochemical microsensors for in vivo analysis. Angew. Chem. Int. Ed. 133, 11880–11884 (2021).
Urban, P. L. Please avoid plotting analytical response against logarithm of concentration. Anal. Chem. 92, 10210–10212 (2020).
Hong, Q., Shen, Y., Liu, S. & Zhang, Y. Re-examination of plotting analytical response against different forms of concentration. Anal. Chem. 93, 11910–11914 (2021).
Hou, Y. et al. Growth of robust carbon nitride films by double crystallization with exceptionally boosted electrochemiluminescence for visual DNA detection. Adv. Opt. Mater 11, 2202737 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22174014 and 22074015), and the Fundamental Research Funds for the Central Universities. We thank Prof. Yingpu Bi (Lanzhou Institute of Chemical Physics) for offering synchronous illumination X-ray photoelectron spectroscopy techniques.
Author information
Authors and Affiliations
Contributions
Y.Z. and Q.H. conceived and designed the experiments. Q.H. performed the synthesis, characterization, activity evaluation, mechanism studies, and intelligent response sensor of CuSAC6N6. H.Y. performed the DFT calculation. Y.F. and W.L. assisted in microwave-assisted condensation. C.Z., Z.W., and X.C. participated in the activity evaluation. All authors contributed to the analysis and discussion of the results. Q.H., H.Y., and Y.Z. co-wrote the manuscript and Y.Z., Z.Z., S.L., and Y.S. revised the manuscript. All authors reviewed the manuscript. Y.Z. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Katarzyna Matras-Postolek, Felix Plasser and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hong, Q., Yang, H., Fang, Y. et al. Adaptable graphitic C6N6-based copper single-atom catalyst for intelligent biosensing. Nat Commun 14, 2780 (2023). https://doi.org/10.1038/s41467-023-38459-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-38459-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
