
Abstract
Gamma aminobutyric acid(GABA) is synthesized by glutamate decarboxylase(GAD) in β-cells. Regarding Type 1 diabetes(T1D), animal/islet-cell studies found that GABA promotes insulin secretion, inhibits α-cell glucagon and dampens immune inflammation, while GAD immunization may also preserve β-cells. We evaluated the safety and efficacy of oral GABA alone, or combination GABA with GAD, on the preservation of residual insulin secretion in recent-onset T1D. Herein we report a single-center, double-blind, one-year, randomized trial in 97 children conducted March 2015 to June 2019(NCT02002130). Using a 2:1 treatment:placebo ratio, interventions included oral GABA twice-daily(n = 41), or oral GABA plus two-doses GAD-alum(n = 25), versus placebo(n = 31). The primary outcome, preservation of fasting/meal-stimulated c-peptide, was not attained. Of the secondary outcomes, the combination GABA/GAD reduced fasting and meal-stimulated serum glucagon, while the safety/tolerability of GABA was confirmed. There were no clinically significant differences in glycemic control or diabetes antibody titers. Given the low GABA dose for this pediatric trial, future investigations using higher-dose or long-acting GABA formulations, either alone or with GAD-alum, could be considered, although GABA alone or in combination with GAD-alum did nor preserve beta-cell function in this trial.
Similar content being viewed by others

Introduction
The pathogenesis of type 1 diabetes mellitus (T1D) entails autoimmune destruction of pancreatic beta cells1,2,3. Once hyperglycemia appears, more than 70% of islet beta cell mass has been eradicated4. Proliferation of surviving β-cells, pancreatic progenitor cells, plus transdifferentiation of alpha, acinar, ductal or hepatic cells, all have the potential to revitalize insulin production5,6.
Multiple immunological abnormalities have been reported in T1D patients including autoantibody production against the insulin molecule, the 65 kD isoform of glutamic acid decarboxylase (GAD65), various islet antigens, and the zinc transporter 8 (ZnT8) as well as decreased regulatory T cell (Treg) capacity to suppress T-cell mediated destruction of the islets of Langerhans3. To date, many studies attempting to ward off or reverse T1D have focused on immune suppression or modulation7,8,9,10,11, which may engender long-term side-effects. However, the recent antiCD3 antibody trials have shown a 3-year delay in clinical diagnosis of T1D12,13.
Animal and in vitro studies maintain that gamma aminobutyric acid (GABA) and glutamic acid decarboxylase (GAD) play fundamental metabolic roles in the pancreas and may be potential therapeutic targets in T1D. As for GAD65 antigen (GAD-alum) treatment per se in new onset T1D, an initial 2008 report of 70 patients was auspicious insofar as residual beta-cell function over 30 months was somewhat preserved. Yet a later, and more comprehensive, study with 334 patients failed to replicate this finding14. However, individual level analysis of these two studies and another15 found that study participants positive for HLA DR3-DQ2, but negative for HLA-DR4-DQ8, demonstrate enhanced beta cell preservation following GAD-alum monotherapy16.
GABA, a major inhibitory neurotransmitter, is abundant within pancreatic islets17,18 and participates in paracrine regulation of β and α cells19,20. GAD, the enzyme that decarboxylates glutamate to form GABA, is a major autoantigen in T1D3,21. In vitro experiments found that isolated human islets treated with GABA receptor blockade have decreased insulin secretion at physiologic glucose concentrations18. Further, GABA-deficient islets did not show appropriate glucagon inhibition in response to increasing glucose concentrations in vitro22, suggesting that GABA is directly involved in the suppression of glucagon secretion in pancreatic alpha cells. GABA activates the Ca2+ - P13K/Akt growth and survival pathway and averts stress-induced apoptosis in islet cell lines treated in vivo with streptozotocin (STZ)19. In vivo, GABA delays diabetes onset in both the non-obese diabetic (NOD) and the STZ-treated mouse if given early in life19. And, if GABA treatment was initiated in NOD and STZ mice after diabetes had already commenced, normoglycemia ensued19. The mechanisms are not fully understood, but are proposed to involve tempering of the pancreatic autoimmune milieu and systemic inflammation.
Apart from demonstrating β-cell regeneration and glucagon suppression with GABA in two distinct diabetic mouse models, Soltani and colleagues described significant decreases in inflammatory cytokine expression19. Functional GABA receptors are present on T-cells and increases expression of splenic T regulatory cells, in turn potentially arresting or slowing T cell mediated beta cell destruction19,23,24. In vivo, GABA inhibits adoptive transfer of T1D following transplant of diabetogenic splenic T cells into a NOD/SCID mouse model. Individually, GABA and GAD-alum promote survival of transplanted beta cells in the NOD mouse, while combination therapy promoted synergistic and dose dependent beta cell survival25. To date, neither GABA alone, nor GABA-GAD in tandem, has been explored as therapeutic agents in study participants with T1D. Here we show, in this human trial of low-dose GABA, alone or as co-therapy GABA/GAD, that while the primary outcome, β-cell function, was not statistically proven, the combination GABA/GAD reduced fasting and meal-stimulated serum glucagon. Glycemic control, proinsulin and diabetes autoantibodies, all secondary outcomes, were similar between GABA/GAD and placebo. Moreover, the safety and tolerability of the treatments was established.
Results
Recruitment and tolerability of intervention
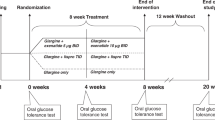
Between March 2015 and June 2018, 350 patients were screened and a total of 97 patients enrolled (Fig. 1). There were six unrelated serious adverse events recorded that required uneventful 1–2 day hospitalizations26.

Participants, aged 4–18 years old, were screened at diagnosis with T1D and enrolled at our tertiary care university center at Children’s Hospital of Alabama in Birmingham, Alabama. Nine study participants were from out-of-state.
Patient characteristics
The baseline patient characteristics for each treatment group are summarized in Table 1. The age-stratified randomization was successful. The ethnic distribution was as follows: 90% Caucasian, 7% African American, 2% Hispanic and 1% Native American. All patients were diabetes antibody positive with most retaining positivity in three. There were no statistical differences regarding initial presentation, including, age, diabetes ketoacidosis, the number of positive diabetes antibodies, body mass index, HbA1c, fasting c-peptide or glucagon. All patients were enrolled by 5–6 weeks post diagnosis of T1D.
Effect of GABA alone and GABA/GAD in combination on c-peptide and glycemic control
There was no statistical effect of oral GABA alone or combination GABA/GAD therapy on the primary outcome measure c-peptide, including both fasting and MMTT-stimulated area under the curve (Fig. 2a, b). The 90 min post MMTT c-peptide values for each study group are shown in Supplementary Fig. 1. As expected, there was a gradual diminution in c-peptide post diagnosis. A tabular summary of the statistical comparisons for the primary and secondary outcomes are presented in Supplementary Table 1. There was no statistical differences in HbA1c outside of a small disparity in GABA versus placebo only at the 5-month visit, and none in GABA/GAD versus placebo at all study visits. To address this further, an analysis of area under the curve (AUC) glucose at baseline and 12-months as well as fasting glucose at baseline, 1 month, 5-months and 12-months showed no differences (Supplementary Fig. 9). Insulin dose-adjusted A1c (IDAA1c)27 was 12% increased in GABA compared to placebo at 5 and 12-months. By contrast, IDAA1c in GABA/GAD was not different from placebo at any time point (Fig. 3a, b). Importantly, applying the gold-standard reference used to establish IDAA1c, namely, a meal-stimulated c-peptide >300 pM27, did not reveal statistical differences between the groups (Supplementary Fig. 8a). Moreover, a sub-analysis of IDAA1c in those participants who transitioned from basal/bolus injections to insulin pumps –which provide far greater accuracy as to total daily insulin dose (TDD) - between the 8–12 study visits revealed no statistical differences in IDAA1c between the groups (Supplementary Fig. 8b).

Fasting c-peptide (a) was measured in the three study groups (GABA-red, GABA/GAD-blue, and placebo-black) at baseline (Time = 0,prior any treatment) and at 1, 5 and 12-months thereafter. AUC c-peptide (b) was calculated over time in the three study groups. Results are given as mean ± 95% CI. No statistical differences were noted by two-sided analysis of covariance (complete statistical data is summarized in Supplementary Table 1). Source data are provided as a Source Data file.

Glycosylated Hemoglobin (HbA1c) (a) and insulin adjusted A1c (IDAA1c) (b) were measured in the three study groups. Results are shown as mean ±95% CI and statistical comparisons were by two-sided analysis of covariance. Regarding HbA1c (a) at 5-months GABA vrs. Placebo **p = 0.003 and GABA vrs GABA/GAD *p = 0.041. For IDAA1c (b) at 5-months, GABA vrs. Placebo **p = 0.007 and GABA vrs. GABA/GAD **p = 0.002. At 12-months, GABA vrs. Placebo *p = 0.020. Source data are provided as a Source Data file.
Effect of GABA alone and GABA/GAD in combination on glucagon
As shown in (Fig. 4a), the mean fasting glucagon in the placebo group increased by 16.8% over the course for the study (baseline to 12- months) in contrast to the two study groups wherein this change over time was curtailed: 0.4% in the GABA group and 0% in the GABA/GAD group. At 5 months, the mean fasting glucagon value in the GABA/GAD group was attenuated by 10.7% compared to placebo (p = 0.086) and 11.1% compared to the GABA group (p = 0.007). By 12-months, the mean fasting glucagon in the GABA/GAD group significantly diminished compared to the placebo patients (p = 0.035), but there were no statistical differences relative to the GABA group.

Fasting glucagon (a) was measured in the three study groups (GABA-red, GABA/GAD-blue, and placebo-black) at baseline (Time=0, prior any treatment) and at 1, 5 and 12-months thereafter. AUC glucagon (b) was calculated over time in the three study groups. Results are given as mean ±95% CI. Statistical differences were by two-sided analysis of covariance. a At 5 month GABA/GAD vrs. GABA, p = 0.007 and GABA/GAD vrs. Placebo, p = 0.086. At 12- months, GABA/GAD vrs. Placebo,**p = 0.035. Regarding AUC glucagon (b), at 12-months GABA/GAD vrs. Placebo, *p = 0.041. Source data are provided as a Source Data file.
Similar to the fasting glucagon data, the mean area under the curve (AUC) glucagon levels increased from baseline to 12-month in all groups (placebo group (24% increase), GABA group (13.7% increase) and in the GABA/GAD group (13.1%). At 12-months, the AUC glucagon in the GABA/GAD group was significantly reduced compared to placebo (p = 0.041) (Fig. 4b).
Based on the association between elevated glucagon and hyperglycemia in T1D28,29,30,31,32, we examined the correlation between glucose and glucagon. At first visit (baseline), both fasting glucose (p = 0.0017) and AUC glucose (p = 0.04) correlated with glucagon (Supplementary Fig. 2a, b). Similar correlations for the 12-month visit were apparent (Supplementary Fig. 2c, d).
Proinsulin levels and diabetes autoantibody titers
Fasting and 90 min post mixed meal plasma proinsulin and the proinsulin/c-peptide ratio was examined in the three study groups at baseline (before treatment), 5 and 12-months. No differences were detected related to treatment (Supplementary Fig. 3). The time course of diabetes autoantibodies (GAD65, ZnT8, ICA512) is presented in Supplementary Fig. 4a–c. The percent positivity for ZnT8 and ICA512 is presented in Supplementary Fig. 4d. Overall, there were no statistical trends or differences in the diabetes antibodies over time.
GABA levels
Plasma GABA levels are presented in Supplementary Fig. 5. Participants swallowed their study drug immediately before the mixed meal. There were no differences between the study groups in the 0 min (fasting) GABA levels at either the baseline (initial) or 12-month visit. Not unexpectedly, given the short half-life of GABA, the morning fasting values were statistically the same in all three cohorts. The veracity of the GABA study drug versus placebo is evidenced by the increase in plasma GABA at 60 and 120 min relative to baseline in the GABA and GABA/GAD groups, with no change in the placebo group.
HLA haplotypes
The primary outcome, fasting and meal-stimulated c-peptide, was re-examined after subdividing each treatment group according to high-risk HLA haplotypes. Results did not show statistically significant differences (Supplementary Table 2).
Discussion
This prospective, randomized, control trial of GABA and combined GABA/GAD in children with new-onset T1D confirmed the safety and tolerability of oral GABA, but did not attain its primary objective, the preservation of β-cell function (Fig. 2). However, a secondary outcome revealed a significant decrease in fasting, as well as nutrient-stimulated, glucagon secretion following 12-months of oral GABA/GAD treatment (Fig. 4). This observation corroborates favorably with animal/cell studies in which GABA (or GABA/GAD) has a paracrine inhibition on α-cells.
GABA, secreted from β-cells, reportedly has both an autocrine effect on insulin secretion as well as a paracrine inhibition of α-cell glucagon production. Whereas a distinct GABA autocrine role remains unsettled, the physiologically-relevant, paracrine inhibition of glucagon secretion or diminution of α-cell mass has been repeatedly documented in isolated cells or islets, perfused or biopsied pancreata, or in vivo animal studies. Upon binding to its cognate chloride channel, GABA begets α-cell membrane hyperpolarization, thereby hampering voltage-dependent calcium channels, which curtails glucagon output. For example, in streptozotocin (STZ)-induced diabetic mice, 12 days of daily intraperitoneal GABA (10 mg/kg) quenched the robust 7- fold increase in α-cell mass, which occurred in controls. And, relevant to β-cells, GABA augmented the proliferation of α-cells expressing GLP–1. The latter, in turn, could plausibly enhance β-cell function and growth33.
In another Type 1 diabetes model (multiple low dose STZ, MDSD), GABA, when added to the drinking water (6 mg/ml), reduced both serum glucagon and α-cell mass34. A similar tandem α and β cellular GABA effect was also found in MDSD mice treated with 20 µmol/kg intraperitoneal GABA prior to diabetes induction, and in a series of mice previously rendered diabetic with severe hyperglycemia19. Islet studies unfailingly corroborate the inhibitory action of GABA on glucagon secretion. In rat islets, GABA was noted to dampen glucose–stimulated glucagon secretion35 and, in normal mice islets or perfused pancreas, an inhibition of glucagon secretion was observed36. Finally, when a non-curative mass of normal human islets was transplanted into diabetic mice (NOD – seid- ϒ), after 5 weeks of drinking water with GABA added (6 mg/ml), serum glucagon was reduced roughly 80%37.
Antagonism of the glucagon receptor, or by genetic knockout, especially in the face of insulin deficiency, promotes normoglycemia. Take, for example, the following observations: (i.) Even without supplemental insulin, by blocking the glucagon receptor in diabetic obese mice, hyperglycemia was normalized38, (ii.) In the high-fat type 2 diabetic mouse, knockout of the glucagon receptor aborted obesity, hyperinsulinemia and abnormal lipogenesis and, notably, prevented hyperglycemia39, (iii.) In glucagon receptor null mice, following massive streptozotocin β−cell destruction, and despite marked hyperglucagonemia (14-fold increase over wild-type), normal blood glucose prevailed40, (iv.) Glucagon receptor antibody alone, i.e., no insulin therapy, can normalize hyperglycemia of type 1 diabetic NOD mice41, and finally, (v.) In humans with T1D, a single subcutaneous dose of a glucagon receptor antibody resulted within days in a 14% reduction in insulin dose and improved glycemic control as assessed by continuous glucose monitoring31. Most recently, a monoclonal glucagon receptor antagonist (Ab-4) corrected both glycemia and provoked restoration of β−cells in type 1 diabetic rodents (NOD and PANIC-ATTAC mouse models) as well as in mouse-implanted human islet xenografts42. Indeed, in the NOD mouse, the Ab-4 antibody increased insulin islet area approximately 900% versus control.
In concordance with previous reports, we found a progressive increase in serum glucagon over the first year following T1D diagnosis (Fig. 4), a phenomena which can persist for 3–5 years43,44,45,46,47. Glucagon may worsen glycemic control28,29 by peripheral effects on hepatic, adipose, and neural metabolism. Even in non-diabetic adults, fasting glucagon correlates inversely with longitudinal β−cell function- inferring that α-cell dysfunction is an incipient stage in disturbed glucose metabolism48. Although suppression in serum glucagon by GABA/GAD was found in our study, the percent lowering may not be sufficient to impact glycemic control (Fig. 3), namely, the insulin-adjusted A1c (IDAA1c) in this group. Of interest, using the reference standard for IDAA1c, a meal stimulated c-peptide >300 pM, there was a trend suggesting improvement in GABA/GAD group at 5-months (Supplementary Fig 8a). As evidenced in Supplementary Fig. 2, serum glucagon correlates positively with serum glucose, which infers a role of glucagon in glucose homeostasis. In our placebo cohort, the AUC glucagon at one year was 24% elevated versus baseline. This compares favorably to the postprandial increases of 37 and 51% previously reported in children with T1D (references45 and43, respectively).
The slight increase in IDAA1c in the GABA group (Fig. 3b) warrants discussion. This calculated metric of glycemic control27 is the least objective index insofar as it incorporates TDD, which in our study depended on participant paper records and recall. Furthermore, TDD is influenced by exercise, carbohydrate load, intercurrent illness and other factors. As aforementioned, using a meal-stimulated c-peptide >300 pmol/l27, there was no difference in glycemic control between the three groups (Supplementary Fig. 8a). Likewise those patients who transitioned to insulin pumps, which provide a more precise digital assessment of TDD, showed no differences in IDAA1c (Supplementary Fig. 8b).Finally, fasting and AUC glucose were not different amongst the groups at 12 months (Supplementary Fig. 9).
This study has many strengths. Foremost, as an adjunct agent, it is the first GABA study conducted in newly diagnosed humans. Further, by studying an exclusively pediatric population, we were able to enroll very young patients with T1D who typically have a more rapid decimation of β cells than adolescents49,50,51,52,53. Forty percent of our study participants were <10 years. Considering the array of confounding factors in β-cell loss, age of onset is the major determinant in the temporal decline in serum c-peptide. Most other potential therapeutics are first investigated in the adult population, making it impossible to reliably exclude patients with latent autoimmune diabetes of adulthood (LADA)54. Thirdly, our study was able to enroll all children within the first 5 weeks after diagnosis, allowing exposure of the pancreatic islets to the intervention before near-total autoimmune β-cell eradication.
Our study established that oral GABA is tolerable. The basis for this “low-dose” designation merits consideration. The daily dose of GABA used in animal studies, mostly mice, are sweeping, ranging 0.25 mg to 1500 mg/kg. Under FDA constraints, our dose of 1 gram/M2 (about 35 mg/kg) was far below nearly all in vivo studies in which salutary outcomes were reported (Supplementary Fig. 6).
It is speculative as to the mechanism whereby the GABA/GAD tandem attenuated glucagon more than GABA alone. However, the combination GABA/GAD strikingly extended, and in a synergistic manner, the time to develop hyperglycemia in diabetic NOD mice with transplanted β-cells25. It is conceivable that GAD-alum may have increased ambient islet cell insulin concentrations - despite no detectible change in the systemic serum levels - thereby reducing adjacent alpha cell glucagon release. To the point, we could have included a GAD-alum group alone, however, we did not because of the previous single and multicenter GAD-alum studies14,15,55,56,57.
Proinsulin and the proinsulin/c-peptide ratio are recognized markers of β-cell stress in T1D, likely related to aberrant proinsulin processing58,59. We investigated whether proinsulin or the proinsulin/c-peptide ratio was modified by treatment with GABA, or the combination GABA with GAD, due to their recognized immunosuppressive actions in diabetes25 (Supplementary Fig. 3), no statistical differences were identified. Likewise, there was no difference in baseline or subsequent diabetes antibody titers or positivity in the treatment groups (Table 1 and Supplementary Fig. 4) which is not unexpected for a one-year T1D trial60.
Considering the role of the DR3-DQ2 haplotypes which confer T1D risk and disease course61, we screened our study cohorts accordingly. Based on previous evidence demonstrating HLA haplotype specificity to GAD-alum therapy16, we examined whether the presence or absence of HLA DR3-DQ2 altered the primary outcome in the three treatment groups. No differences were detected; however, a larger cohort may be required to detect statistical distinctions (Supplementary Table 2).
The study has limitations, most notably the unpropitious compliance (assessed by pill counts and recall,) as is commonly encountered in the real-clinic setting (Supplementary Fig. 7). Based on in vivo animal trials, the dose of GABA (alone) may have been inadequate, namely, beneath a threshold response (Supplementary Fig. 6). As aforementioned, a further weakness of our study was that the GABA preparation was relatively short acting and taken only twice daily (Supplementary Fig. 5). Alternatively, long-acting preparations of GABA and/or currently available GABA-ergic drugs that have longer half-lives of action offer promise. And, based on affirmative β-cell studies in human islets, co-treatment of GABA with an allosteric positive modulator (Ly49) of its cognate receptor is an ingenious notion62.
To sum, in this prospective, randomized controlled trial of twice-daily GABA, or co-treatment with GABA/GAD, in humans with T1D, we demonstrate a significant decrease in fasting and AUC glucagon in the GABA/GAD group, with a non-significant reduction in the GABA group at 12-months. There were no statistically significant changes in the primary outcome, namely, fasting and meal-stimulated c-peptide between the cohorts. Notwithstanding the necessarily low GABA dose for this trial in TID children, in combination with the compliance challenge, the reduction in serum glucagon augurs well for further studies to conceivably preserve β-cell function or mass. Indeed, in the sole study using co-therapy with GABA/GAD, β-cell preservation was dependent on the dose25. Lastly, bearing in mind that GABA/GAD attenuated glucagon production, this could in turn expand β-cell mass and/or improve glucose homeostasis. Case in point, in diabetic mice, blocking glucagon action begets a nearly 8-fold increase in insulin-positive islet cell mass and mediates β-cell regeneration42,63. Insofar as GABA tempers immune inflammation at higher doses in rodents, and our study was constrained to relatively low-dose GABA dosing in this pediatric trial in T1D, it is plausible that increased GABA doses, or long-acting preparations, could offer sufficiently prolonged, above-threshold GABA concentrations to preserve islet cells, particularly during stage 1 diabetes.
Methods
The detailed rationale and methods for this study have been described elsewhere with minor modification26. A succinct summary follows:
Study design and treatment
This is a prospective, one-year randomized, double blind, placebo controlled trial to evaluate the safety and efficacy of GABA alone and combination GABA/GAD-alum® in children with newly diagnosed T1D (https://clinicaltrials.gov/ct2/show/NCT02002130). Patients were randomized into one of three study arms (Fig. 1). The original clinicaltrial.gov posting (2013) predates the final protocol submission (2015). We had a protracted period (2 years) prior to study launch in order to obtain FDA approval to administer GABA in children (first human trial). The formal study protocol approvals and funding were in place by 2015 and the first patient enrolled 3/2/2015. Suboptimally, we noticed the documentation discordance from 2013 and updated the clinicaltrial.gov outcomes in July 2019 to align with the 2015 study protocol.
Participants and eligibility criteria
Participants were screened at the time of diagnosis with T1D, as defined by ADA criteria. All patients were enrolled from the clinics and in-patient wards at Children’s of Alabama (CoA), a tertiary care university-associated referral center. The majority of patients were residents of the state of Alabama. There were 11 out of state participants (AZ, GA, MS, MO, NC, ND, TX, VA). The first participant was enrolled 3/2/2015 and the last study visit was 6/24/2019. Inclusion criteria: children 4–18 years of age, positivity for autoantibody GAD65, and enrollment within 5 weeks of diagnosis. If the participant was female and not abstinent, two forms of contraception were required. Exclusion criteria: pregnancy, systemic or inhaled steroid use, neurologic/seizure disorders, adjunct oral therapies that might affect glucose or GABA metabolism26. Six randomized patients were excluded from analysis because all c-peptide values, fasting and MMTT stimulated, were <0.6 ng/ml at the initial baseline study visit64,65. Participants received a $60 gift card as compensation for every blood draw.
Randomization
Patients were randomized into one of three regimens (GABA, GABA/GAD-alum, or placebo) stratified by age and in balanced blocks of three (1:1:1) for the first 75 patients using a pre-set randomization list (generated by using a computerized procedure) known only to the un-blinded pharmacist. This second protocol was a consequence of unanticipated additional funding that afforded trial extension for the GABA versus placebo groups only.
Study drugs
Oral gamma-aminobutyric acid (GABA)
GABA and placebo capsules were prepared commercially (NOW Foods, Bloomingdale, IL). GABA or placebo was administered using premeasured capsules (1 gram/M2/day up to maximum of 1.5 gram/day) divided into two daily doses (morning and evening). The purity of both the GABA and placebo products was verified by LC/MS/MS prior to study enrollment. The control GABA that was used for mass spectroscopy analysis was obtained from Sigma-Aldrich Chemical Company (St. Louis, MO). Placebo and GABA capsules were, taste-wise and visually, indistinguishable.
Glutamic acid decarboxylase (GAD-alum)
GAD-alum and placebo were prepared as a suspension with recombinant GAD enzyme and the vaccine adjuvant Alhydrogel ® (alum) by Diamyd Medical (Stockholm, Sweden). The subcutaneous GAD-alum injections (20 μg/dose), or placebo, were given in clinic by the research nurse.
Mixed meal tolerance testing (MMTT)
MMTT occurred according to the visit schedule outlined in Table 2 and as described previously26.
Safety monitoring
Safety assessments included observations of reactions at the injection site, occurrence of all adverse events (AEs)/serious adverse events (SAEs), laboratory measurements (chemistry panel, complete blood counts with differential, and urinalysis), neurological assessments, and physical examination.
Adherence and retention measures
Treatment adherence of the oral capsules was assessed subjectively by patient recall, and objectively by calculating the unused capsule count at each visit. Study participants were asked to return any unused study drug for safe disposal and queried whether any capsules were destroyed or lost.
Investigative endpoints
The primary outcome measure was the effect of GABA or GABA/GAD on fasting and meal-stimulated serum c-peptide compared to placebo at baseline,1-month, 5-months and 12-months. Secondary endpoints included (1) fasting and meal-stimulated glucagon and proinsulin (2) glycemic control (HbA1c, IDAA1c27), (3) diabetes autoantibodies, and (4) immune studies in peripheral blood mononuclear cells (to be presented in a separate manuscript). Exploratory endpoints included plasma GABA levels and the proinsulin/c-peptide ratio before and after meal-stimulation. Also, we examined the effect of diabetes-related HLA risk haplotype on the primary outcome.
Endocrine assays
C-peptide, glucose and glucagon were measured in the University of Alabama Core Metabolic Laboratory and as previously noted26. C-peptide was measured by a two-site immunoenzymometric analyzer (900 AIA-Pack, TOSOH, San Francisco, CA) and glucagon by radioimmunassay (Millipore Sigma, Burlington, MA). Antibodies to GAD65, IA512, and Zinc 8 Transporter were assayed commercially by Labcorp (Burlington, NC) as standard of care.
Plasma GABA
Plasma GABA levels were obtained during mixed meal tolerance test (MMTT) at both the baseline (initial study visit) and 12 month visits. Patients swallowed oral study drug dose at 0 min, immediately prior to ingesting mixed meal drink. GABA levels were determined at 0, 60 and 120 min.
GABA analysis
Materials and sample preparation
Solid stocks of GABA and GABA-d6 were purchased from Sigma & CDN Isotopes respectively. Standards were reconstituted in methanol. The analytical range was 1–5000 ng/ml over 8 calibrators. Plasma samples were thawed on ice and spiked with 10 µl of 500 ng/ml GABA-d6. They were transferred quantitively to 1 cc Phree SPE cartridges (Phenomenex, Torrance, CA) containing 600 µl of 1% formic acid acetonitrile, incubated at room temp for 5 min and centrifuged at 1000 g for 5 min. The flow-through was retained and transferred to a Biotage N2 evaporator to dry. Samples and standards were reconstituted in 100 µl of 1.0% formic acid before analysis. LC-MS Conditions. Separation and detection were carried out by Shimadzu Prominence 20 series HPLC in tandem with a Sciex API 4000 triple quadrupole mass spectrometer (MS) utilizing a modified method from Imtakt66. Chromatographic separation occurred with a Intrada Amino Acid column 3 µM 50 ×3 mm at 60 degrees C. Mobile phases were A) 0.3% formic acid in MeCN and B) 100 mM ammonium formate. Gradient schedule was as follows: 0 min 30% B, 4 min 35% B, 5 min 100% B, 5.1 min 30% B, and 5.5 min stop. The flowrate of 0.6 ml/min. Injection volume was 5 µl. Analyst v1.7.2 was used for instrument control & data acquisition. The MS was operated in positive polarity electrospray ionization. MS source parameters were as follows: collision gas 5, curtain gas 25, GS1 40, GS2 45, IS 2000, and temperature 600. Compound mass transitions were 104.1 m/z à 87 m/z & 110 m/z à 93 m/z for GABA and GABA-d6 respectively. Compound parameters were as follows: collision energy 15, cell exit potential 6, and declustering potential 60. Data processing occurred in MultiQuant v3.0.3. The standard curve was regressed linear with 1/x2 weighting
HLA genotyping in study participants
The Histocompatibility and Immunogenetics Laboratory at the University of Alabama at Birmingham performed HLA typing on genomic DNA that was isolated from frozen peripheral blood mononuclear cells (PBMC).
Statistical analysis
Baseline demographic and other clinical characteristics were compared between the treatment groups using t- and chi-square tests (or their non-parametric equivalents) for continuous and categorical variables, respectively. Analysis of covariance was used to compare changes in C-peptide levels between the treatment groups. For these analyses, the 12-month measurement served as the dependent variable with two independent variables: (1) a categorical variable for treatment group and (2) the baseline C-peptide measurement. A similar analytical approach was used for the other study outcomes of interest including glucagon, hemoglobin A1C, IDAA1C, and total daily insulin dose. Mixed statistical models were used to conduct longitudinal analyses of C-peptide and hemoglobin A1C measurements, and daily insulin requirements, incorporating all three measurements. This study utilized REDCap (Research Electronic Data Capture, version 12.3.3 https://www.project-redcap.org), a software toolset and workflow methodology for electronic collection and management of clinical and research data. Data analysis of for primary, secondary and exploratory outcomes used SAS/STAT software, version 9.4 of the SAS System. Copyright, SAS Institute Inc. Cary, NC, USA. Graphs were prepared with GraphPad Prism 9.0 for Windows, GraphPad Software, San Diego, CA, USA, www.graphpad.com. Correlations and Fisher’s exact analyses were by GraphPad.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Following de-identification, all of the individual participant data collected during this trial, as well as data dictionaries, will be available to any researcher who provides a methodologically-sound proposal for academic purposes. Requests should be directed to the corresponding author and is subject to a material transfer agreement. Proposals may be submitted up to 36-months following publication. Source data are provided with this paper. The study protocol is available online (https://clinicaltrials.gov/ct2/show/NCT02002130). Source data are provided with this paper.
References
Eizirik, D. L., Colli, M. L. & Ortis, F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 5, 219–226 (2009).
Notkins, A. L. & Lernmark, A. Autoimmune type 1 diabetes: resolved and unresolved issues. J. Clin. Invest. 108, 1247–1252 (2001).
Atkinson, M. A. & Eisenbarth, G. S. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 358, 221–229 (2001).
Pipeleers, D. et al. Restoring a functional beta-cell mass in diabetes. Diabetes Obes. Metab. 10, 54–62 (2008).
Kim, H. S. & Lee, M. K. β-Cell regeneration through the transdifferentiation of pancreatic cells: Pancreatic progenitor cells in the pancreas. J. Diabetes Investig. 7, 286–296 (2016).
Zhang, J. & Liu, F. The De-, Re-, and trans-differentiation of β-cells: Regulation and function. Semin Cell Dev. Biol. 103, 68–75 (2020).
Rewers, M. & Gottlieb, P. Immunotherapy for the prevention and treatment of type 1 diabetes: human trials and a look into the future. Diabetes Care.32, 1769–1782 (2009).
Bresson, D. & von Herrath, M. Immunotherapy for the prevention and treatment of type 1 diabetes: optimizing the path from bench to bedside. Diabetes Care. 32, 1753–1768 (2009).
Greenbaum, C. & Atkinson, M. A. Persistence is the twin sister of excellence: an important lesson for attempts to prevent and reverse type 1 diabetes. Diabetes 60, 693–694 (2011).
Skyler, J. S. & Ricordi, C. Stopping type 1 diabetes: attempts to prevent or cure type 1 diabetes in man. Diabetes 60, 1–8 (2011).
Orban, T. et al. Costimulation modulation with abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment. Diabetes Care. 37, 1069–1075 (2014).
Herold, K. C. et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 381, 603–613 (2019).
Sims, E. K. et al. Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci. Transl. Med. 13, abc8980 (2021).
Ludvigsson, J. et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N. Engl. J. Med. 359, 1909–1920 (2008).
Wherrett, D. K. et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet 378, 319–327 (2011).
Hannelius, U., Beam, C. A. & Ludvigsson, J. Efficacy of GAD-alum immunotherapy associated with HLA-DR3-DQ2 in recently diagnosed type 1 diabetes. Diabetologia 63, 2177–2181 (2020).
Reetz, A. et al. GABA and pancreatic beta-cells: colocalization of glutamic acid decarboxylase (GAD) and GABA with synaptic-like microvesicles suggests their role in GABA storage and secretion. EMBO J. 10, 1275–1284 (1991).
Braun, M. et al. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 59, 1694–1701 (2010).
Soltani, N. et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc. Natl Acad. Sci. 108, 11692–11697 (2011).
Xu, E. et al. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab. 3, 47–58 (2006).
Baekkeskov, S. et al. Identification of the 64 K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 347, 151–156 (1990).
Li, C. et al. Regulation of glucagon secretion in normal and diabetic human islets by gamma-hydroxybutyrate and glycine. J. Biol. Chem. 288, 3938–3951 (2013).
Alam, S., Laughton, D. L., Walding, A. & Wolstenholme, A. J. Human peripheral blood mononuclear cells express GABAA receptor subunits. Mol. Immunol. 43, 1432–1442 (2006).
Tian, J. et al. Homotaurine Treatment Enhances CD4(+) and CD8(+) Regulatory T Cell Responses and Synergizes with Low-Dose Anti-CD3 to Enhance Diabetes Remission in Type 1 Diabetic Mice. Immunohorizons 3, 498–510 (2019).
Tian, J., Dang, H. & Kaufman, D. L. Combining antigen-based therapy with GABA treatment synergistically prolongs survival of transplanted ss-cells in diabetic NOD mice. PLoS One. 6, e25337 (2011).
Choat, H. M. et al. Effect of gamma aminobutyric acid (GABA) or GABA with glutamic acid decarboxylase (GAD) on the progression of type 1 diabetes mellitus in children: Trial design and methodology. Contemp. Clin. Trials. 82, 93–100 (2019).
Mortensen, H. B. et al. New definition for the partial remission period in children and adolescents with type 1 diabetes. Diabetes Care. 32, 1384–1390 (2009).
Salehi, A., Vieira, E. & Gylfe, E. Paradoxical stimulation of glucagon secretion by high glucose concentrations. Diabetes 55, 2318–2323(2006).
Cryer, P. E. Minireview: Glucagon in the pathogenesis of hypoglycemia and hyperglycemia in diabetes. Endocrinology 153, 1039–1048 (2012).
Davidson, J. A. et al. Glucagon therapeutics: Dawn of a new era for diabetes care. Diabetes Metab. Res Rev. 32, 660–665 (2016).
Pettus, J. et al. Effect of a glucagon receptor antibody (REMD-477) in type 1 diabetes: A randomized controlled trial. Diabetes Obes. Metab. 20, 1302–1305 (2018).
Finan, B., Capozzi, M. E. & Campbell, J. E. Repositioning Glucagon Action in the Physiology and Pharmacology of Diabetes. Diabetes 69, 532–541 (2020).
Feng, A. L. et al. Paracrine GABA and insulin regulate pancreatic alpha cell proliferation in a mouse model of type 1 diabetes. Diabetologia 60, 1033–1042 (2017).
Liu, W. et al. Combined Oral Administration of GABA and DPP-4 Inhibitor Prevents Beta Cell Damage and Promotes Beta Cell Regeneration in Mice. Front Pharm. 8, 362 (2017).
Wendt, A. et al. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes 53, 1038–1045 (2004).
Gilon, P., Bertrand, G., Loubatieres-Mariani, M. M., Remacle, C. & Henquin, J. C. The influence of gamma-aminobutyric acid on hormone release by the mouse and rat endocrine pancreas. Endocrinology 129, 2521–2529 (1991).
Purwana, I. et al. GABA promotes human beta-cell proliferation and modulates glucose homeostasis. Diabetes 63, 4197–4205 (2014).
Okamoto, H. et al. Glucagon Receptor Blockade With a Human Antibody Normalizes Blood Glucose in Diabetic Mice and Monkeys. Endocrinology 156, 2781–2794 (2015).
Lee, Y. et al. Hyperglycemia in rodent models of type 2 diabetes requires insulin-resistant alpha cells. Proc. Natl Acad. Sci. 111, 13217–13222 (2014).
Lee, Y., Wang, M. Y., Du, X. Q., Charron, M. J. & Unger, R. H. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 60, 391–397 (2011).
Wang, M. Y. et al. Glucagon receptor antibody completely suppresses type 1 diabetes phenotype without insulin by disrupting a novel diabetogenic pathway. Proc. Natl Acad. Sci. 112, 2503–2508 (2015).
Wang, M. Y. et al. Glucagon blockade restores functional beta-cell mass in type 1 diabetic mice and enhances function of human islets. Proc Natl Acad Sci. 118, 2021–2031 (2021).
Fredheim, S. et al. The influence of glucagon on postprandial hyperglycaemia in children 5 years after onset of type 1 diabetes. Diabetologia 58, 828–834 (2015).
Porksen, S. et al. Meal-stimulated glucagon release is associated with postprandial blood glucose level and does not interfere with glycemic control in children and adolescents with new-onset type 1 diabetes. J. Clin. Endocrinol. Metab. 92, 2910–2916 (2007).
Brown, R. J., Sinaii, N. & Rother, K. I. Too much glucagon, too little insulin: time course of pancreatic islet dysfunction in new-onset type 1 diabetes. Diabetes Care. 31, 1403–1404 (2008).
Urakami, T. et al. Influence of plasma glucagon levels on glycemic control in children with type 1 diabetes. Pediatr. Int. 53, 46–49 (2011).
Sherr, J. et al. Evolution of abnormal plasma glucagon responses to mixed-meal feedings in youth with type 1 diabetes during the first 2 years after diagnosis. Diabetes Care. 37, 1741–1744 (2014).
Adams, J. D. et al. Fasting glucagon concentrations are associated with longitudinal decline of beta-cell function in non-diabetic humans. Metabolism. 105, 154175 (2020).
Barker, A. et al. Age-dependent decline of beta-cell function in type 1 diabetes after diagnosis: a multi-centre longitudinal study. Diabetes Obes. Metab. 16, 262–267 (2014).
Davis, A. K. et al. Prevalence of detectable C-Peptide according to age at diagnosis and duration of type 1 diabetes. Diabetes Care. 38, 476–481 (2015).
Hao, W., Gitelman, S., DiMeglio, L. A., Boulware, D. & Greenbaum, C. J. Fall in C-Peptide During First 4 Years From Diagnosis of Type 1 Diabetes: Variable Relation to Age, HbA1c, and Insulin Dose. Diabetes Care. 39, 1664–1670 (2016).
Marino, K. R. et al. A predictive model for lack of partial clinical remission in new-onset pediatric type 1 diabetes. PLoS One. 12, e0176860 (2017).
Johnson, M. B. et al. Type 1 diabetes can present before the age of 6 months and is characterised by autoimmunity and rapid loss of beta cells. Diabetologia 63, 2605–2615 (2020).
American Diabetes, A. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2020. Diabetes Care. 43, S14–S31 (2020).
Ludvigsson, J. et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N. Engl. J. Med. 366, 433–442 (2012).
Ludvigsson, J. et al. GAD-treatment of children and adolescents with recent-onset type 1 diabetes preserves residual insulin secretion after 30 months. Diabetes Metab. Res Rev. 30, 405–414 (2014).
Beam, C. A. et al. GAD vaccine reduces insulin loss in recently diagnosed type 1 diabetes: findings from a Bayesian meta-analysis. Diabetologia 60, 43–49 (2017).
Sullivan, C. A., Cacicedo, J. M., Rajendran, I. & Steenkamp, D. W. Comparison of proinsulin and C-peptide secretion in healthy versus long-standing type 1 diabetes mellitus cohorts: A pilot study. PLoS One. 13, e0207065 (2018).
Watkins, R. A. et al. Proinsulin and heat shock protein 90 as biomarkers of beta-cell stress in the early period after onset of type 1 diabetes. Transl. Res. 168, 96–106 e101 (2016).
Warshauer, J. T., Bluestone, J. A. & Anderson, M. S. New Frontiers in the Treatment of Type 1 Diabetes. Cell Metab. 31, 46–61 (2020).
Noble, J. A. & Valdes, A. M. Genetics of the HLA region in the prediction of type 1 diabetes. Curr. Diabetes Rep. 11, 533–542 (2011).
Untereiner, A. et al. GABA stimulates β-cell proliferation through the mTORC1/p70S6K pathway, an effect amplified by Ly49, a novel GABA(A) -R positive allosteric modulator. Diabetes Obes. Metab. 11, 2021–2031 (2020).
Xi, Y. et al. Glucagon-receptor-antagonism-mediated beta-cell regeneration as an effective anti-diabetic therapy. Cell Rep. 39, 110872 (2022).
Quattrin, T. et al. Golimumab and Beta-Cell Function in Youth with New-Onset Type 1 Diabetes. N. Engl. J. Med. 383, 2007–2017(2020).
Raz, I. et al. Treatment of recent-onset type 1 diabetic patients with DiaPep277: results of a double-blind, placebo-controlled, randomized phase 3 trial. Diabetes Care. 37, 1392–1400 (2014).
LC-MS: Glutamic acid and GABA isomers. in Innovative Chromatography Columns for every application, Vol. Technical information (ed. Imtakt) 1 (Imtakt USA, Portland, OR, https://www.imtaktusa.com/, 2021).
Acknowledgements
This study was funded by the Juvenile Diabetes Research Foundation (#201303440) K.L.M., Diamyd Medical (Stockholm, Sweden), Janssen Pharmaceuticals (Raitan,NJ), and NIH R01 grant awards: DK126456(HMT) and DK127497(H.M.T.). NOW foods (Bloomingdale, IL) supplied the GABA and the placebo capsules. We are grateful to the children and families for their participation in this clinical trial and Sharon May, our research clinician specialist. Purchase of the AB Sciex 4000 mass spectrometer in the Targeted Metabolomics and Proteomics Laboratory came from the University of Alabama Birmingham Health Services General Endowment Fund. Sponsors were not involved in the study design, data collection and analysis or manuscript writing.
Author information
Authors and Affiliations
Contributions
M.A., K.L.M and G.J.M designed the studies, wrote protocols and enrolled and cared for patients, K.L.M and A.A.L obtained the Federal IND for GABA, H.M.C. was responsible for patient care and protocol oversight, G.G.M. provided statistical analysis. K.L.M., G.J.M. and H.M.T. acquired data and analyzed the results, and K.L.M., G.J.M., and M.A. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical standard
The protocol and consent documents were approved by the University of Alabama at Birmingham (UAB) Institutional Review Board.
Informed consent
Written informed consent was obtained from each participant or from the participant’s parent or legal guardian. Also, each participant assented.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martin, A., Mick, G.J., Choat, H.M. et al. A randomized trial of oral gamma aminobutyric acid (GABA) or the combination of GABA with glutamic acid decarboxylase (GAD) on pancreatic islet endocrine function in children with newly diagnosed type 1 diabetes. Nat Commun 13, 7928 (2022). https://doi.org/10.1038/s41467-022-35544-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-35544-3
This article is cited by
-
Supplementation with high-GABA-producing Lactobacillus plantarum L5 ameliorates essential tremor triggered by decreased gut bacteria-derived GABA
Translational Neurodegeneration (2023)
-
Typ-1-Diabetes – neue Aspekte in Therapie und Technologie 2023
Die Diabetologie (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
