
Abstract
Phytochrome proteins detect red/far-red light to guide the growth, motion, development and reproduction in plants, fungi, and bacteria. Bacterial phytochromes commonly function as an entrance signal in two-component sensory systems. Despite the availability of three-dimensional structures of phytochromes and other two-component proteins, the conformational changes, which lead to activation of the protein, are not understood. We reveal cryo electron microscopy structures of the complete phytochrome from Deinoccocus radiodurans in its resting and photoactivated states at 3.6 Å and 3.5 Å resolution, respectively. Upon photoactivation, the photosensory core module hardly changes its tertiary domain arrangement, but the connector helices between the photosensory and the histidine kinase modules open up like a zipper, causing asymmetry and disorder in the effector domains. The structures provide a framework for atom-scale understanding of signaling in phytochromes, visualize allosteric communication over several nanometers, and suggest that disorder in the dimeric arrangement of the effector domains is important for phosphatase activity in a two-component system. The results have implications for the development of optogenetic applications.
Similar content being viewed by others


Introduction
Light is essential for life and all living organisms have developed intricate systems to detect and adapt to it. At the molecular level, this is achieved by photoreceptor proteins. Phytochromes are a protein superfamily in bacteria, plants, and fungi, and function by photoswitching between states that absorb red light (Pr) and far-red light (Pfr)1,2,3,4,5,6. These states have differential signaling activity. Conserved over the entire superfamily and exemplified by the phytochrome from D. radiodurans, phytochromes share a photosensory core module, consisting of PAS, GAF, and PHY domains and a bilin chromophore (Fig. 1), which is connected to a variable effector module via helical linkers3. The structure of the photosensory core is very similar in plant and bacterial phytochromes7,8,9,10,11,12,13 but diverges in the context of full-length proteins14. Phytochromes are highly interesting targets for gene optimization of plants, are used as near-infrared fluorescent markers in microscopy15, and have been utilized in optogenetic applications16,17.

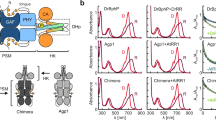
a The domain organization and the linker configuration are shown with the position of the biliverdin (BV) chromophore indicated. CA catalytic ATP-binding, DHp dimerization and histidine phosphotransfer, GAF cGMP phosphodiesterase-adenylate cyclase FhlA, HK histidine kinase, PAS Per-ARNT-Sim, PHY phytochrome-specific, PSM photosensory module, REC receiver domain, RR response regulator. b Absorption spectra of both proteins recorded in dark (D) or after saturating 655 nm red light (R). The spectra are normalized at the peak at 700 nm of the corresponding dark spectrum. Source data are provided as a Source Data file. c Dark reversion of DrBphP and DrBphP-DrRR. Response regulator fusion increases the DrBphP dark reversion, which resembles the addition of soluble DrRR to DrBphP (see Supplementary Fig. 1b)35. Source data are provided as a Source Data file. d Phos-tag assay of phosphatase activity35. In the assay, the retention of phosphorylated protein is slower than its non-phosphorylated counterparts. Here, all samples were supplemented with the same amount of phospho-DrRR (p-DrRR) and the change in its final amount implies either kinase (increase) or phosphatase (decrease) activity. The gel shows that DrBphP-DrRR dephosphorylates p-DrRR in its red light-illuminated state (lane R) and the activity is stalled in the darkness (lane D). The phosphatase activity of the fusion protein resembles that of DrBphP, although is generally weaker because of competition between the fused and free DrRR. See Supplementary Fig. 1c for an extended gel. Source data are provided as a Source Data file.
Many bacteriophytochromes are histidine kinases or phosphatases in two-component systems (TCSs)18,19. TCSs sense environmental cues in all kingdoms of life. They are particularly important in microorganisms and plants, where they provide for cell growth, survival, and pathogenicity, and are implicated in multidrug and antibiotic resistance20. TCSs consist of sensor kinases, which send phospho-signals to so-called response regulators. A highly pertinent question is how the kinases achieve this signal transduction at the atomic level. Crystal structures of proteins involved in TCS have resulted in a number of partially conflicting conformational mechanisms for signal transduction21,22,23,24,25,26, one of which is that the region around the active site for phosphate transfer is destabilized27. Opposing the notion that one clearly defined cascade of structural changes leads to signaling, it has been proposed that allosteric coupling between the different modules controls the activity of the effector domains25,28, but a structural understanding of this concept is missing.
Reminiscent of the state-of-the-art for TCSs, the conformational changes that guide phytochrome activation are not understood. Structures have been obtained for the photosensory core fragment of bacteriophytochromes in Pr and Pfr states10,11,12, pinpointing isomerization of the D-ring of the bilin cofactor and refolding of the so-called PHY tongue, which is a hairpin structure that extends from the PHY domain onto the chromophore region, as outstanding features of the photoconversion13. Finer mechanistic insight near the chromophore has been obtained using spectroscopy and crystallography29,30,31,32. Signal transduction from the photosensory core to the effector domains has been proposed to be guided by a monomerization mechanism33, or by a shift of register in the dimeric binding interface of the sensor-effector linker34. However, solid progress is limited by a lack of structures of full-length phytochromes in active and resting states.
In this work, we use single-particle cryo-electron microscopy (cryo-EM) to solve the structure of the full-length phytochrome from D. radiodurans (DrBphP) in the resting (Pr) and photoactivated (Pfr) states. We present models in both states, revealing a mechanism of photoactivation.
Results
For the single-particle cryo-EM experiment, we used a fusion of DrBphP with its response regulator protein (abbreviated DrBphP-DrRR) via a 12 amino acid tether (Fig. 1a). This gave a better spatial resolution in the reconstructed maps compared to the unfused DrBphP. We assume that DrRR stabilizes the DrBphP structure by transiently interacting with its cognate binding site, but that the structure of DrBphP is otherwise unaffected. Compared to the wild type, the fusion protein has similar absorption spectra (Fig. 1b), a slightly enhanced dark reversion rate (Fig. 1c), and similar but reduced phosphatase activity in Pfr (Fig. 1d)35.
Figure 2 shows the reconstructed electron density map from 117,297 single-particle images of DrBphP-DrRR in Pr, with grids prepared from monodisperse protein in darkness (Supplementary Fig. 1a). The selection and refinement process is summarized in Supplementary Fig. 2. Incomplete particles were removed based on 2D classifications. Three 3D classes were obtained from ab-initio classification. Classes 2 and 3 contained incomplete densities and were discarded. Homogeneous refinement of class 1 followed by local refinement implemented in cryoSPARC v3.1.0 gave a resolution of 3.6 Å, and local refinement with a mask over the PAS-GAF-PHY-neck region resulted from a resolution of 3.4 Å. Both maps had a local difference in resolution (Supplementary Fig. 6). The clear densities corresponding to PAS-GAF-PHY-DHp, including the “neck”-linker between PHY and DHp (Supplementary Fig. 6), allowed us to build a model with confidence for these domains (Table 1). The modeled structure shows the signatures expected for the Pr state, including the PHY tongue in a β-sheet conformation (Fig. 2b) and the biliverdin in 15Z conformation (Fig. 2c and Supplementary Fig. 5c)12,13. The DHp domains show a dimeric and symmetric conformation, which is often found in histidine kinases23. The two helices of a DHp domain can be connected in two ways in the four helical bundles of the dimer. We chose to link the two DHp helices following the crystal structure of the Thermotoga maritima histidine kinase26, but note that the reconstructed electron densities would also support the alternative linkage of the helices. The structure of the PAS-GAF-PHY domains is almost identical to the crystal structure of the corresponding truncation of DrBphP (Supplementary Fig. 3e)13,36. We also find two strong and two weaker patches of densities, which are arranged symmetrically around the DHp domains. They resemble the crystal structure of the Thermotoga maritima histidine kinase in complex with its response regulator (pdb entry 3DGE)36, and we assign the patches to the REC and CA domains (Supplementary Fig. 5a).

a The reconstructed electron density of Pr full-length DrBphP-DrRR is shown with the structural model which was refined up to residue Leu591. The electron density of the CA domains is below the presented contour level. See Supplementary Fig. 5a for CA and REC domain assignment. b The PHY tongue has adopted a β-sheet form characteristic of Pr-state phytochromes. c The density of the BV chromophore supports a 15Z-conformation (compare Supplementary Fig. 5c). The densities of the tongue and BV are from the cryo-EM map with the local refinement of the PSM and neck region (Pr PSM, see Supplementary Fig. 2).
We continued by solving the structure of DrBphP-DrRR in its Pfr state. The grids were prepared as for Pr, but under illumination with red light (see Materials and Methods). These conditions yield proteins in a photoequilibrium of Pr/Pfr. The selection and refinement process is summarized in Supplementary Fig. 4. Pfr and Pr conformations were identified and separated as 3D classes in the data (see Supplementary Fig. 4 for details), yielding a reconstructed electron density map for Pfr from 98,050 single-particle images of DrBphP-DrRR (Fig. 3). From the reconstruction we estimated that ~50% of intact proteins on the grid were in Pfr (Supplementary Fig. 4). Although we did not impose any symmetry in the classification, no mixed Pr/Pfr conformations were resolved as judged by the densities at the tongue region. We used local refinement with a mask of the region up to an approximate residue number of 521, which comprises the PAS-GAF-PHY domains and the helical neck to obtain a map with an average resolution of 3.5 Å (Supplementary Fig. 7). In these densities, we built a protein model consisting of PAS-GAF-PHY-neck (residues 22–521) (Fig. 3a, Table 1). The protein is confirmed to be in Pfr as the PHY tongue appears as an α−helix (Fig. 3b)13 and the chromophore is best modeled in 15E conformation (Fig. 3c and Supplementary Fig. 5d)11. Unexpectedly, the 3D arrangement of the PAS-GAF-PHY domains is notably different from the crystal structures of the PAS-GAF-PHY truncation in Pfr (Supplementary Fig. 3f)13. The electron density in the histidine kinase output domains is asymmetric and weaker compared to the corresponding density in the Pr state. This made an assignment of the output domains challenging and our assignment of the CA and REC effector domains remains tentative (Supplementary Fig. 5b). However, visual inspection clearly reveals that the dimer interface at the DHp domain is modified in Pfr (Fig. 3a and Supplementary Fig. 5).

a The reconstructed electron density of Pfr full-length is shown with the structural model, which was refined up to residue Ala521 based on the cryo-EM map of the photosensory core module in Pfr (Pfr PSM). Patches of electron density were assigned to the CA and REC domains (see Supplementary Fig. 5b). b The tongue is in α-helical form characteristic of Pfr-state phytochrome structures. c The density of the BV chromophore supports a 15E conformation (compare to Supplementary Fig. 5d). The densities of the PHY tongue and BV are extracted from the cryo-EM map with the local refinement of the PSM and neck in Pfr (Pfr PSM, see Supplementary Fig. 4).
Now, we are in a position to compare the Pr and Pfr structures (Fig. 4). A strikingly similar positioning of the PHY domains in Pr and Pfr is observed, despite the change in the fold of the PHY tongue. The distance between the center of mass of PHY domains in Pfr is only 0.4 Å larger compared to the same measure in Pr (Fig. 4a). Yet, a clear change is observed in the densities of the output domains (Figs. 2a and 3a). The change is apparent in the neck region, which shows a parallel arrangement of the helical domains in Pr but spreads apart in Pfr by ~4 Å at the top (Fig. 4b). The two helices of the coiled-coil neck are held together by hydrophobic packing interactions and hydrogen bonds. Notable hydrophobic interactions are shared by Leu502, Leu506, and Ile509 in the lower part of the neck. Asn513, Ser519, and Asn520 form hydrogen bonds across the dimer (Fig. 4c, insets). In the Pfr state, the dimer interface is broken C-terminal of Asn513, which leads to the apparent unzipping of the coiled-coil neck under red light.

a The entire structure is compared. The CA domains in the Pr-state structure are shown as transparent. They were not refined against the data and are taken from a homology model against the structure of a histidine kinase from Thermotoga maritima (pdb entry 2C2A)52. The black dots indicate the center of mass of the PHY domains. b The graph shows the distance between Cα atoms across the dimer interface in the neck region. Source data are provided as a Source Data file. c The neck region with the densities is shown for Pr (blue) and Pfr (magenta). A zipper-like opening of the dimerization interface is observed. The insets show detailed interactions between sister residues Ser519 and Asn520 (upper), Asn513 (middle), and Leu502, Leu506 and Ile509 (lower). The structures are colored according to their state, the hydrogen bonds are shown in yellow dashed lines, and the distances are in Å.
Discussion
From the single particle cryo-EM structures of full-length DrBphP in Pr and Pfr, the following model of photoactivation arises: (1) Red light leads to isomerization of the D-ring in BV. This triggers a change in the chromophore binding pocket and a transition of the PHY tongue from a β-sheet into an α-helix13. (2) The PHY domains do not splay apart in a dramatic way, as has been suggested by the crystal and solution structures of truncated DrBphP13. Instead, we find that these changes are much smaller in the context of the full-length protein. (3) The neck opens up in a zipper-like fashion in Pfr, (4) introducing conformational flexibility in the DHp and CA output domains.
In consequence, the dimer interface across the DHp domains breaks, which is consistent with a chromatography study of DrBphP37. The change of the shape of the protein is moderate, which is consistent with small-angle X-ray scattering data on the same phytochrome and pulsed electron paramagnetic resonance spectroscopy on a homologous bacterial phytochrome38,39. Our structures contradict a previously suggested model of photoconversion based on low-resolution cryo-EM structures36. Although the zipper-like opening does not imply a change-of-register in the dimer binding interface of the linker as it has been proposed for phytochrome based on analysis of a phytochrome-regulated diguanylyl cyclases34, it is still consistent with heptad periodicity of buried residues in the dimer interface40.
We evaluated the applicability of the mechanism in cyanobacterial, fungal, and plant phytochromes by generating homology models of our Pr structure with the phytochromes from Synechocystis PCC6803 (Cph1), Emericella nidulans (EnPhy), and Arabidopsis thaliana (AtPhyA)41, respectively (Supplementary Fig. 3). We find that all of these phytochromes show potential for a homologous neck region and thus the mechanism, or a variation thereof, may also apply for other phytochromes. However, plant phytochromes signal through light-dependent protein–protein interactions and have recently been shown to have a different dimer arrangement compared to bacterial phytochromes14. Other mechanisms of signal transduction may therefore apply in plant phytochromes.
Several crystal structures of truncated phytochromes have both head-to-head and head-to-tail dimer arrangements. In Pr, a bent conformation of the photosensory core module is most often found, whereas varying arrangements have been observed in Pfr10,11,13,42,43,44. These structures may be affected by packing artefacts in the crystal and by the truncation of the phytochrome. The cryo-EM structures presented here are free of these biases. They firmly confirm a head-to-head arrangement of the dimer for bacterial phytochromes and indicate that the bent conformation of the photosensory core module previously detected in Pr crystals is the one that is present in the solution. Surprisingly, we found that the same bent arrangement of the module holds for both the Pr and Pfr states.
Like most crystal structures, our cryo-EM models are homodimeric with respect to Pr and Pfr states, and Pr/Pfr heterodimers were not detected. This may be caused by the particle selection process in the EM data processing being unable to assign a class on its own for the Pr/Pfr heterodimers. We believe that the algorithm and data should be accurate enough to do so and have allowed this by requesting more than three classes out of ab-initio refinement and by not imposing any symmetry in the refinement. Thus, the lack of Pr/Pfr heterodimers may indicate their absence on the grids. This could be explained by fast Pr/Pfr heterodimer reversion to the Pr state during blotting, or that the quantum yield for the photoreaction from Pr to Pfr is allosterically increased in the heterodimer. However, these explanations remain somewhat speculative and we believe that the issue requires further investigation.
The structures are the first full-length and dimeric structures of a TCS histidine kinase in active and resting states. Firstly, the structures show evidence of a zipper-like mechanism to guide signal transduction into the effector domains. To the best of our knowledge, this is the first observation of such a mechanism and adds to earlier proposals for various histidine kinase architectures on scissoring piston-like movements, or rotation of the helical bundles21,23. Several mechanisms of signal transduction into the effector domains may coexist25, reflecting that histidine kinases integrate many different signals in a variety of architectures and have evolved through domain swapping45. Similarly, the photosensory modules of phytochromes may adapt their structural changes to the output domains, which may explain why they can form a variety of domain fusions with distinct effectors16,17,40,46. Secondly, our data provide direct and strong evidence that the effector histidine kinase module loses symmetry and order when its biochemical activity changes. Crystal structures of enzymatically active HKs do not show disorder, which is however likely due to that diffracting crystals require ordered packing. The present cryo-EM structures are free of such restrictions representing the structure of the proteins in solution much more closely.
Finally, our structures provide insight into how allosteric communication across the entire bacteriophytochrome occurs. We observe structural changes around the chromophore and in the output domains. Apart from the secondary structural changes of the tongue region, the PHY domains, which connect the chromophore region to the effector domains, undergo only a rather small change of position. The center of mass of the PHY domains moves apart by only 0.4 Å in Pfr compared to Pr. We conclude that the small structural changes in the PHY domains are sufficient to tip the scales and break the dimer interface across the neck, which then controls the enzymatic activity of the effector domain. The dimer interface at the DHp domains thereby amplifies the structural changes in the PHY domain and plays a key part in signaling along the phytochromes. This interpretation is in line with allostery where coupled equilibria between domains transduce information across the entire phytochrome25,28. Realizing that coupling across the entire phytochrome exists—and persists after photoexcitation—provides a structural rationale for that the dark reversion time back to the resting state changes when the protein is truncated at the neck30.
The structures of a prototypical full-length bacteriophytochrome in active and resting states reveal a zipper-like mechanism of how the signal is transduced into the effector domains in phytochromes, explaining how bacteria, plants, and fungi sense red/far-red light. This mechanism serves as a template for understanding two-component signaling and can inspire the development of new near-infrared optogenetic applications16,17,47.
Methods
Protein preparation and purification
The phytochrome from Deinococcus radiodurans strain R1 (DrBphP, gene DR_A0050) in pET21b(+) plasmid (Novagen) was a kind gift from Prof. Richard Vierstra48. The response regulator from Deinococcus radiodurans strain R1 (DrRR, gene DR_A0049) is described elsewhere35. The DrBphP-DrRR fusion protein was generated by introducing the DrRR gene (residues 1–149) after DrBphP (residues 1–755) and before the XhoI restriction site of the pET21b(+) with Gibson assembly (NEBuilder HiFi DNA assembly cloning kit, New England Biolabs). A linker of 12 residues (ASSAGGSAGSAG), inspired by the Agp2 sequence, was also introduced between the DrBphP and DrRR. The resulting 938-aa construct had a PAS-GAF-PHY-DHp-CA-REC domain composition flanked by an N-terminal T7-tag and a C-terminal His6-tag.
DrBphP-DrRR and DrBphP were expressed and purified as described below and adapted from ref. 35. The expression was conducted in Escherichia coli strain BL21 (DE3) overnight at 24 °C. After cell lysis with EmulsiFlex®, the sample was incubated with a molar excess of biliverdin hydrochloride (Frontier Scientific) overnight on ice. The His6-tagged protein was purified with NiNTA affinity purification (HisTrap™, GE Healthcare), followed by size-exclusion chromatography (HiLoad™ 26/600 Superdex™ 200 pg, GE Healthcare) in buffer (30 mM Tris, pH 8.0). Finally, the purified protein was concentrated to 12 mg/ml and flash-frozen. Prior to application to the grids, the protein was thawed and purified by size exclusion chromatography (Supplementary Fig. 1a) to ensure sample homogeneity.
UV–vis spectroscopy and Phos-tag assay
The absorption spectra of phytochromes were measured with Agilent Cary 8454 UV–visible spectrophotometer (Agilent). Phytochrome samples were diluted with (30 mM Tris/HCl pH 8.0) to 1.0 µM concentration and an approximate A700 value of 0.1 cm−1. The samples were illuminated with saturating (662 ± 1) nm red light and (782 ± 1) nm far-red light to populate Pfr and Pr states, respectively. In the dark reversion experiments, phytochromes were first driven to the Pfr-state population with 3-min saturating red light, after which the reversion data were recorded at room temperature. Data points were taken at 2-min intervals for the first 10 min, 5-min intervals until 30 min, 10-min intervals until 60 min, and 20-min intervals until 120 min. The exponential fits from dark reversion data were calculated with Matlab R2020b (9.9.0.1467703) (MathWorks Inc.) using Eq. (1). Two components were used for fitting in all samples.
where t is time, A700 and A750 are absorption values, A1 and A2 are the decay amplitudes of the absorbance ratio, and τn the time constant of the decay component.
The Phos-tag assay was carried out with a similar method as in Ref. 3. Namely, phosphorylated DrRR (p-DrRR) was generated by incubating DrRR in the assay buffer (25 mM Tris/HCl pH 7.8, 5 mM MgCl2, 4 mM 2-mercaptoethanol, 5% ethylene glycol) supplemented with 90 mM acetyl phosphate at 37 °C for 30 min. For the phosphatase reactions in the assay buffer, p-DrRR (0.3 mg/ml) was mixed with phytochrome samples (0.3 mg/ml each), and the reactions were initiated with 1.67 mM ATP. The reactions were incubated either in dark or under saturating 650-nm light (LED Spot Light, 3 W Deep Red 657 nm, Lens-C) for 20 min after which the reaction was stopped and analyzed according to Zn2+-Phos-tag® SDS–PAGE assay (Wako Chemicals)35.
Single-particle cryo-EM grid preparation and data acquisitions
Cryo-EM grids were prepared under dim green safe light using a Vitrobot (FEI) with the sample chamber at 4 °C and 100% humidity. Before grids preparation, CaCl2, MgCl2, and AMP-PNP were added to the sample to 10, 10, and 4 mM concentrations, respectively. To prepare grids with proteins in Pr state, three microliter protein samples (1–1.5 mg/ml) were applied to glow-discharged Quantifoil R 2/2 Cu300 grids and pre-illuminated with 780 nm far-red light for Pr state and 660 nm red light for Pfr state on the grid before plunge-freezing in liquid ethane. The particles were imaged using a Titan Krios operated at 300 kV and a magnification of 105k. The images were recorded on a Gatan K3 BioQuantum detector with a pixel size of 0.8617 Å and an exposure rate of 48.3 electrons per Å2 for a total of 40 frames. The targeted defocus range was varied from –0.6 to –2.6 µm using the EPU software (Thermo Fisher).
Analysis of cryo-EM data
A total of 6392 movies were collected from a grid prepared in far-red light illuminated (Pr) state and 7251 movies from a grid prepared in red light illuminated (Pfr) state, see Supplementary Fig. 2 and Supplementary Fig. 4 for the scheme of image processing. Movies (Supplementary Fig. 8a, b) were motion-corrected and contrast transfer function (CTF)-estimated using cryoSPARC v349. After blob-picker, extracted particles were subjected to 2D classification to remove the junk particles. Good particles were used in Topaz deep pick50. Extracted particles were further cleaned up with more rounds of 2D classification (Supplementary Fig. 8c, d). From a far-red light-illuminated grid, a set of 316,139 particles was used to reconstruct three initial 3D models using Ab-initio in cryoSPARC. These three models were used as initial volumes for heterogeneous refinement and the class 1 model with 117,297 particles showed the best features and was chosen to further homogeneous and local refinement without any symmetry applied. The final model of the Pr state has an overall resolution of 3.6 Å (Supplementary Fig. 6). From the grid prepared in the red light illuminated state, good particles with side view only from the blob-picker went through an extra round of Topaz deep pick. After duplicated particles got removed, a set of 387,312 particles was used to reconstruct three initial 3D models. After heterogeneous refinement, the class 2 model was identified to be Pfr state and the class 1 model Pr state. After two more rounds of heterogeneous refinement to clean up particles of Pr state from Pfr state, a total of 98,050 particles were used for further homogeneous, local refinement, and sharpening.
Model building and refinement
The crystal structure of DrPAS-GAF-PHY fragment (pdb code 4Q0J) was first fit into the Pr full-length map (Pr full-length, see Supplementary Fig. 2), then built and refined further in real space with Coot 0.9.6. We were able to build the DrBphP up to residue Leu591, covering the PAS-GAF-PHY-DHp domains. The Pfr structure was initiated by fitting the DrPAS-GAF-PHY crystal structure (pdb code 4Q0J) into the map of the photosensory core module (Pfr PSM, see Supplementary Fig. 4), followed by modeling the Pfr-specific traits at the chromophore surroundings and the PHY tongue based on a Pfr-state structure of DrPAS-GAF-PHY (pdb code 5C5K). The model was built up to Ala521, covering the neck part of the DHp. Due to the poor density in the histidine kinase module, model building was not conducted with the Pfr full-length map (Pfr full-length, see Supplementary Fig. 4). Once the Pr and Pfr models were built and refined, the structures were refined with REFMAC5 (version 5.8.0267) of the CCP-EM software suite (version 1.5.0) with tight (0.0002–0.001) manual weights51. The model and refinement parameters are collected in Table 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon reasonable request. The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-15684 (DrBphP(PSM) in PR), EMD-15685 (DrBphP in Pr), and EMD-15686 (DrBphP in Pfr). The coordinated have been deposited in the Protein Data Bank (PDB) under accession codes 8AVV (DrBphP(PSM) in PR), 8AVW (DrBphP in Pr), and 8AVX (DrBphP in Pfr). Source data are provided with this paper.
References
Quail, P. H. et al. Phytochromes: Photosensory Perception and Signal Transduction. Science Vol. 268 675–680 (American Association for the Advancement of Science, 1995).
Legris, M., Ince, Y. Ç. & Fankhauser, C. Molecular mechanisms underlying phytochrome-controlled morphogenesis in plants. Nat. Commun. 10, 1–15 (2019).
Rockwell, N. C., Su, Y.-S. & Lagarias, J. C. Phytochrome structure and signaling mechanisms. Annu. Rev. Plant Biol. 57, 837–858 (2006).
Butler, W. L., Norris, K. H., Siegelman, H. W. & Hendricks, S. B. Detection, assay, and preliminary purification of the pigment controlling photoresponsive development of plants. Proc. Natl Acad. Sci. USA 45, 1703–1708 (1959).
Davis, S. J., Vener, A. V. & Vierstra, R. D. Bacteriophytochromes: phytochrome-like photoreceptors from nonphotosynthetic eubacteria. Science 286, 2517–2520 (1999).
Hughes, J. et al. A prokaryotic phytochrome [6]. Nature 386, 663 (1997).
Burgie, E. S., Bussell, A. N., Walker, J. M., Dubiel, K. & Vierstra, R. D. Crystal structure of the photosensing module from a red/far-red light-absorbing plant phytochrome. Proc. Natl Acad. Sci. USA 111, 10179–10184 (2014).
Nagano, S. et al. Structural insights into photoactivation and signalling in plant phytochromes. Nat. Plants 6, 581–588 (2020).
Song, C. et al. 3D Structures of plant phytochrome A as Pr and Pfr from solid-state NMR: implications for molecular function. Front. Plant Sci. 9, 1–15 (2018).
Essen, L.-O., Mailliet, J. & Hughes, J. The structure of a complete phytochrome sensory module in the Pr ground state. Proc. Natl Acad. Sci. USA 105, 14709–14714 (2008).
Yang, X., Kuk, J. & Moffat, K. Crystal structure of Pseudomonas aeruginosa bacteriophytochrome: photoconversion and signal transduction. Proc. Natl Acad. Sci. USA 105, 14715–14720 (2008).
Wagner, J. R., Brunzelle, J. S., Forest, K. T. & Vierstra, R. D. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature 438, 325–331 (2005).
Takala, H. et al. Signal amplification and transduction in phytochrome photosensors. Nature 509, 245–248 (2014).
Li, H., Burgie, E. S., Gannam, Z. T. K., Li, H. & Vierstra, R. D. Plant phytochrome B is an asymmetric dimer with unique signalling potential. Nature 2022 604:7904 604, 127–133 (2022).
Shu, X. et al. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science (1979) 324, 804–807 (2009).
Gasser, C. et al. Engineering of a red-light-activated human cAMP/cGMP-specific phosphodiesterase. Proc. Natl Acad. Sci. USA 111, 8803–8808 (2014).
Ryu, M. H. et al. Engineering adenylate cyclases regulated by near-infrared window light. Proc. Natl Acad. Sci. USA 111, 10167–10172 (2014).
Stock, A. M., Robinson, V. L. & Goudreau, P. N. Two-component signal transduction. Annu. Rev. Biochem. 69, 183–215 (2000).
Montgomery, B. L. & Lagarias, J. C. Phytochrome ancestry: sensors of bilins and light. Trends Plant Sci. 7, 357–366 (2002).
Nizet, V. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr. Issues Mol. Biol. 8, 11–26 (2006).
Gushchin, I. et al. Mechanism of transmembrane signaling by sensor histidine kinases. Science 356, 6342 (2017).
Berntsson, O. et al. Sequential conformational transitions and α-helical supercoiling regulate a sensor histidine kinase.Nat. Commun. 8, 284 (2017).
Jacob-Dubuisson, F., Mechaly, A., Betton, J. M. & Antoine, R. Structural insights into the signalling mechanisms of two-component systems. Nat. Rev. Microbiol. 16, 585–593 (2018).
Neiditch, M. B. et al. Ligand-induced asymmetry in histidine sensor kinase complex regulates quorum sensing. Cell 126, 1095–1108 (2006).
Mensa, B. et al. Allosteric mechanism of signal transduction in the two-component system histidine kinase PhoQ. Elife 10, e73336 (2021).
Casino, P., Rubio, V. & Marina, A. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139, 325–336 (2009).
Dikiy, I. et al. Insights into histidine kinase activation mechanisms from the monomeric blue light sensor EL346. Proc. Natl Acad. Sci. USA 116, 4963–4972 (2019).
Takala, H. et al. On the (un)coupling of the chromophore, tongue interactions, and overall conformation in a bacterial phytochrome. J. Biol. Chem. 293, 8161–8172 (2018).
Claesson, E. et al. The primary structural photoresponse of phytochrome proteins captured by a femtosecond x-ray laser. Elife 9, e53514 (2020).
Velazquez Escobar, F. et al. A protonation-coupled feedback mechanism controls the signalling process in bathy phytochromes. Nat. Chem. 7, 423–430 (2015).
Toh, K. C., Stojkovic, E. A., van Stokkum, I. H. M., Moffat, K. & Kennis, J. T. M. Proton-transfer and hydrogen-bond interactions determine fluorescence quantum yield and photochemical efficiency of bacteriophytochrome. Proc. Natl Acad. Sci. USA 107, 9170–9175 (2010).
Song, C. et al. Two ground state isoforms and a chromophore D-ring photoflip triggering extensive intramolecular changes in a canonical phytochrome. Proc. Natl Acad. Sci. USA 108, 3842–3847 (2011).
Otero, L. H. et al. Structural basis for the Pr–Pfr long-range signaling mechanism of a full-length bacterial phytochrome at the atomic level. Sci. Adv. 7, 1097 (2021).
Gourinchas, G., Heintz, U. & Winkler, A. Asymmetric activation mechanism of a homodimeric red light-regulated photoreceptor. Elife 7, e34815 (2018).
Multamäki, E. et al. Comparative analysis of two paradigm bacteriophytochromes reveals opposite functionalities in two-component signaling. Nat. Commun. 2021 12:1 12, 1–14 (2021).
Burgie, E. S. et al. Crystallographic and electron microscopic analyses of a bacterial phytochrome reveal local and global rearrangements during photoconversion. J. Biol. Chem. 289, 24573–24587 (2014).
Takala, H., Björling, A., Linna, M., Westenhoff, S. & Ihalainen, J. A. Light-induced changes in the dimerization interface of bacteriophytochromes. J. Biol. Chem. 290, 16383–16392 (2015).
Björling, A. et al. Structural photoactivation of a full-length bacterial phytochrome. Sci. Adv. 2, e1600920 (2016).
Kacprzak, S. et al. Intersubunit distances in full-length, dimeric, bacterial phytochrome Agp1, as measured by pulsed electron–electron double resonance (PELDOR) between different spin label positions, remain unchanged upon photoconversion. J. Biol. Chem. 292, 7598–7606 (2017).
Möglich, A. Signal transduction in photoreceptor histidine kinases. Protein Sci. 28, 1923 (2019).
Yuan Wahlgren, W., Golonka, D., Westenhoff, S. & Möglich, A. Cryo-electron microscopy reveals the head-to-head homodimeric architecture of Arabidopsis thaliana phytochrome A. Front. Plant Sci. 12, 694 (2021).
Burgie, E. S., Zhang, J. & Vierstra, R. D. Crystal structure of Deinococcus phytochrome in the photoactivated state reveals a cascade of structural rearrangements during photoconversion. Structure 24, 448–457 (2016).
Bellini, D. & Papiz, M. Z. Structure of a bacteriophytochrome and light-stimulated protomer swapping with a gene repressor. Structure 20, 1436–1446 (2012).
Nagano, S. et al. The crystal structures of the N-terminal photosensory core module of Agrobacterium phytochrome Agp1 as parallel and anti-parallel dimers. J. Biol. Chem. 291, 20674–20691 (2016).
Capra, E. J. & Laub, M. T. Evolution of two-component signal transduction systems. Annu. Rev. Microbiol. 66, 325–347 (2012).
Levskaya, A. et al. Engineering Escherichia coli to see light. Nature 2005 438:7067 438, 441–442 (2005).
Deisseroth, K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci. 2015 18:9 18, 1213–1225 (2015).
Wagner, J. R., Zhang, J., Brunzelle, J. S., Vierstra, R. D. & Forest, K. T. High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J. Biol. Chem. 282, 12298–12309 (2007).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A.cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 2019 16:11 16, 1153–1160 (2019).
Burnley, T., Palmer, C. M. & Winn, M. Recent developments in the CCP-EM software suite. Acta Crystallogr. D Struct. Biol. 73, 469–477 (2017).
Marina, A., Waldburger, C. D. & Hendrickson, W. A. Structure of the entire cytoplasmic portion of a sensor histidine-kinase protein. EMBO J. 24, 4247–4259 (2005).
Acknowledgements
Cryo-EM samples were prepared at the Centre for Cellular Imaging at the University of Gothenburg. Cryo-EM sample screening and data collection were performed at the cryo-EM Swedish National Facility in Stockholm, Sweden funded by the Knut and Alice Wallenberg, Family Erling Persson and Kempe Foundations, SciLifeLab, Stockholm University, and Umeå University. We thank Julian Conrad, Karin Walldén, Michael Hall, and Marta Carroni for the technical assistance in data collection and the advice on the reconstruction of the electron density maps. We thank Dr. Dmitry Morozov (University of Jyväskylä) for help in modeling the structure and fruitful discussions, as well as Vilma Tarkiainen and Roosa Vanhatalo (University of Jyväskylä) for help with the sample preparation. S.W. acknowledges support from the European Research Council, ERC grant to Westenhoff, MolStrucDyn, 725642. This work was supported by Academy of Finland grants 332742 (J.A.I.) and 330678 (H.T.).
Funding
Open access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
S.W., J.A.I., H.T., and E.C. perceived the project; S.W., W.Y.W., E.C., H.T., and J.A.I. devised the methodology; W.Y.W., S.T.-M., and I.T. performed the investigation; W.Y.W., S.B., and H.T. prepared the figures; S.W., H.T., and J.A.I. secured the funding and supervised the project; S.W., H.T., S.B., and W.Y.W. wrote the paper with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wahlgren, W.Y., Claesson, E., Tuure, I. et al. Structural mechanism of signal transduction in a phytochrome histidine kinase. Nat Commun 13, 7673 (2022). https://doi.org/10.1038/s41467-022-34893-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-34893-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
