
Abstract
We are currently witnessing transformative change for people with cystic fibrosis with the introduction of small molecule, mutation-specific drugs capable of restoring function of the defective protein, cystic fibrosis transmembrane conductance regulator (CFTR). However, despite being a single gene disorder, there are multiple cystic fibrosis-causing genetic variants; mutation-specific drugs are not suitable for all genetic variants and also do not correct all the multisystem clinical manifestations of the disease. For many, there will remain a need for improved treatments. Those patients with gene variants responsive to CFTR modulators may have found these therapies to be transformational; research is now focusing on safely reducing the burden of symptom-directed treatment. However, modulators are not available in all parts of the globe, an issue which is further widening existing health inequalities. For patients who are not suitable for- or do not have access to- modulator drugs, alternative approaches are progressing through the trials pipeline. There will be challenges encountered in design and implementation of these trials, for which the established global CF infrastructure is a major advantage. Here, the Cystic Fibrosis National Research Strategy Group of the UK NIHR Respiratory Translational Research Collaboration looks to the future of cystic fibrosis therapies and consider priorities for future research and development.
Similar content being viewed by others
Introduction
Cystic fibrosis (CF) affects ~100,000 people globally, with >10,000 on the UK’s patient registry (https://www.cysticfibrosis.org.uk/the-work-we-do/uk-cf-registry/reporting-and-resources). For decades regarded as a fatal inherited disease of childhood, prognosis has improved greatly so that there are now more adults than children in many regions. CF is however still associated with reduced life expectancy and a high treatment burden related to its multisystem manifestations. Many people living with CF now have access to CFTR modulator therapies which, in many cases, have dramatically improved outcomes. Access to these high-cost drugs is not universal, and even in regions where drugs are reimbursed, a proportion of people cannot benefit due to possessing non-responsive gene variants. Here, the CF National Research Strategy group, part of the National Institutes of Health and Care Research (NIHR) Respiratory Translational Research Collaborative review the status of CF today and outline challenges and priority areas for future research.
The cystic fibrosis landscape today
The last few decades: steadily improving outcomes
Many factors over the last few decades have contributed to greatly improved outcomes experienced by people with (pw)CF today (Fig. 1) including (i) timely (early postnatal life) diagnosis, (ii) centre-based, multidisciplinary care delivery models underpinned by a (iii) co-ordinated infrastructure based on (inter)national patient registries, clinical trials networks and partnering patient organisations.

From the CF Trust Patient Registry Report (https://www.cysticfibrosis.org.uk/sites/default/files/2022-03/2020%20Annual%20data%20report%20-%20Version%203.pdf).
Newborn screening (NBS) programmes, in which heel-prick blood tests are obtained in the first few days of life, have been in place for decades for conditions such as hypothyroidism and phenylketonuria. In the UK and many other countries, these programmes now include CF, leading to a significantly reduced time to diagnosis1. Currently, most programmes incorporate DNA analysis as a second stage test to improve the specificity and performance of the protocol and a positive screen will then be confirmed as a CF diagnosis with the gold-standard test, sweat chloride2. For the majority of CF infants recognised through NBS, this has been a positive intervention facilitating early access to therapies. There is good evidence that earlier diagnosis has a profound impact on clinical outcomes such as weight gain and survival and is associated with reductions in treatment burden and costs1,3.
Long before the recent advances in drug development discussed later in this article, the development of internationally agreed, evidence-based Standards of Care (SoC), underpinned by clinical trials and systematic reviews, drove substantial improvements in clinical outcomes for people with pwCF4,5. One major advance was the adoption of centre-based, highly specialised multidisciplinary teams. Respiratory disease remains the predominant manifestation of CF with a progressive decrease in lung function, most commonly assessed with the spirometric measurement of forced expiratory volume in the 1st second (FEV1) standardised for height and sex and expressed as percent predicted (pp) of normal. Conventional management strategies6,7,8,9,10 include early initiation of airway clearance with physiotherapy and mucus-clearing, inhaled drugs; antimicrobials through a variety of routes; escalation of treatment for episodes of increased chest symptoms (pulmonary exacerbations, PEx); supplementing pancreatic enzymes and fat-soluble vitamins, which are deficient due to pancreatic ductal obstruction, thereby optimising growth and nutrition; and routine surveillance for developing complications such as liver disease, CF-related diabetes and bone disease. The major cause of premature death in CF is lung disease progression with the eventual development of respiratory failure. At this stage, lung transplantation has been shown to confer a survival benefit for pwCF, but is limited by organ availability11.
Progress in CF has been aided by a strong global infrastructure supporting clinical care and accelerating research. CF patient registries of patient-consented demographic and clinical data are valuable resources for studying natural history/ long‐term health outcomes12, understanding impacts of treatments and informing future research. As examples, the UK CF Registry13 contains data from 10,655 people, an estimated 99% of individuals with CF in the UK under the care of specialist CF centres and clinics. The European Cystic Fibrosis Society Patient Registry (ECFSPR) (https://www.ecfs.eu/projects/ecfs-patient-registry/intro) was developed over a decade ago and now collects data from 38 countries and > 50,000 patients (www.ecfs.eu/projects/ecfs-patient-registry/annual-reports). Clinical trial networks have been key in catalysing clinical research for rare diseases. Several CF trials networks exist worldwide, the largest being the CF Foundation’s (CFF) Therapeutics Development Network (CFF TDN; https://www.cff.org/researchers/therapeutics-development-network) in North America. The European Cystic Fibrosis Society (ECFS) Clinical Trials Network (CTN; https://www.ecfs.eu/ctn) comprises 57 trials sites in 17 countries14. CTN works closely with national networks such as the UK CF Clinical Trial Accelerator Platform (https://www.cysticfibrosis.org.uk/the-work-we-do/clinical-trials-accelerator-platform) amongst others. Patient organisations (PO), such as the CFF (https://www.cff.org/), the UK CF Trust (https://www.cysticfibrosis.org.uk/) and the federation of European national CF Associations, CF Europe (https://www.cf-europe.eu/), are another key aspect of global CF infrastructure; several newer organisations are growing in regions such as the Middle East (https://www.mecfa.org/) and Latin America15. Regular engagement through these organisations builds strong, open relationships with the CF lay community enabling representation of and advocacy for people with CF and their families. The combined impact of the advances above led to mortality rates decreasing annually in the UK by 2% during 2006–2015. For pwCF born today survival into the fifth decade was predicted, even prior to the arrival of cystic fibrosis transmembrane conductance regulator (CFTR) modulators, the focus of the next section16.
The last few years: transformational change from CFTR modulator drugs
Building on this substantial progress in outcomes, the last decade has witnessed the development of new drugs targeting the basic defect in CF, with substantial health and quality of life benefits. The cause of CF is a reduction in the amount or function of CFTR protein at the surface of epithelial cells in the lungs and other organs. This directly reduces epithelial chloride and bicarbonate transport through CFTR and also perturbs function of a range of other ion channels and intracellular pathways with which CFTR interacts.
There are more than 350 recognised CF-causing mutations in the CFTR gene, from a total of >2000 identified variants (https://cftr2.org/). CFTR mutations have historically been grouped into six classes, based on how they impact CFTR transcription/ translation, intracellular trafficking or function. In practice, this distinction is not clear-cut, and the same mutation can cause defects in multiple aspects of CFTR expression17. For example, the most common mutation, found on at least one allele in around 90% of UK CF patients, is p.Phe508del, a deletion of three base pairs resulting in the deletion of phenylalanine at codon 508. This is generally regarded as a class II mutation leading to aberrant folding/ trafficking of CFTR and proteasome-mediated degradation; however, p.Phe508del CFTR also demonstrates reduced channel open probability (class III), and instability leading to rapid turnover at the cell membrane (class VI)18,19. Knowledge of different CFTR mutation classes has underpinned the development of new molecular therapies. An important initial step was the identification of ivacaftor, a CFTR potentiator that increases channel opening and ion transport20. This is highly effective in mutations where apical cell surface CFTR is present but dysfunctional (e.g., class III)21. Where mutations are mixed in class, such as p.Phe508del, ivacaftor has been combined with CFTR correctors, which improve folding, reduce protein degradation and facilitate protein trafficking to the cell surface. The initial dual drug combinations (lumacaftor/ ivacaftor and tezacaftor/ ivacaftor) led to modest clinical improvements in patients22,23, but most recently, a triple combination of two correctors targeting different aspects of CFTR misfolding (elexacaftor and tezacaftor) plus ivacaftor (ETI) has been developed. ETI is highly effective in patients with even just one copy of the p.Phe508del mutation, regardless of the second mutation24,25,26. This combination, licensed as Kaftrio® or Trikafta®, therefore offers an even greater possibility of effective CFTR modulation for ~ 90% of CF patients.
Clinically, CFTR correction can be measured by sweat chloride concentrations. In the ivacaftor and ETI trials, sweat chloride was reduced to levels below the diagnostic threshold for CF (60 mmol/L) in many subjects25,26. Trials of ETI have shown mean increases in ppFEV1 of around 14-15%25,26, beyond anything seen previously in CF. These drugs are also associated with other important clinical improvements, including in body mass index (BMI), reported well-being, and pulmonary exacerbation rates. There is also an increasing amount of data emerging from the use of CFTR modulators in routine clinical practice. The European Medicines Agency has strengthened the role of registries in data reporting by granting a favourable qualification opinion for the use of the ECFSPR as an appropriate data source for post-authorisation studies in monitoring effectiveness and safety of new drugs27; this will help ensure good quality evidence is gathered moving forward. Several large, real-world studies of ETI are underway. The PROMISE study, recently reported 6-month data, which largely mirrored efficacy and safety in clinical trials;28 improvements were observed in FEV1, BMI, quality of life and sweat chloride concentration. In Ireland and UK, the RECOVER study is underway, although at the time of writing, results have not been published. (clinicaltrials.gov NCT04602468)
However, drug costs are high: drug prices differ by geography and regional costs may not be fully transparent, but likely exceed £100,000 per patient annually in most regions. Drug discovery to proof of safety and efficacy in randomised controlled trials take up to 15 years and the rare disease space further drives high prices. Health technology appraisals by government bodies assess cost-effectiveness of new drugs, often using a cost per quality-adjusted life-year (QALY) in their deliberations. These economic evaluations require the use of health state utility values and the evidence for these in CF is sparse29. What monetary cost should be given to a longer or better-quality life is where controversy can arise, particularly when a new drug is not approved to be prescribed within a nation’s health care system.
Current status of drug development: clinical trials pipelines
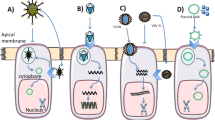
Despite the therapeutic advances of the last few decades and the recent acceleration of these with modulator therapies, the community recognises that more will continue to be needed. It is encouraging that a pipeline of new drugs trials remains active and varied (https://apps.cff.org/trials/pipeline/; https://www.ecfs.eu/ctn/clinical-trials; https://www.cysticfibrosis.org.uk/get-involved/clinical-trials/trialstracker). In this section, we briefly outline progress to date in pulmonary therapies (Fig. 2 includes references for further reading) before focussing on remaining challenges and proposed research priorities.

There are many individuals who cannot be helped by current modulator therapy because their specific CFTR mutations are not amenable to the ‘making the most of a mutant protein’ approach, the hallmark of current modulator therapy. For these patients, alternative therapies are needed such as ribosomal readthrough agents, RNA-specific approaches (transfer (t)RNAs, mRNA stabilisers and repair). Both mRNA and DNA replacement are at clinical trial stages, whilst gene editing remains an area of active research.
For patients with class I (nonsense) mutations there is hope that small molecules will be identified that can facilitate premature truncation codon (PTC) read-through and/or impede mRNA decay allowing for clinically relevant levels of functional CFTR. Ribosomal read-through agents have a chequered history. Ataluren, the most extensively developed, was promising in preclinical studies and early trials but failed to demonstrate clinical benefit in larger phase three trials30,31. The drug is licensed for certain variants of Duchenne muscular dystrophy, but a recent systematic review confirmed modest impacts on disease progression, but flagged the need for further work on quality of life and cost utility32. Other ribosomal read-through drugs are in preclinical or early phase clinical trial stages33,34.
An alternative strategy to overcome PTC mutations is engineered transfer RNAs (tRNAs) (https://apps.cff.org/Trials/Pipeline/details/10162/ReCode-Therapeutics), designed to introduce an amino acid to an elongating peptide in place of the termination codon; early preclinical studies are encouraging35. PTCs lead to short-lived mRNA due to nonsense-mediated decay (NMD) mechanisms. General NMD inhibitors would be undesirable, as this is a normal gene expression regulatory mechanism, so specific antisense oligonucleotides are being developed; these stabilise mRNA leading to restored CFTR mediated chloride current in vitro36. Oligonucleotides to repair CFTR mRNA have also been explored. One such agent, Eluforsen, was a 33 base antisense oligonucleotide (ASO) targeting p.Phe508del CFTR. Treatment in vitro and in animal studies showed restoration of CFTR function using an array of electrophysiological assays37. In human trials, Eluforsen was well tolerated with a favourable safety profile and patients who were homozygous for the p.Phe508del mutation showed improvement in their nasal potential difference (NPD) a surrogate for CFTR restoration38. Whilst this drug is no longer being developed by ProQR, a similar approach is being used to target CFTR mutations that are more challenging to overcome such as splicing mutations and nonsense mutations. Splisense has several ASOs in preclinical development for 3849 + 10KbC > T and W1282X CFTR, which should reach clinical trials in the near future39.
All of the above will be specific to certain mutations which will disadvantage some, especially those with the rarest of variants. The development of ‘mutation agnostic’ treatments suitable for anyone with CF could be achieved with DNA or mRNA replacement strategies. the major challenge of which is delivery to the respiratory epithelium. An ongoing clinical trial investigating the potential of mRNA delivery for CF has reported interim results twice in the last eighteen months. Translate Bio uses a specialized lipid-based nanoparticle carrier for mRNA delivery (MRT5005) which is aerosolized for inhalation. The first interim results showed that subjects demonstrated a > 10% change in ppFEV1; however, this was not replicated in a subsequent treatment cohort. Other companies are taking a similar approach (https://apps.cff.org/Trials/Pipeline/details/10159/Arcturus-Therapeutics; https://www.cysticfibrosis.org.uk/news/vertex-announced-that-their-mrna-therapy-ind-has-been-cleared-by-the-fda). In terms of gene (DNA) transfer, to date, there have been several clinical trials of both viral and non-viral approaches, the vast majority of which were designed as early phase proof of principal studies without clinical efficacy read-outs40. The UK CF Gene Therapy Consortium (GTC) undertook a large, phase 2b trial of lipid-mediated CFTR gene transfer in 2014, reporting a statistically significant, but clinically modest, impact on FEV141. Currently at the preclinical stage, the GTC is working in partnership with Boehringer Ingelheim and Oxford Biomedica towards a first-in-man trial using a pseudotyped lentiviral vector42. 4D Molecular Therapeutics and Spirovant are using adeno-associated vectors to transfer CFTR DNA and are currently at phase I and pre-clinical stages respectively (https://apps.cff.org/Trials/Pipeline/details/10161/4D-710; https://apps.cff.org/Trials/Pipeline/details/10160/Spirovant-Sciences). Despite decades of research, drugs that specifically target CF inflammation remain elusive. The inflammatory process in CF is complicated43, characterised by early and sustained neutrophil influx and high levels of elastase which correlate with structural damage on imaging even in early life. There is some evidence that normal resolution mechanisms are impaired44, so that inflammation can persist even if an infective insult is cleared45. There is certainly evidence that inflammation persists when CFTR function is corrected46. The presence of excessive neutrophilia, pro-inflammatory macrophages and a range of pro-inflammatory mediators provide numerous targets for therapeutic development47. One approach has been to target the neutrophil chemoattractant leukotriene B4 (LTB4) to inhibit neutrophil recruitment to the lung. Amebulant, an LTB4 antagonist, showed promise in preclinical evaluation but was associated with increased pulmonary exacerbations in a phase II study48. In contrast, the LTA4 hydroxylase inhibitor acebilustat (which reduces LTB4 levels rather than blocking the effect of LTB4 completely) was well tolerated and led to reduced lung neutrophil numbers in early phase studies49. It did not however meet its primary endpoint of improvement in FEV1, nor did it reduce exacerbations in a 48-week phase 2 trial50. The cannabinoid receptor agonist, lenabasum, reduces IL-6 transcription in macrophages in vitro51, suggesting direct effects on the inflammatory potential of these cells. A phase II clinical trial of lenabasum in pwCF showed modest clinical effects but a significant reduction in sputum interleukin-852, and further studies are now underway. Despite significant effects on lung function, the impacts of CFTR modulators on lung inflammation are not yet known. A key early step in the process of tissue repair is the resolution of inflammation and the interaction of immune cells such as macrophages (known to be dysfunctional in CF) with resident stem cells may be key to this process53,54. Thus, the prospect of lung tissue repairing itself once CFTR function has been restored is intriguing. The ability of healthy lung tissue to regenerate after damage is well described and is contingent on the activity of basal cells in the airway (resident lung stem cells) that proliferate and differentiate in response to injury55. Whether this process occurs normally in CF is unknown but radiological improvements in inflammatory changes such as bronchiectasis have been reported in patients after prolonged modulator treatment56. We could speculate that by combining CFTR correction and anti-inflammatory strategies we may provide the perfect environment for tissue repair to thrive.
Infection (bacteria, mycobacteria and fungi) is a major problem in the CF lung. No published data are yet available on the impacts of ETI on infection, although there is a focussed substudy as part of PROMISE28. However, as many patients are already chronically infected at the time of commencing these therapies, we consider it likely that anti-infectives will continue to be required. The armamentarium available, limited both in scope and efficacy, means new agents must continue to be developed for these common organisms and also rarer, but more challenging pathogens such as the non-tuberculous mycobacterium (NTM) M. abscessus57. A wide range of drugs is passing through the CFF TDN pipeline, including antibiotic adjuvants, biofilm targeted approaches and bacteriophages.
Airway surface rehydration and reducing mucus viscosity are additional targets of new therapeutic approaches. Sodium channel (ENaC) blockers have been under development for some time58, although none has yet progressed through pivotal trials to licensing59. An agonist of an alternative chloride channel, TMEM16A, termed EDT002 is in early phase trials (https://apps.cff.org/Trials/Pipeline/details/10172/ETD002)60. OligoG, a seaweed-derived oligonucleotide, acts both on mucus and bacterial biofilms and is currently in phase 2 trials61. Whether these agents will continue to be needed by patients experiencing substantial health benefits from CFTR modulators remains to be seen, but there certainly remains an unmet need in those for whom modulators are not appropriate; such drugs may also be useful outside CF for other forms of bronchiectasis and chronic obstructive pulmonary disease (COPD) for which treatments are currently lacking62.
Current challenges in CF care
The growing CF population and changing care delivery needs
Improved health for pwCF has resulted in a significant increase in numbers of adults living with the condition63. With the introduction of CFTR modulators, morbidity and life expectancy are anticipated to improve further and the adult population will continue to expand in number for decades. Although respiratory and gastrointestinal morbidity may remain central to CF care (at least until children commence modulators very early in life), new and emerging complications associated with increased longevity means the CF care community must continually adapt and respond to the needs of this changing population. For example, microvascular complications including retinopathy, peripheral neuropathy and chronic kidney disease are now being identified in people with longstanding CF-related diabetes64, and patients who developed CF-related low bone mineral density in childhood/ early adult life are at greater risk of fracture from age-related bone loss, particularly following the menopause65. New health problems are also being identified as pwCF age, one example being an increased risk of gastrointestinal cancer, in particular of the bowel66. As a consequence, surveillance colonoscopy is now recommended for patients over the age of 40 years and in some regions for transplant recipients over the age of 30 years67. Although speculative, the high fat/high-calorie diet recommended for decades for pancreatic insufficient patients with CF may result in hypercholesterolaemia, type 2 diabetes and coronary artery disease in later life68. Even before the advent of CFTR modulators, pregnancy in female pwCF was increasing, and we expect this to continue69. The safety of modulators in pregnancy is now the subject of observational studies (Mayflowers, clinicaltrials.gov NCT04828382) and there will likely be a requirement for expanded antenatal and obstetric services for women with CF.
Following the introduction of CFTR modulators and the consequent reduction in exacerbation frequency and hospitalisation, the most appropriate model of care for pwCF is having to be reconsidered. The traditional outpatient model is time inefficient for healthcare teams and pwCF, and may not be well suited to those with mild or stable disease. Catalysed by the COVID-19 pandemic, virtual clinics using remote monitoring equipment (such as home spirometry, weight and oxygen saturation) and secure telephone/video conference platforms are becoming increasingly common68.
Remaining issues for those receiving CFTR modulator drugs
A number of issues remain, even for those people successfully established on modulators and demonstrating clinical benefits:
Disease will progress, albeit more slowly, and will be more challenging to monitor
Effective CFTR modulators will likely slow or at best, halt disease progression, but will not reverse a disease that has already become fixed, examples being pancreatic destruction in the majority, bronchiectasis and absence of the vas deferens. Pulmonary exacerbations still recur albeit less frequently, chronic infections are not fully cleared and there is persistent airway inflammation46,70,71. It is essential that we do not become complacent about disease progression in this population and yet monitoring will likely become more challenging. Effective surveillance for infection is critical in asymptomatic patients and underpins the management of young healthy children with CF who demonstrate disease progression despite a lack of symptoms72,73,74,75. Many patients previously able to expectorate sputum become non-productive on CFTR modulators, so pathogen surveillance will increasingly rely on alternative samples. Oropharyngeal sampling lacks sensitivity76 and bronchoalveolar lavage is invasive and time-consuming. Recently, interest has focussed on sputum-induction, which has been shown in several studies to outperform oropharyngeal sampling77,78,79. It is well tolerated, quick and relatively repeatable, even in young children. Similarly, spirometry may become less useful than more sensitive, but more time-consuming, tests such as lung clearance index or imaging.
Responses to treatments, including CFTR modulators, vary
There is considerable variability in patient response to modulators, much of which remains poorly understood, examples being FEV1 and sweat chloride responses. For lung disease, this may not be surprising: at least 50 candidate modifier genes have been proposed80,81, as well as several non-genetic influences on lung disease such as exposure to tobacco smoke and chronic P. aeruginosa infection82,83. Both of these may reduce modulator efficacy84,85. It seems likely that there will also be genetic influences on the absorption and metabolism of the drugs, an area ripe for further research86.
Adherence to CFTR modulators (and other treatments) may be challenging
Adherence to CF medications is often suboptimal with some studies suggesting that modulators may not be an exception in this regard87,88. For subjects experiencing great benefit, continuing to adhere to their symptom-directed therapies may be challenging; modulator trials to date have been ‘on top of’ such drugs and impact could be lessened if they are dropped. Advances are being made in supporting patients with adherence in other areas, such as mobile health applications, smart inhalers and chipped pill containers89,90.
Some people will experience side effects or tolerability issues
The long-term data available on ETI is limited, so we need to remain vigilant for emerging side effects, particularly those that are rare. Post-market surveillance and registry studies are invaluable in this regard. In addition to adverse effects reported in clinical trials, and in contrast to the group improvements in structured quality of life questionnaire scores, there is some evidence to link CFTR modulator therapies with worsening mental health in some individuals91, meaning adequate psychological and social support needs to be in place. Reporting of side effects in younger children is of paramount importance given the length of time they may be exposed to these agents.
Issues for patients not receiving CFTR modulators
Even in regions where modulators are funded, up to 10–15% pwCF do not have access to these drugs due to either unsuitable CFTR mutations or drug intolerances. Rather than confirmed as non-responsive, many CFTR mutations are simply so rare that they are poorly understood; ex vivo testing approaches may be helpful as discussed further in the next section. Other people may live in areas which have not yet approved reimbursement; the adverse impact of delayed access on health outcomes is substantial and has recently been modelled92. The team demonstrated that immediate access as compared with a 4-year delay would reduce the number of individuals with severe lung disease by 60%, increase the number with mild lung disease by 18% and reduce the number of pulmonary exacerbations by 19%. Over a 10-year period, deaths would be reduced by 15% and median age of survival increased by >9 years92. Over time therefore, the gap in physical health between people who are and are not receiving modulators will widen. In parallel, there may also be impacts on mental health and well-being if patients feel ‘left behind’. There are already trials of novel therapies targeted specifically at the non-modulator group and as these grow in number, competition for this small population may emerge and there may be perceived pressure to participate. In addition to those currently in the pipeline mentioned above, there is substantial preclinical work in genetic-based therapies such as gene editing93,94,95, which may in time translate into trials. This group of patients will also have an even greater need than the modulator-treated group for drugs targeting downstream consequences of CF such as anti-inflammatories, mucoactive agents or anti-infectives so it is essential we maintain momentum in these areas.
Future directions & research priorities
Identifying priorities with the CF community
The James Lind Alliance Priority Setting Partnership (JLA PSP) in CF (https://www.jla.nihr.ac.uk/priority-setting-partnerships/cystic-fibrosis/), a list of top health priorities in CF, was compiled based on inputs from >600 contributors from >30 countries, including pwCF, family members and clinical care teams96. The 2017 partnership publication has been influential, stimulating both research and funding to address several of the priority questions. The top research priority in both the JLA PSP and in a large US survey was ‘reducing the burden of treatment’97. Focus on the role of the more burdensome conventional treatments (such as nebulised therapies) is increasingly relevant with the widespread availability of CFTR modulators. This is the foundation for the CF STORM (www.cfstorm.org.uk) and SIMPLIFY (clinicaltrials.gov NCT04378153) trials which aim to assess the real-world effects of stopping mucoactive agents; the latter has recently been published confirming non-inferiority of stopping either nebulised hypertonic saline or DNase over the short-term98. The CF community has also identified airway clearance techniques as the most burdensome of treatments;99 the question ‘Can exercise replace chest physiotherapy for people with CF?’ ranked highly96. Trial design here will be a major challenge, particularly in relation to ensuring compliance with the allocation arm and adherence to remaining treatments. The JLA PSP exercise was undertaken before the advent of increasing access to ETI. Priorities may now be very different for those with and without access to CFTR modulators. For this reason the CF JLA PSP has recently been refreshed (https://www.cysticfibrosis.org.uk/news/refreshed-top-10-research-priorities-for-cf-revealed).
Maintaining pace in development of- and access to- new therapies
The ECFS Task Force for speeding up access to new therapies has highlighted several priorities in this area100,101,102. Theratypes are groups of CFTR variants categorised according to their effect on CFTR protein103. Theranostics describes a personalised medicine approach to determine whether these variants respond to existing (or novel) drugs by pre-assessing agents directly on a patient’s tissue ex vivo101. This has been proposed as a pathway for testing of CFTR modulator therapies in individuals who are unlikely to be included in conventional clinical trials. There are multiple preclinical model systems for theratyping;104 in one example, spheroids are grown from epithelial cells (most commonly intestinal, but also respiratory) in which the apical cell surface faces into the lumen. Activation of CFTR leads to quantifiable swelling, which can serve as a personalised efficacy read-out for therapies restoring CFTR function105. The HIT-CF (www.hitcf.org) program is testing drug candidates on rectal organoids from pwCF with rare mutations. A cohort will then be selected for a clinical trial of various CFTR function restoring therapies. An ex vivo testing approach has already led the US Food & Drug Administration (FDA) to expand its licensed indication for modulators, although to date, the European Medicines Agency (EMA) has not adopted the same position.
Future clinical trials in CF will face very different challenges to those experienced to date102, likely different depending on whether they seek to recruit pwCF on or not on CFTR modulators. Those with mutations unsuitable for modulators represent generally small populations. Modifications to trial design may be required therefore to maximise power, such as adaptive designs, use of historical (including within-subject) controls, and seamless phase 2-3 protocols106. An in vitro theranostics approach as mentioned above may enable enrichment for those most likely to respond. Trial networks can prove valuable hub and spoke models allowing patients from these small subgroups to access trials being conducted outside their own centre107. Design of gene therapy studies poses some additional, specific challenges: there will be a need for long-term follow up, particularly for integrating viral vectors with a theoretical oncogenic risk (https://www.fda.gov/vaccines-blood-biologics/biologics-guidances/cellular-gene-therapy-guidances); previous participants could find themselves excluded from new trials of similar agents if immune response is a concern108,109. Providing comprehensive information as part of the consent process will be essential but is difficult in the face of known unknowns. Any currently plausible gene therapy/ editing approach will be topically targeted to the lungs, so there will not be the systemic benefits seen in some people on modulators. Finally, in terms of outcome measures, assays to confirm gene expression may be invasive (e.g., requiring bronchoscopy), prone to sampling error or lacking in sensitivity/ precision. As an example of the latter, even with ivacaftor, nasal potential difference measurements were a poor reflection of CFTR function in the sweat gland or of clinical benefit110, and the assay has not been used since in trials programmes for more recent modulators. With regard to clinical efficacy, the unprecedented success of ETI- in particular the large improvements achievable in FEV1 - has perhaps led to expectations for newer drugs which may be unrealistic and lead to disappointment. In our opinion, it would be unfortunate if a drug was rejected in its early stages based on more modest efficacy which still provided clinical benefit for this population with a high unmet need. When progression of a product is dependent on early/ mid-phase efficacy signal, academic investigators and commercial companies may differ in magnitude considered to be required.
In studies wishing to assess new drugs given to patients already receiving CFTR modulators a major challenge will be in assessing efficacy and selection of outcome measures. Patients receiving modulators are likely to have higher baseline FEV1, better BMI and fewer pulmonary exacerbations than previously111. This could mean that seeking further significant improvements in these clinical parameters is more challenging, requiring longer study times or larger patient numbers. Patient-reported outcome measures may also need to be revised, since they exhibit a ceiling effect in those with milder disease112. Alternative outcome measures may offer greater sensitivity to residual disease (Table 1). For example lung clearance index (LCI) has been especially useful to date in children and those with earlier-stage disease113. LCI has been used as a primary outcome in paediatric CFTR modulator studies, where FEV1 was within the normal range114, and will likely now find greater application in older patients on modulators. Chest computed tomography (CT) imaging provides detail about structural changes in the lungs and can provide proxy measures of small airway change such as gas trapping and mucus plugging115. The effect of widespread ETI treatment may also however make such findings harder to identify. New radiation-free magnetic resonance imaging (MRI) techniques, in particular those using hyperpolarised tracer gases to visualise ventilation distribution or other functional adjuncts such as oxygen-enhancement116,117, appear to be a sensitive measure of early airway change, and also provide regional detail. They may have a particular role in early phase studies or ‘n of 1’ trials in patients with unusual genotypes. Other outcome measures may need to be tailored to specific questions, such as gastrointestinal symptoms or imaging. If a study is placebo controlled, or requires washout of an existing modulator, recruitment may be an issue. Patients may not be willing to give up their proven modulators to trial an experimental drug with an uncertain chance of clinical efficacy118. Coming off modulators carries risks119, so placebo-controlled studies may be both ethically challenging and hard to recruit to. Short washout periods which are adequate for a change in sweat chloride and will minimise risk for patients may be useful in this context, with safety data collected subsequently from open-label, single arm extension phases106.
Infants and children with CF make up the group with the chance of maximal benefit from new treatments for CF, as they may not already have irreversible organ disease. However, their advantage in this regard is also a problem in that measuring ‘improvement’ in any outcome measure from a ‘normal’ baseline is difficult. Children over the age of around 3 years are able to perform LCI and certain imaging modalities also show promise113. To date, however, regulatory approval for ivacaftor in the youngest cohorts has been based on pharmacokinetics, pharmacodynamics (sweat chloride) and safety, with efficacy extrapolated from older cohorts. Surprisingly large changes in faecal elastase, a biomarker of pancreatic exocrine function, have been demonstrated120,121, and this assay should be included in future trials of systemic CFTR-targeted approaches.
Given these challenges, it is essential that the patient community’s opinions are factored into trial design. The first step is to better understand what motivates people with CF to enrol into and remain in trials. A recent survey from Canada122, at a time when ~50% of the CF population had access to older modulator drugs but not yet to ETI, reported that adults with CF, or parents of children with CF, were most interested in trials that targeted the root or underlying cause of CF, inflammation and infection. In the UK, feedback from workshops facilitated by the CF Trust has revealed that pwCF are motivated to take part in research not only by the possibility of personal benefit but also in advancing treatment for others with CF and driving development of future options. Barriers to participation include adding to an already burdensome regime, fitting in around life (work, school, family), and avoidance of certain procedures. A UK based Delphi study confirmed these findings and identified a number of other aspects as facilitators or barriers123. Time required and missing out on other activities was a common theme, lending support for home-based or decentralised trial designs, for at least some visits. The COVID-19 pandemic has driven these developments faster than may have otherwise occurred124. Much is still to be learned regarding data/ sample quality, participants’ opinions and regulatory agencies’ positions, but we consider this type of approach an important consideration for some studies.
In addition to the patients’/ families’ involvement, collaboration between key stakeholders involved in the design, planning and delivery of clinical trials will improve efficiencies and increase chances of successful delivery. Interdisciplinary collaboration is particularly important for early phase translational studies where knowledge exchange is essential125. In the UK for example, the supporting research infrastructure includes our NIHR Respiratory Translational Research Collaboration (https://www.nihr.ac.uk/explore-nihr/support/respiratory-trc.htm), the National CF Research Strategy Group (authors of this article) and the CF Clinical Trials Accelerator Platform (https://www.cysticfibrosis.org.uk/the-work-we-do/clinical-trials-accelerator-platform); all of these are supported by the CF Community Involvement workstream of the CF Trust. Finally, the UK CF Registry has a well-established role in pharmacovigilance, including post marketing authorisation studies for safety and real-world effectiveness of CFTR modulators70,126. The registry can also support clinical trials with adaptations for eligibility assessment, recording consent, randomisation and outcome collection. It is currently hosting two randomised controlled drug trials, CF START (https://www.cfstart.org.uk/) and CF STORM (www.cfstorm.org.uk), both of which have been pragmatically designed to fit in with routine care, adding relatively little burden to either patient or clinical research team.
Addressing inequity in access to care
Authors of this article are largely from UK and deliver services in well-resourced healthcare systems. Even within the UK though, there have been prolonged periods of negotiation over new, high-cost drugs which in some cases have led to delays in access. These are nothing compared to the challenges faced by CF care teams in low and middle-income countries; even before high-cost CFTR modulators were developed, there were large differences globally in the provision of funding and expertise for CF care which was broadly reflected in survival94,95. For example, many patients in eastern European countries have limited access to specialist services and modulator therapies are not funded at all96. There is increasing recognition that CF is not the rarity it was previously considered in countries with non-white populations; many of these are in low and middle income brackets meaning the establishment of multidisciplinary services poses a major challenge. Rare CFTR mutations are commoner in these populations, posing a major unmet need with regard to modulator therapies. Even within high-income countries such as US and UK, patients’ and families’ socioeconomic factors are known to play a substantial role in disease expression of CF95,97, with particular impact in the early years of childhood;98 the impact- if any- of modulators on this gap requires research. Whilst tackling systemic, economic issues is a daunting challenge, there are ways in which the CF community can help: European Cystic Fibrosis Society (ECFS) has recently launched a twinning project (https://www.ecfs.eu/ecfs-standards-care/twinning), linking up highly experienced centres with those in less well-resourced areas for the sharing of clinical expertise, protocols and training materials. Participation in the ECFS Patient Registry where appropriate, may aid countries demonstrating unmet need to their funders. This was a major theme in the recent Lancet Respiratory Medicine Commission which took a global perspective on CF care, and in which several potential fruitful areas for future development are outlined127. As stated therein: ‘A concerted effort is needed to ensure that all patients with cystic fibrosis have access to high-quality health care in the future.’ Evidence-based standards of care guidelines produced by the European CF Society have facilitated access to resources across developing CF services.
Conclusions
The collaborative working model adopted by the CF community to date, which has underpinned substantial progreess, needs to further continue to address health inequities and to ensure that the pipeline of new treatments remains active and successful. The NIHR Respiratory Translational Research Collaboration established the CF National Research Strategy Group specifically to identify areas of unmet need and provide potential solutions to challenges. The group of people with genetic variants unsuitable for CFTR modulator drugs remains a high priority and trials of genetic therapies will be a major focus of the next few years. The recent JLA Priority setting partnership (PSP) illustrated this, identifying treatment for this subgroup as the top priority research area. Knowledge arising from the PSP will be used to support the design and funding of both national and international research efforts to ensure that the progress made to date is further built upon, until (to quote the CF Trust) the hope for ‘a life unlimited’ by CF becomes a reality.
References
Castellani, C., Massie, J., Sontag, M. & Southern, K. W. Newborn screening for cystic fibrosis. Lancet Respir. Med. 4, 653–661 (2016).
Barben, J. et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J. Cyst. Fibros. 16, 207–213 (2017).
Dijk, F. N., McKay, K., Barzi, F., Gaskin, K. J. & Fitzgerald, D. A. Improved survival in cystic fibrosis patients diagnosed by newborn screening compared to a historical cohort from the same centre. Arch. Dis. Child. 96, 1118–1123 (2011).
Kerem, E., Conway, S., Elborn, S., Heijerman, H. & Consensus, C. Standards of care for patients with cystic fibrosis: a European consensus. J. Cyst. Fibros. 4, 7–26 (2005).
Castellani, C. et al. ECFS best practice guidelines: the 2018 revision. J. Cyst. Fibros. 17, 153–178 (2018).
Yang, C. & Montgomery, M. Dornase alfa for cystic fibrosis. Cochrane Database Syst. Rev. 3, CD001127 (2021).
Wark, P. & McDonald, V. M. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst. Rev. 9, CD001506 (2018).
Nevitt, S. J., Thornton, J., Murray, C. S. & Dwyer, T. Inhaled mannitol for cystic fibrosis. Cochrane Database Syst. Rev. 5, CD008649 (2020).
Ratjen, F. et al. Inhaled hypertonic saline in preschool children with cystic fibrosis (SHIP): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 7, 802–809 (2019).
Tiddens, H. et al. The effect of inhaled hypertonic saline on lung structure in children aged 3-6 years with cystic fibrosis (SHIP-CT): a multicentre, randomised, double-blind, controlled trial. Lancet Respir. Med. 10, 669–678 (2022).
Thabut, G. et al. Survival benefit of lung transplant for cystic fibrosis since lung allocation score implementation. Am. J. Respiratory Crit. Care Med. 187, 1335–1340 (2013).
Dasenbrook, E. C. & Sawicki, G. S. Cystic fibrosis patient registries: a valuable source for clinical research. J. Cyst. Fibros. 17, 433–440 (2018).
Trust, C. C. F. Trust registry annual report data 2020 (2020).
De Boeck, K., Bulteel, V. & Fajac, I. Disease-specific clinical trials networks: the example of cystic fibrosis. Eur. J. Pediatr. 175, 817–824 (2016).
Silva Filho, L. V., Castanos, C. & Ruiz, H. H. Cystic fibrosis in Latin America-Improving the awareness. J. Cyst. Fibros. 15, 791–793 (2016).
Keogh, R. H., Szczesniak, R., Taylor-Robinson, D. & Bilton, D. Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data. J. Cyst. Fibros. 17, 218–227 (2018).
Veit, G. et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 27, 424–433 (2016).
Meng, X., Clews, J., Kargas, V., Wang, X. & Ford, R. C. The cystic fibrosis transmembrane conductance regulator (CFTR) and its stability. Cell Mol. Life Sci. 74, 23–38 (2017).
Ward, C. L., Omura, S. & Kopito, R. R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 (1995).
Van Goor, F. et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl Acad. Sci. USA 106, 18825–18830 (2009).
Ramsey, B. W. et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 365, 1663–1672 (2011).
Wainwright, C. E. et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 373, 220–231 (2015).
Donaldson, S. H. et al. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 197, 214–224 (2018).
Keating, D. et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 379, 1612–1620 (2018).
Middleton, P. G. et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 381, 1809–1819 (2019).
Heijerman, H. G. M. et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 394, 1940–1948 (2019).
Agency, E. M. Qualification Opinion on The European Cystic Fibrosis Society Patient Registry (ECFSPR) and CF Pharmaco-epidemiology Studies, https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/qualification-opinion-european-cystic-fibrosis-society-patient-registry-ecfspr-cf-pharmaco_en.pdf (2018).
Nichols, D. P. et al. Clinical effectiveness of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis: a clinical trial. Am. J. Respir. Crit. Care Med. 205, 529–539 (2022).
Mohindru, B. et al. Health state utility data in cystic fibrosis: a systematic review. Pharmacoecon. Open 4, 13–25 (2020).
Zainal Abidin, N., Haq, I. J., Gardner, A. I. & Brodlie, M. Ataluren in cystic fibrosis: development, clinical studies and where are we now. Expert Opin. Pharmacother. 18, 1363–1371 (2017).
Konstan, M. W. et al. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF. J. Cyst. Fibros. 19, 595–601 (2020).
Pascual-Morena, C. et al. Restorative treatments of dystrophin expression in Duchenne muscular dystrophy: a systematic review. Ann. Clin. Transl. Neurol. 7, 1738–1752 (2020).
Crawford, D. K. et al. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros. 20, 436–442 (2021).
Liang, F. et al. High-throughput screening for readthrough modulators of CFTR PTC mutations. SLAS Technol. 22, 315–324 (2017).
Ko, W., Porter, J. J., Sipple, M. T., Edwards, K. M. & Lueck, J. D. Efficient suppression of endogenous CFTR nonsense mutations using anticodon-engineered transfer RNAs. Mol. Ther. Nucleic acids 28, 685–701 (2022).
Kim, Y. J. et al. Gene-specific nonsense-mediated mRNA decay targeting for cystic fibrosis therapy. Nat. Commun. 13, 2978 (2022).
Beumer, W. et al. Evaluation of eluforsen, a novel RNA oligonucleotide for restoration of CFTR function in in vitro and murine models of p.Phe508del cystic fibrosis. PloS one 14, e0219182 (2019).
Sermet-Gaudelus, I. et al. Antisense oligonucleotide eluforsen improves CFTR function in F508del cystic fibrosis. J. Cyst. Fibros. 18, 536–542 (2019).
Oren, Y. S. et al. Antisense oligonucleotide-based drug development for Cystic Fibrosis patients carrying the 3849+10 kb C-to-T splicing mutation. J. Cyst. Fibros. 20, 865–875 (2021).
Alton, E. W. et al. Genetic medicines for CF: Hype versus reality. Pediatr. Pulmonol. 51, S5–S17 (2016).
Alton, E. W. et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 3, 684–691 (2015).
Alton, E. W. et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 72, 137–147 (2017).
Perrem, L. & Ratjen, F. Designing clinical trials for anti-inflammatory therapies in cystic fibrosis. Front Pharm. 11, 576293 (2020).
Briottet, M., Shum, M. & Urbach, V. The role of specialized pro-resolving mediators in cystic fibrosis airways disease. Front Pharm. 11, 1290 (2020).
Garratt, L. W. et al. Changes in airway inflammation with pseudomonas eradication in early cystic fibrosis. J. Cyst. Fibros. 20, 941–948 (2021).
Harris, J. K. et al. Changes in airway microbiome and inflammation with ivacaftor treatment in patients with cystic fibrosis and the g551d mutation. Ann. Am. Thorac. Soc. 17, 212–220 (2020).
Roesch, E. A., Nichols, D. P. & Chmiel, J. F. Inflammation in cystic fibrosis: an update. Pediatr. Pulmonol. 53, S30–S50 (2018).
Konstan, M. W. et al. A randomized double blind, placebo controlled phase 2 trial of BIIL 284 BS (an LTB4 receptor antagonist) for the treatment of lung disease in children and adults with cystic fibrosis. J. Cyst. Fibros. 13, 148–155 (2014).
Elborn, J. S. et al. Phase I studies of acebilustat: biomarker response and safety in patients with cystic fibrosis. Clin. Transl. Sci. 10, 28–34 (2017).
Elborn, J. S. et al. Empire-CF study: A phase 2 clinical trial of leukotriene A4 hydrolase inhibitor acebilustat in adult subjects with cystic fibrosis. J. Cyst. Fibros. 20, 1026–1034 (2021).
Parker, J. et al. Suppression of human macrophage interleukin-6 by a nonpsychoactive cannabinoid acid. Rheumatol. Int 28, 631–635 (2008).
Chmiel, J. F. et al. Safety and efficacy of lenabasum in a phase 2 randomized, placebo-controlled trial in adults with cystic fibrosis. J. Cyst. Fibros. 20, 78–85 (2021).
Lucas, A., Yasa, J. & Lucas, M. Regeneration and repair in the healing lung. Clin. Transl. Immunol. 9, e1152 (2020).
Lechner, A. J. et al. Recruited Monocytes and Type 2 Immunity Promote Lung Regeneration following Pneumonectomy. Cell Stem Cell 21, 120–134.e127 (2017).
Donne, M. L., Lechner, A. J. & Rock, J. R. Evidence for lung epithelial stem cell niches. BMC Dev. Biol. 15, 32 (2015).
Hoare, S. et al. Ivacaftor imaging response in cystic fibrosis. Am. J. Respir. Crit. Care Med. 189, 484 (2014).
Martiniano, S. L. et al. Challenging scenarios in nontuberculous mycobacterial infection in cystic fibrosis. Pediatr. Pulmonol. 55, 521–525 (2020).
Shei, R. J., Peabody, J. E., Kaza, N. & Rowe, S. M. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr. Opin. Pharmacol. 43, 152–165 (2018).
Mall, M. A. ENaC inhibition in cystic fibrosis: potential role in the new era of CFTR modulator therapies. The European respiratory journal 56, https://doi.org/10.1183/13993003.00946-2020 (2020).
Danahay, H. & Gosling, M. TMEM16A: An alternative approach to restoring airway anion secretion in cystic fibrosis? Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21072386 (2020).
van Koningsbruggen-Rietschel, S. et al. Inhaled dry powder alginate oligosaccharide in cystic fibrosis: a randomised, double-blind, placebo-controlled, crossover phase 2b study. ERJ Open Res 6, https://doi.org/10.1183/23120541.00132-2020 (2020).
Tarrant, B. J. et al. Mucoactive agents for chronic, non-cystic fibrosis lung disease: a systematic review and meta-analysis. Respirology 22, 1084–1092 (2017).
McCormick, J. et al. Comparative demographics of the European cystic fibrosis population: a cross-sectional database analysis. Lancet 375, 1007–1013 (2010).
Alves, C., Della-Manna, T. & Albuquerque, C. T. M. Cystic fibrosis-related diabetes: an update on pathophysiology, diagnosis, and treatment. J. Pediatr. Endocrinol. Metab. 33, 835–843 (2020).
Putman, M. S., Anabtawi, A., Le, T., Tangpricha, V. & Sermet-Gaudelus, I. Cystic fibrosis bone disease treatment: current knowledge and future directions. J. Cyst. Fibros. 18, S56–S65 (2019).
Yamada, A. et al. Risk of gastrointestinal cancers in patients with cystic fibrosis: a systematic review and meta-analysis. Lancet Oncol. 19, 758–767 (2018).
Hadjiliadis, D. et al. Cystic fibrosis colorectal cancer screening consensus recommendations. Gastroenterology 154, 736–745 e714 (2018).
Shteinberg, M., Haq, I. J., Polineni, D. & Davies, J. C. Cystic fibrosis. Lancet 397, 2195–2211 (2021).
Cohen-Cymberknoh, M. et al. Baseline Cystic fibrosis disease severity has an adverse impact on pregnancy and infant outcomes, but does not impact disease progression. J. Cyst. Fibros. 20, 388–394 (2021).
Volkova, N. et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. J. Cyst. Fibros. 19, 68–79 (2020).
Hisert, K. B. et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med. 195, 1617–1628 (2017).
Thia, L. P. et al. Is chest CT useful in newborn screened infants with cystic fibrosis at 1 year of age? Thorax 69, 320–327 (2014).
Aurora, P. et al. Lung clearance index at 4 years predicts subsequent lung function in children with cystic fibrosis. Am. J. respiratory Crit. care Med. 183, 752–758 (2011).
Ramsey, K. A. et al. Early respiratory infection is associated with reduced spirometry in children with cystic fibrosis. Am. J. respiratory Crit. care Med. 190, 1111–1116 (2014).
Sly, P. D. et al. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 368, 1963–1970 (2013).
Rosenfeld, M. et al. Diagnostic accuracy of oropharyngeal cultures in infants and young children with cystic fibrosis. Pediatr. Pulmonol. 28, 321–328 (1999).
Ronchetti, K. et al. The CF-Sputum Induction Trial (CF-SpIT) to assess lower airway bacterial sampling in young children with cystic fibrosis: a prospective internally controlled interventional trial. Lancet Respir. Med. 6, 461–471 (2018).
Zampoli, M., Pillay, K., Carrara, H., Zar, H. J. & Morrow, B. Microbiological yield from induced sputum compared to oropharyngeal swab in young children with cystic fibrosis. J. Cyst. Fibros. 15, 605–610 (2016).
Ferreira, A. C. M. et al. Hypertonic saline as a useful tool for sputum induction and pathogen detection in cystic fibrosis. Lung 195, 431–439 (2017).
Shanthikumar, S., Neeland, M. N., Saffery, R. & Ranganathan, S. Gene modifiers of cystic fibrosis lung disease: A systematic review. Pediatr. Pulmonol. 54, 1356–1366 (2019).
Dang, H. et al. Mining GWAS and eQTL data for CF lung disease modifiers by gene expression imputation. PloS one 15, e0239189 (2020).
Kopp, B. T. et al. The impact of secondhand smoke exposure on children with cystic fibrosis: a review. Int. J. Environ. Res. Public Health 13, https://doi.org/10.3390/ijerph13101003 (2016).
Emerson, J., Rosenfeld, M., McNamara, S., Ramsey, B. & Gibson, R. L. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol. 34, 91–100 (2002).
Baker, E., Harris, W. T., Rowe, S. M., Rutland, S. B. & Oates, G. R. Tobacco smoke exposure limits the therapeutic benefit of tezacaftor/ivacaftor in pediatric patients with cystic fibrosis. J. Cyst. Fibros., https://doi.org/10.1016/j.jcf.2020.09.011 (2020).
Stanton, B. A., Coutermarsh, B., Barnaby, R. & Hogan, D. Pseudomonas aeruginosa Reduces VX-809 Stimulated F508del-CFTR Chloride Secretion by Airway Epithelial Cells. PloS one 10, e0127742 (2015).
Sakon, C. et al. Opportunity for pharmacogenomic testing in patients with cystic fibrosis. Pediatric pulmonology, https://doi.org/10.1002/ppul.25809 (2021).
Mehta, Z., Kamal, K. M., Miller, R., Covvey, J. R. & Giannetti, V. Adherence to cystic fibrosis transmembrane conductance regulator (CFTR) modulators: analysis of a national specialty pharmacy database. J. Drug Assess. 10, 62–67 (2021).
Mitchell, R. M., Jones, A. M., Stocking, K., Foden, P. & Barry, P. J. Longitudinal effects of ivacaftor and medicine possession ratio in people with the Gly551Asp mutation: a 5-year study. Thorax 76, 874–879 (2021).
Mikulski, B. S., Bellei, E. A., Biduski, D. & De Marchi, A. C. B. Mobile Health Applications and Medication Adherence of Patients With Hypertension: A Systematic Review and Meta-Analysis. Am. J. Prev. Med. https://doi.org/10.1016/j.amepre.2021.11.003 (2021).
Blakey, J. D. et al. Digital technologies and adherence in respiratory diseases: the road ahead. The European respiratory journal 52, https://doi.org/10.1183/13993003.01147-2018 (2018).
Talwalkar, J. S. et al. Cystic fibrosis transmembrane regulator modulators: implications for the management of depression and anxiety in cystic fibrosis. Psychosomatics 58, 343–354 (2017).
Stanojevic, S. et al. Projecting the impact of delayed access to elexacaftor/tezacaftor/ivacaftor for people with Cystic Fibrosis. J. Cyst. Fibros. 20, 243–249 (2021).
Schwank, G. et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13, 653–658 (2013).
Geurts, M. H. et al. CRISPR-based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell 26, 503–510 (2020).
Suzuki, S. et al. Highly efficient gene editing of cystic fibrosis patient-derived airway basal cells results in functional CFTR correction. Mol. Ther.: J. Am. Soc. Gene Ther. 28, 1684–1695 (2020).
Rowbotham, N. J. et al. The top 10 research priorities in cystic fibrosis developed by a partnership between people with CF and healthcare providers. Thorax 73, 388–390 (2018).
Gifford, A. H., Mayer-Hamblett, N., Pearson, K. & Nichols, D. P. Answering the call to address cystic fibrosis treatment burden in the era of highly effective CFTR modulator therapy. J. Cyst. Fibros. 19, 762–767 (2020).
Mayer-Hamblett, N. et al. Discontinuation versus continuation of hypertonic saline or dornase alfa in modulator treated people with cystic fibrosis (SIMPLIFY): results from two parallel, multicentre, open-label, randomised, controlled, non-inferiority trials. The Lancet. Respir. Med. https://doi.org/10.1016/S2213-2600(22)00434-9 (2022).
Davies, G. et al. Characterising burden of treatment in cystic fibrosis to identify priority areas for clinical trials. J. Cyst. Fibros. 19, 499–502 (2020).
Davies, J. C. et al. Speeding up access to new drugs for CF: considerations for clinical trial design and delivery. J. Cyst. Fibros. 18, 677–684 (2019).
Amaral, M. D. et al. Theranostics by testing CFTR modulators in patient-derived materials: the current status and a proposal for subjects with rare CFTR mutations. J. Cyst. Fibros. 18, 685–692 (2019).
De Boeck, K. et al. Cystic fibrosis drug trial design in the era of CFTR modulators associated with substantial clinical benefit: stakeholders’ consensus view. J. Cyst. Fibros. 19, 688–695 (2020).
Cutting, G. R. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat. Rev. Genet 16, 45–56 (2015).
Clancy, J. P. et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 18, 22–34 (2019).
Noordhoek, J., Gulmans, V., van der Ent, K. & Beekman, J. M. Intestinal organoids and personalized medicine in cystic fibrosis: a successful patient-oriented research collaboration. Curr. Opin. Pulm. Med. 22, 610–616 (2016).
Mayer-Hamblett, N. et al. Building global development strategies for cf therapeutics during a transitional cftr modulator era. J. Cyst. Fibros. 19, 677–687 (2020).
Dobra, R. et al. Optimising equity of access: how should we allocate slots to the most competitive trials in Cystic Fibrosis (CF). J. Cyst. Fibros. 20, 978–985 (2021).
Batty, P. & Lillicrap, D. Advances and challenges for hemophilia gene therapy. Hum. Mol. Genet 28, R95–R101 (2019).
Masat, E., Pavani, G. & Mingozzi, F. Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Disco. Med 15, 379–389 (2013).
Accurso, F. J. et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 363, 1991–2003 (2010).
Nichols, D. P. et al. PROMISE: working with the CF community to understand emerging clinical and research needs for those treated with highly effective CFTR modulator therapy. J. Cyst. Fibros. 20, 205–212 (2021).
Moss, R. B. et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir. Med. 3, 524–533 (2015).
Nissenbaum, C., Davies, G., Horsley, A. & Davies, J. C. Monitoring early stage lung disease in cystic fibrosis. Curr. Opin. Pulm. Med. 26, 671–678 (2020).
Davies, J. C. et al. A phase 3, double-blind, parallel-group study to evaluate the efficacy and safety of tezacaftor in combination with ivacaftor in participants 6 through 11 years of age with cystic fibrosis homozygous for F508del or heterozygous for the F508del-CFTR mutation and a residual function mutation. J. Cyst. Fibros. 20, 68–77 (2021).
Mondejar-Lopez, P. et al. A multimodal approach to detect and monitor early lung disease in cystic fibrosis. Expert Rev. Respir. Med 15, 761–772 (2021).
Marshall, H. et al. Detection of early subclinical lung disease in children with cystic fibrosis by lung ventilation imaging with hyperpolarised gas MRI. Thorax 72, 760–762 (2017).
Martini, K. et al. Volumetric dynamic oxygen-enhanced MRI (OE-MRI): comparison with CT Brody score and lung function in cystic fibrosis patients. Eur. Radio. 28, 4037–4047 (2018).
VanDevanter, D. R., Mayer-Hamblett, N. & Boyle, M. Feasibility of placebo-controlled trial designs for new CFTR modulator evaluation. J. Cyst. Fibros. 16, 496–498 (2017).
Trimble, A. T. & Donaldson, S. H. Ivacaftor withdrawal syndrome in cystic fibrosis patients with the G551D mutation. J. Cyst. Fibros. 17, e13–e16 (2018).
Davies, J. C. et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir. Med. 4, 107–115 (2016).
Rosenfeld, M. et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir. Med. 6, 545–553 (2018).
Lee, M. et al. Factors influencing clinical trial participation for adult and pediatric patients with cystic fibrosis. J. Cyst. Fibros. 20, 57–60 (2021).
Dobra, R. et al. Guiding the rational design of patient-centred drug trials in Cystic Fibrosis: a Delphi study. J. Cyst. Fibros. 20, 986–993 (2021).
van Koningsbruggen-Rietschel, S. et al. Protecting clinical trials in cystic fibrosis during the SARS-CoV-2 pandemic: risks and mitigation measures. Trials 22, 578 (2021).
Fudge, N. et al. Optimising Translational Research Opportunities: a Systematic Review and Narrative Synthesis of Basic and Clinician Scientists’ Perspectives of Factors Which Enable or Hinder Translational Research. PloS one 11, e0160475 (2016).
Bessonova, L. et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax 73, 731–740 (2018).
Bell, S. C. et al. The future of cystic fibrosis care: a global perspective. Lancet Respir. Med. 8, 65–124 (2020).
King, J. A., Nichols, A. L., Bentley, S., Carr, S. B. & Davies, J. C. An Update on CFTR Modulators as New Therapies for Cystic Fibrosis. Paediatr. drugs 24, 321–333 (2022).
Christopher Boyd, A. et al. New approaches to genetic therapies for cystic fibrosis. J. Cyst. Fibros. 19, S54–S59 (2020).
Fajac, I. & Sermet-Gaudelus, I. Therapeutic pipeline for individuals with cystic fibrosis with mutations nonresponsive to current cystic fibrosis transmembrane conductance regulator modulators. Curr. Opin. Pulm. Med. 27, 567–574 (2021).
Galietta, L. J. V. TMEM16A (ANO1) as a therapeutic target in cystic fibrosis. Curr. Opin. Pharmacol. 64, 102206 (2022).
Figueira, M. F., Ribeiro, C. M. P. & Button, B. Mucus-targeting therapies of defective mucus clearance for cystic fibrosis: a short review. Curr. Opin. Pharmacol. 65, 102248 (2022).
Manos, J. Current and emerging therapies to combat cystic fibrosis lung infections. Microorganisms 9, https://doi.org/10.3390/microorganisms9091874 (2021).
Antos, N. J. & Savant, A. P. Cystic fibrosis year in review 2020: section 2 pulmonary disease, infections, and inflammation. Pediatr. Pulmonol. 57, 347–360 (2022).
Acknowledgements
Jane C Davies is supported by the NIHR through a Senior Fellowship, the Imperial College London Biomedical Research Centre (BRC) and the Royal Brompton Hospital Clinical Research Facility. The clinical site receives funding from the CF Trust’s Clinical Trials Accelerator Platform (CTAP) to support clinical trial delivery. She leads the European Lung Clearance Index Core Facility on behalf of the European CF Society’s Clinical Trials Network. Gwyneth Davies is funded by a UK Research & Innovation Future Leaders Fellowship (grant ref: MR/T041285/1), and all research at UCL GOS ICH is supported by the NIHR via the GOSH BRC. Alexander Horsley is supported by the NIHR Manchester Biomedical Research Centre. The clinical site receives funding from the CF Trust’s Clinical Trials Accelerator Platform to support clinical trial delivery. Alan R Smyth is part of the NIHR Nottingham Biomedical Research Centre & the Nottingham CTAP centre. He is co-ordinating editor of the Cochrane CF & Genetic Disorders Group. Julian T Forton has received funding from a Health and Care Research Wales-Academic Health Science Collaboration, The Wellcome Trust and Cystic Fibrosis Foundation, and is part of the Cardiff CTAP centre. The authors wish to acknowledge the Respiratory Translational Research Collaborative Chair, Ling Pei Ho, and the NIHR for establishing and supporting the activities of the CF National Research Strategy Group. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. Editorial assistance was provided by Gina Rivellini, with thanks.
Author information
Authors and Affiliations
Contributions
Lu. A., Lo. A., S.B.C., G.D., D.D., M.E., J.T.F., R.G., C.H., A.H., A.R.S., K.W.S. and J.C.D. are members of the NIHR Respiratory-TRC CF National Research Strategy Group and discussed and agreed on the structure of this article. sections were drafted based on interests and expertise. Sections were merged by J.C.D. All authors reviewed and commented on the final submitted version. Post-review amendments were made by J.C.D. on behalf of the group.
Corresponding author
Ethics declarations
Competing interests
Lu. Allen, Lo. Allen, J.T.F., and K.W.S. declare no competing interests. S.B.C. has received grants from the NIHR and undertaking consultancy work for Vertex Pharmaceuticals and Chiesi. She has performed advisory roles for Profile Pharma, Pharmaxis and Vertex Pharmaceuticals. Siobhan is the chair of the UK CF registry steering committee and the European CF society patient registry scientific committee. G.D. has performed clinical trial leadership roles and received speaker honoraria from Vertex Pharmaceuticals, and speaker honoraria from Chiesi Ltd for educational events. She holds current grants from UKRI, NIHR and CF Trust. D.D. has received honoraria and/or grants from, Vertex, Proteostasis, Chiesi, Gilead and Insmed. M.E. is a consultant for Xanadu Bio Sciences. R.G. has received speaker fees from Vertex and Chiesi and and provided consultancy work for Chiesi. C.H. has performed clinical trial leadership roles, and educational and/or advisory activities for 30 Technology, Aradigm, Chiesi, CSL Behring, Gilead, Grifols, GSK, Insmed, Janssen, Meiji, Mylan, Novartis, Pneumagen, Shionogi, Teva, Vertex and Zambon. A.H. has performed clinical trial leadership roles, and educational or advisory activities for the following: Boehringer Ingelheim Pharma GmbH, Celtaxys Pharmaceuticals, Flatley Labs, Galapagos NV, Roche-Genentech, Krystal Biotech, Novabiotics, Pulmocide, Vertex Pharmaceuticals. He holds current grants from EPSRC, UKRI, CF Trust, JP Moulton charity, North West Lung Centre Charity. A.R.S. has received research grants; honoraria for lectures; and support for attending meetings from Vertex Pharmaceuticals (all outside the submitted work). A.R.S. has patents issued (Camara M, Williams P, Barrett D, Halliday N, Knox A, Smyth A, Fogarty A, Barr H, Forrester D. Alkyl quinolones as biomarkers of Pseudomonas aeruginosa infection and uses thereof (US-2016131648-A1), outside the submitted work. J.C.D. has performed clinical trial leadership roles, and educational and/or advisory activities for the following: Abbvie, Algipharma AS, Bayer AG, Boehringer Ingelheim Pharma GmbH & Co. KG, Eloxx, Enterprise, Galapagos NV, Genentech, ImevaX GmbH, Ionis, LifeArc, Nivalis Therapeutics, Inc., Krystal Biotech, Novartis, PARI Medical Holding GmbH, ProQR Therapeutics III B.V., Proteostasis Therapeutics INC., Pulmocide, Raptor Pharmaceuticals Inc, Recode, Vertex Pharmaceuticals.
Peer review
Peer review information
Nature Communications thanks Anne Chang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Allen, L., Allen, L., Carr, S.B. et al. Future therapies for cystic fibrosis. Nat Commun 14, 693 (2023). https://doi.org/10.1038/s41467-023-36244-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-36244-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
