
Abstract
The enantioselective pyridinylation is important for providing chiral compounds bearing heterocycles of pharmaceutical interests. 4-CN-pyrinde is extensively applied in the radical pyridinylation reaction, however, its’ enantioselective application is highly challenging. To achieve this goal, we propose an electrochemical catalytic activation of 4-CN-pyridine with a chiral transition metal complex instead of direct cathodic reduction. The chiral catalyst acts as the electron mediator and the transition metal catalysis in turn. The radical species from 4-CN-pyridine is captured via radical rebound by chiral catalyst, and undergoes enantioselective pyridinylation reaction. Here, we show the first method for catalytic asymmetric allylic 4-pyridinylation reactions using 4-CN-pyridine under electrochemical conditions.
Similar content being viewed by others
Introduction
Controlling the enantioselectivity during catalytic electron-transfer reactions is a challenging task1,2,3,4,5,6,7,8. Electron transfer can lead to open-shell species that have different bonding characteristics than species generated by close-shell reactions involving chiral catalysts. Rationale enantioselective reactions that use the tremendous driving force of an electric potential are in high demand.
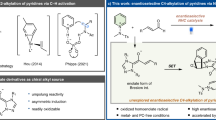
4-CN-pyridine is a typical reagent in open-shell chemistry. It is able to accept electrons under various conditions, and it has been shown to be a versatile pyridinylation reagent in photoredox catalysis9,10,11,12,13,14,15,16, electrochemistry17,18,19,20,21, and thermal chemistry22,23 to build molecules with Csp3 centers (Fig. 1a). In these reactions, it forms a persistent radical that undergoes C–C bond formation reactions with the substrates22. This chemistry can be used to build pyridine-bearing stereogenic centers, which are present in many pharmaceutical compounds24,25,26. Despite these highly important achievements, to date, the method to control the key open shell radical is a challenging task.27 In addition, our previous findings22 suggested the persistent radical derived from 4-CN-pyridine localizes the major spin density at its tertiary carbon center, which does not fall into the paradigm established for controlling the stereochemistry of radical reaction28,29,30,31,32,33,34,35,36,37,38,39. At this point, it would be highly desirable to develop an enantioselective pyridinylation reaction with 4-CN-pyridine, which can not only achieve an enantioselective model regulating persistent tertiary radical center, but also provide a class of chiral pyridine compounds. The protocol would provide the complementary to successfully enantioselective 4-pyrdinylation methods via 2-electron or single electron pathways.40

a Established chemistry with 4-CN-pyridine for racemic/achiral compounds. b This work: enantioselective electroreductive palladium-catalyzed pyridinylation via allylic substitution.
Asymmetric allylic alkylation is a powerful strategy for converting readily available allylic compounds to a diverse array of chiral products41,42,43,44,45,46,47,48,49,50,51,52,53,54,55. Notably, however, asymmetric allylic pyridinylation has not been achieved, partly because of the difficulty in generating and regulating the nucleophilic planar close-shell pyridinyl species. In this regard, we conceive a pathway involving the persistent radical would be a solution, in which an enantioselective catalysis design for both allyl partner and tertiary radical is the key.
Here, we show the first example for asymmetric allyl pyridinylation with 4-CN-pyridine driven by electrochemistry, in which the chiral Pd complex functions as both an electron-transfer mediator and chiral catalyst in the relay model to introduce persistent radical into the stereogenic center via a Pd-effected radical rebound,56,57,58,59 which has not been explored (Fig. 1b).60,61,62,63,64
Results
Reaction optimization
To begin our study, we chose cinnamyl acetate 1a as a substrate for reactions with 4-CN-pyridine (2a) under constant-current electrolysis conditions in an undivided cell with a graphite felt (GF) cathode and a Zn anode (Table 1). When PdCl2 was the catalyst precursor and (R)-SEGPHOS (L1) was the chiral ligand, reaction in 5:1 (v/v) MeCN/MeOH at 35 °C and a current density of 5 mA/cm3 afforded a 8:1 mixture of desired branched (B) product 3a and linear (L) product 3a’ (entry 1); the enantiomeric excess (ee) for 3a was 70%. Changing to a single solvent (MeCN or DMF) decreased the yield, B/L ratio, and enantioselectivity (entries 2 and 3). If Et4NBF4 was used as supporting electrolyte, the reaction gave 3a in 60% yield, with a B/L of 5:1 and 65% ee (entry 4). When (R)-DTBM-SEGPHOS (L2) was used as the ligand, the B/L ratio and the ee value increased to 13:1 and 93%, respectively; and the isolated yield of 3a was 76% (entry 5). X-ray analysis of a hydroxylative derivative of 3a indicated that it had a R configuration (Supplementary Fig. 162). Subsequently, chiral ligand (R)-BINAP (L3) and (R)-SPD (L4) were evaluated, and these experiments showed that a ligand with a small dihedral angle gave better results (entries 1, 6, and 7). (R)-DTBM-GARPHOS (L5, entry 8), which has a small dihedral angle, gave an ee similar to that obtained with L2. In contrast, trialkyl phosphine ligand L6 gave a moderate yield and relatively low enantioselectivity (entry 9). Reactions involving (S, R)-Josiphos (L7) and monodentate ligand L8 (entries 10 and 11, respectively) did not give measurable amounts of the desired product. When a stoichiometric metal reductant (Zn or Mg) was used instead of electricity, no reaction took place (entry 12). Finally, when Pd and chiral ligand were absent, no conversion of both 1a and 2a were observed (entry 13).
Substrate scope
Using the optimized conditions (Table 1, entry 5), we explored the substrate scope of this asymmetric allylic pyridinylation reaction (Fig. 2). We began by systematically assessing the effect of the substituent (X) on the phenyl ring of cinnamyl acetate 1. The steric bulk of the substituent did not markedly affect the regio- or stereoselectivity, as indicated by the results obtained for branched products 3b–3 g with good to excellent ees. Products 3h–3 m, all of which have an electron-donating alkoxide group at the ortho position, were obtained in moderate to good yields, with ee values typically around 90%. Next, the standard conditions were used to test a series of halogenated substrates, which gave products 3n–3r with good to excellent enantioselectivities (85%–94%). We were pleased to find that even an aryl bromide moiety survived the reaction: product 3p was obtained with 94% ee. Products 3 s and 3t, which have para electron-donating groups, were obtained with 89% and 86% ee values, respectively; and dihalogenated product 3 u was obtained with 91% ee. Products 3 v and 3w, which have electron-withdrawing substituents, had low yield due to low regioselectivity, and the enantioselectivies were moderate. Compounds with cyclopropyl, carboxylate, and dihydro-benzofuran moieties (3x–3z, respectively) were generated in acceptable yields with good to excellent ee values. A substrate with a secondary allyl acetate group afforded branched product 3aa in 62% isolated yield with an ee of 90%; the internal alkene with the E configuration was the only detectable product. Moreover, products 3ab–3al were generated smoothly with good to excellent enantioselectivities.

aConditions: 1 (0.2 mmol), 2a (0.6 mmol), PdCl2 (10 mol%), L2 (12 mol%), MeCN/MeOH 5:1 (v/v), Et4NCl (0.1 M), Zn(+)|GF(-), 5 mA/cm3, 5 h, 35 °C; isolated yields of branched product are reported; B/L and E/Z ratios were determined by 1H NMR analysis of crude reaction mixtures; ee values were determined by HPLC on a chiral stationary phase. bCurrent density, 8 mA/cm3. conly E configuration was detected. MOM, methoxylmethyl; PMB, 4-methoxybenzyl; TBDPS, t-butyldiphenylsilyl; Ts, tosyl; Boc, t-butyloxycarbonyl.
To further explore the scope, we prepared acetate 4a from benzaldehyde and vinyl Grignard reagent and allowed it to react with 2a under the standard conditions (Fig. 3). The outcome of this reaction was almost identical to that of the reaction of 1a and 2a (Fig. 2). We also tested several other 3-acetoxy-3-aryl propenes. Specifically, product 3am and 3an, which has an alkoxyl or phenoxyl group on the phenyl ring, was obtained with 93% and 91% ee. Other branched substrates also gave corresponding product 3ao–3at in good enantioselectivities. Compound 3au–3aw bearing two heterocycles were prepared with the same protocol in 81%–85% ee.

aConditions: 4 (0.2 mmol), 2a (0.6 mmol), PdCl2 (10 mol%), L2 (12 mol%), MeCN/MeOH 5:1 (v/v), Et4NCl (0.1 M), Zn(+)|GF(-), 5 mA/cm3, 5 h, 35 °C; isolated yields of branched products are reported; B/L were determined by with 1H NMR analyses of crude reaction mixture; Ee values were determined by HPLC on a chiral stationary phase. bCurrent density, 8 mA/cm3.
Subsequently, we explored reactions of 1a with different N-heterocycles 2 (Fig. 4). A broad scope of both electronic and sterically differentiated substituted on pyridine ring were accommodated well. However, the steric hindrance of 3-substituted substrate on pyridine ring has a certain effect on the enantioselectivity of the reaction. The greater the steric hindrance, the lower the corresponding enantioselectivity (3ax-3bb). The steric effect of pyridine 2-position only slightly influenced the enantioselectivity: products 3bc–3be were obtained with 85–86% ee. Finally, reaction between 1-CN-isoquinoline and 1a gave corresponding product 3bf in 49% isolated yield with an ee of 87%.

Conditions: 1a (0.2 mmol), 2 (0.6 mmol), PdCl2 (10 mol%), L2 (12 mol%), 5:1 (v/v) MeCN/MeOH, Et4NCl (0.1 M), Zn(+)|GF(-), 8 mA/cm3, 3 h, 35 °C; isolated yields are reported; ee values were determined by HPLC on a chiral stationary phase.
Next, the reaction of 1a and 2a was conducted at 10 mmol scale under standard conditions. It was glad to find 1.29 g of 3a was isolated in 66% yield with 91% ee (Fig. 5a). We demonstrated the synthetic utility of this method by carrying out a reaction between 2 mmol of 1r and 2a under the standard conditions; this reaction afforded desired product 3r in 45% yield with 85% ee. Further elaboration of 3r gave 5 (43% yield over three steps) with 87% ee. This product is an intermediate in a known procedure for the synthesis of tachykinin/serotonin-reuptake inhibitors (Fig. 5b)65. In addition, reaction of 2 mmol of 1a gave product 3a (75% yield, 93% ee), which was converted to amine 6, an intermediate in the synthesis of a calcium receptor modulator66, with almost complete enantioretention (Fig. 5c).

a Reaction at 10 mmol scale. b Synthesis of chiral precursor of tachykinin/serotonin reuptake inhibitor. c Synthesis of chiral precursor of calcium receptor modulator. 9-BBN, 9-borabicyclo[3.3.1]nonane; DMP: Dess-Martin periodinane; DIAD, diisopropyl azodicarboxylate.
Study of mechanism
Next, we conducted some control experiments to gain information about the reaction mechanism. At first, we used 2-naphthaldehyde, carbon dioxide, and perfluoropyridine as electrophiles instead of 4-CN-pyridine under otherwise identical conditions. These reactions did not afford the corresponding adducts, implying that allylic nucleophilic addition was not the predominant pathway (Fig. 6a). Then we measured the high-resolution mass spectrum of a reaction mixture containing L1 under the standard conditions after 10 min of reaction time, which revealed that the major Pd species was an L1-Pd(cinnamyl) complex (Fig. 6b, more details in Supplementary Fig. 5). Furtherly, a positive charge tagged BINAP ligand was prepared and subjected to the standard reaction conditions. In addition to the allyl complex, the two Pd-CN rather than Pd-Cl complexes were identified by HRMS, suggesting the stronger coordinative ability of cyanide group (Fig. 6c, more details in Supplementary Fig. 6).

a Control experiment with reactive electrophiles. b High resolution mass spectrometry (HRMS) analysis of reaction mixture under standard conditions. c HRMS analysis of reaction mixture using positive charge-tagged BINAP as chiral ligand. d Cyclic voltammetry (CV) analysis of Pd-cinnamyl complex. e CV analysis of reactants 1a and 2a. f Catalytic current of Pd complex. g Experiment under controlled cathodic potential. h The role of Zn in the reaction. SCE, saturated calomel electrode.
Therefore, we prepared an L1-Pd(cinnamyl)–BF4 complex and subjected it to cyclic voltammetry. Two significant reduction peaks were present in the voltammogram: one at −0.8 V and the other at −1.7 V vs SCE (labelled with a and b in Fig. 6d); these CV data are similar to data reported by Lei in their study on Pd(MeCN)2Cl2/P(p-Tol)3 in MeCN/nBuOH67. In addition, chiral phosphine ligand L1 along did not undergo cathodic reduction even at −2.2 V vs SCE. This resulted suggested an open-shell Pd species might exist after the first cathodic electron transfer at −0.8 V. In comparison, cathodic electron transfer to cinnamyl acetate (1a) and 4-CN-pyridine (2a) took place at a cathode potential around −1.8 V vs SCE, which is more difficult than L1-Pd(cinnamyl)–BF4 (Fig. 6e). Subsequently, we studied the CV of the L1-Pd(cinnamyl)–BF4 complex at −0.8 V vs SCE using 2a as an additive. A catalytic current around 10 μA scale (Fig. 6f) was detected, similar to the catalytic current observed in the reported Cu-catalyzed68,69 and Rh-catalyzed70 electrochemical asymmetric syntheses. This observation revealed that the reduced Pd species instantly transfers an electron to 2a to generate intermediate E, which again accepts an electron from the cathode. In this manner, the Pd complex not only serves as a transition-metal catalyst but also mediates an electron transfer. To further verify the mediation, the reaction was conducted with controlled cathodic potential (Fig. 6g). It was found with −1.2 V vs SCE cathodic potential was adequate to effect the conversation to desired product along with pyridine as side product. On the other hand, if PdCl2 and phosphine ligand were removed, no reaction took place at −1.2 V vs SCE, and the substrate 1a and pyridine 2a were recovered almost quantitatively. In addition, the role of Zn in the reaction was examined. When the sacrificial anode was changed from Zn to Al, the reaction did not take place (Fig. 6h). In comparison, if 0.2 equiv of ZnCl2 was used as additive in the reaction with Al as anode, the reaction worked smoothly to give product 3a in 58% yield.
On the basis of the above-described results, a plausible pathway is outlined in Fig. 7. First, PdCl2–diphosphine ligand complex A is reduced to [L*Pd0] complex B. Oxidative addition of cinnamyl acetate 1 to B gives rise to η3 allyl PdII complex C. This complex undergoes cathodic reduction at −0.8 V (vs SCE) to form Pd species D, which readily donates an electron to 4-CN-pyridine (2a), generating Pd species E; this single-electron transfer occurs readily because of the ability of 2a to stabilize anionic radicals with zinc counter ion. Complex E quickly accepts another electron from the cathode to produce Pd species F, generating a catalytic current. Subsequently, η3 allyl pyridinyl PdII complex G forms from F via radical rebound and then undergoes reductive elimination to afford desired product 3 and regenerate Pd0 complex B. The Zn2+ from the sacrificial anode both activates 2a and quenches the CN− anion.

Precursor A is reduced to complex B and in turn to allyl intermediate C. C undergoes rebound of 2a under cathodic reductions twice via D and E to give species F and in turn G. The reductive elimination of G furnishes the product and regenerates catalyst B.
By using the reaction of 3aa as the model, the regio-, and stereoselectivities were studied with density functional theory (DFT) computation study at B3LYP-3D level of theory (Fig. 8a). The free energy profile starting from intermediate F shown in Fig. 7. The atomic spin population analysis shows the single electron delocalizes on Pd and allyl group of intermediate F. (Supplementary Fig. 24). At first, the complex F undergoes the extrusion of Zn salt gives rise to complex G as exothermal process of −12.8 kcal/mol, since the formation of aromatic pyridinyl group. At this stage, the reductive elimination offers the product 3aa and establishes regio- and enantioselectivity. To compare the regioselectivity, two transition state TS-R and TS-R-L corresponding to branch and linear products were located. In TS-R, the phenyl ring connected to allyl group participates in the complexation with Pd, leading to an aromatic stacking between this phenyl ring and one phenyl ring of chiral ligand. In comparison, in TS-R-L there is allyl group coordinating to Pd metal, spacing the phenyl groups and diminishing the aromatic interaction. This difference between TS-R and TS-R-L may result in the favor on branched product, though the 6.0 kcal/mol gap exceeds the experimental ratio (B/L = 8:1). Subsequently, the comparison of TS-R and TS-S corresponding to R and S products showed the aromatic stacking stabilization is absent in TS-S. As a result, TS-R is 1.9 kcal/mol lower than TS-S in terms of energy. Within the precision range of DFT calculation, this result is confirmed by the experimental data (86% ee). In this reaction, only alkene with E configuration was detected and isolated substrate 1ac with either E or Z configuration (Supplementary Fig. 19). This observation was investigated with DFT calculation of intermediate C in Fig. 7, showing the syn-allyl-Pd-diphosphine complex is predominant over the interconvertible anti-counterpart with a stabilization of 7.6 kcal/mol due to steric effect (Fig. 8b). A 31P NMR tracking of reaction mixture also confirmed this predominance (Supplementary Fig. 21). In turn, the syn-intermediate can lead to product with E configuration.

a Reaction profile of reaction pathway to determine the regio- and enantioselectivity. b The difference of energy between syn- and anti- isomers of intermediate C.
In summary, we developed the first method for asymmetric allylic 4-pyridinylation reactions under electrochemical conditions. These reactions are catalyzed by a chiral diphosphine Pd complex and use 4-CN-pyridine as a pyridine source. The Pd complex not only acts as a chiral catalyst but also mediates electron transfer between the cathode and 4-CN-pyridine. More than 50 examples are reported, giving products with ee values as high as 94%; and the method was used to synthesize chiral pharmaceutical intermediates. Under these catalytic conditions, the 4-CN-pyridine is activated in solution below its reduction potential instead of at interface of electrode directly, avoiding the generation of uncontrollable species.
Methods
General procedure for the synthesis of 3
A 10 mL three-necked flask was charged with PdCl2 (0.02 mmol, 0.1 equiv), (R)-DTBM-SEGPHOS (0.024 mmol, 0.12 equiv) and a magnetic stir bar. The flask was evacuated and backfilled with argon for three times, and anhydrous MeCN (2 mL) was added via syringe. The mixture was stirred under room temperature for 30 min. Then the substrate 1 (0.2 mmol, 1.0 equiv), 2 (0.6 mmol, 3.0 equiv), Et4NCl (0.6 mmol, 3.0 equiv), anhydrous MeCN (3 mL) and MeOH (1 mL) was added. The flask was equipped with a rubber stopper, graphite felt (2 cm × 1 cm x 0.5 cm) as cathode, Zn (1.5 cm × 1 cm x 0.2 cm) as anode. The Zn anode attached to a platinum wire and graphite felt cathode attached to a titanium wire. The mixture was stirred under 35 °C and constant current electrolysis (5 mA). After the reaction completed (TLC or GC-MS analysis), the mixture was extracted with ethyl acetate. The organic layers were washed with brine, dried over Na2SO4, filtered and concentrated. The residue was purified by chromatography on silica gel to afford the desire product.
Data availability
The HPLC, NMR, HRMS and DFT compuation data generated in this study are provided in the Supplementary Information. Cartesian coordinates of the calculated structures are available from the Supplementary Data 1. Crystal structure data generated in this study is also available in the CCDC database under accession code 2118567.
References
Meggers, E. Asymmetric catalysis activated by visible light. Chem. Commun. 51, 3290–3301 (2015).
Ghosh, M., Shinde, V. S. & Rueping, M. A review of asymmetric synthetic organic electrochemistry and electrocatalysis: concepts, applications, recent developments and future directions. Beilstein J. Org. Chem. 15, 2710–2746 (2019).
Lin, Q., Li, L. & Luo, S. Asymmetric Electrochemical Catalysis. Chem. Eur. J. 25, 10033–10044 (2019).
Chang, X., Zhang, Q. & Guo, C. Asymmetric Electrochem. Transform. Angew. Chem. Int. Ed. 59, 12612–12622 (2020).
Lu, J., Wang, Y., McCallum, T. & Fu, N. Harnessing Radical Chemistry via Electrochemical Transition Metal Catalysis. iScience 23, 101796 (2020).
Phillips, A. M. F. & Pombeiro, A. J. L. Electrochemical asymmetric synthesis of biologically active substances. Org. Biomol. Chem. 18, 7026–7055 (2020).
Zhou, Z., Xu, S., Zhang, J. & Kong, W. Nickel-catalyzed enantioselective electroreductive cross-couplings. Org. Chem. Front. 7, 3262–3265 (2020).
Yamamoto, K., Kuriyama, M. & Onomura, O. Asymmetric electrosynthesis: Recent advances in catalytic transformations. Curr. Opin. Electrochem. 28, 100714 (2021).
McNally, A., Prier Christopher, K. & MacMillan David, W. C. Discovery of an α-Amino C–H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science 334, 1114–1117 (2011).
Lima, F. et al. Visible Light Activation of Boronic Esters Enables Efficient Photoredox C(sp2)–C(sp3) Cross-Couplings in Flow. Angew. Chem. Int. Ed. 55, 14085–14089 (2016).
Lipp, B., Nauth, A. M. & Opatz, T. Transition-Metal-Free Decarboxylative Photoredox Coupling of Carboxylic Acids and Alcohols with Aromatic Nitriles. J. Org. Chem. 81, 6875–6882 (2016).
Betori, R. C. & Scheidt, K. A. Reductive Arylation of Arylidene Malonates Using Photoredox Catalysis. ACS Catal. 9, 10350–10357 (2019).
Miao, M., Liao, L.-L., Cao, G.-M., Zhou, W.-J. & Yu, D.-G. Visible-light-mediated external-reductant-free reductive cross coupling of benzylammonium salts with (hetero)aryl nitriles. Sci. Chin. Chem. 62, 1519–1524 (2019).
Zhu, S., Qin, J., Wang, F., Li, H. & Chu, L. Photoredox-catalyzed branch-selective pyridylation of alkenes for the expedient synthesis of Triprolidine. Nat. Commun. 10, 749 (2019).
Qi, J. et al. New Radical Borylation Pathways for Organoboron Synthesis Enabled by Photoredox Catalysis. Angew. Chem. Int. Ed. 59, 12876–12884 (2020).
Shi, J., Yuan, T., Zheng, M. & Wang, X. Metal-Free Heterogeneous Semiconductor for Visible-Light Photocatalytic Decarboxylation of Carboxylic Acids. ACS Catal. 11, 3040–3047 (2021).
Lehnherr, D. et al. Electrochemical Synthesis of Hindered Primary and Secondary Amines via Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 142, 468–478 (2020).
Zhang, S. et al. Electroreductive 4-Pyridylation of Electron-deficient Alkenes with Assistance of Ni(acac)2. Org. Lett. 22, 3570–3575 (2020).
Wen, J. et al. Electroreductive C3 Pyridylation of Quinoxalin-2(1H)-ones: An Effective Way to Access Bidentate Nitrogen Ligands. Org. Lett. 23, 1081–1085 (2021).
Zhang, X. et al. Reductive Arylation of Aliphatic and Aromatic Aldehydes with Cyanoarenes by Electrolysis for the Synthesis of Alcohols. Org. Lett. 23, 3472–3476 (2021).
Ding, W., Sheng, J., Li, J. & Cheng, X. Electroreductive 4-pyridylation of unsaturated compounds using gaseous ammonia as a hydrogen source. Org. Chem. Front. 9, 2634–2639 (2022).
Wang, G. et al. Metal-Free Synthesis of C-4 Substituted Pyridine Derivatives Using Pyridine-boryl Radicals via a Radical Addition/Coupling Mechanism: A Combined Computational and Experimental Study. J. Am. Chem. Soc. 139, 3904–3910 (2017).
Gao, L. et al. Lewis Acid-Catalyzed Selective Reductive Decarboxylative Pyridylation of N-Hydroxyphthalimide Esters: Synthesis of Congested Pyridine-Substituted Quaternary Carbons. ACS Catal. 9, 10142–10151 (2019).
Tucker, T. J. et al. Synthesis of a Series of Potent and Orally Bioavailable Thrombin Inhibitors That Utilize 3,3-Disubstituted Propionic Acid Derivatives in the P3 Position. J. Med. Chem. 40, 3687–3693 (1997).
Alexander, R. P. et al. CDP840. A prototype of a novel class of orally active anti-Inflammatory phosphodiesterase 4 inhibitors. Bioorg. Med. Chem. Lett. 12, 1451–1456 (2002).
Huang, H.-C. et al. Discovery of Potent, Nonsystemic Apical Sodium-Codependent Bile Acid Transporter Inhibitors (Part 2). J. Med. Chem. 48, 5853–5868 (2005).
Druing the revison of this work, a report of photoredox asymmetric pyridinylaton using 4-CN-pyridine is published by Jiang group. et al. Catalytic Asymmetric Reductive Azaarylation of Olefins via Enantioselective Radical Coupling. J. Am. Chem. Soc. 144, 7805–7814 (2022).
Renaud, P. & Gerster, M. Use of Lewis Acids in Free Radical Reactions. Angew. Chem. Int. Ed. 37, 2562–2579 (1998).
Sibi, M. P., Manyem, S. & Zimmerman, J. Enantioselective Radical Processes. Chem. Rev. 103, 3263–3296 (2003).
Lu, H. & Zhang, X. P. Catalytic C–H functionalization by metalloporphyrins: recent developments and future directions. Chem. Soc. Rev. 40, 1899–1909 (2011).
Fu, G. C. Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes. ACS Cent. Sci. 3, 692–700 (2017).
Miyabe, H., Kawashima, A., Yoshioka, E. & Kohtani, S. Progress in Enantioselective Radical Cyclizations. Chem. Eur. J. 23, 6225–6236 (2017).
Zhang, L. & Meggers, E. Stereogenic-Only-at-Metal Asymmetric Catalysts. Chem. Asian J. 12, 2335–2342 (2017).
Wang, F., Chen, P. & Liu, G. Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res. 51, 2036–2046 (2018).
Li, Z.-L., Fang, G.-C., Gu, Q.-S. & Liu, X.-Y. Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev. 49, 32–48 (2020).
Prentice, C., Morrisson, J., Smith, A. D. & Zysman-Colman, E. Recent developments in enantioselective photocatalysis. Beilstein J. Org. Chem. 16, 2363–2441 (2020).
Proctor, R. S. J., Colgan, A. C. & Phipps, R. J. Exploiting attractive non-covalent interactions for the enantioselective catalysis of reactions involving radical intermediates. Nat. Chem. 12, 990–1004 (2020).
Chan, A. Y. et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 122, 1485–1542 (2022).
Großkopf, J., Kratz, T., Rigotti, T. & Bach, T. Enantioselective Photochemical Reactions Enabled by Triplet Energy Transfer. Chem. Rev. 122, 1626–1653 (2022).
Guo, L. et al. General Method for Enantioselective Three-Component Carboarylation of Alkenes Enabled by Visible-Light Dual Photoredox/Nickel Catalysis. J. Am. Chem. Soc. 142, 20390–20399 (2020).
Trost, B. M. & Van Vranken, D. L. Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem. Rev. 96, 395–422 (1996).
Trost, B. M. & Crawley, M. L. Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev. 103, 2921–2944 (2003).
Alexakis, A. et al. The Copper-Catalyzed Asymmetric Allylic Substitution. Chimia 60, 124–130 (2006).
Harutyunyan, S. R., den Hartog, T., Geurts, K., Minnaard, A. J. & Feringa, B. L. Catalytic Asymmetric Conjugate Addition and Allylic Alkylation with Grignard Reagents. Chem. Rev. 108, 2824–2852 (2008).
Hartwig, J. F. & Stanley, L. M. Mechanistically Driven Development of Iridium Catalysts for Asymmetric Allylic Substitution. Acc. Chem. Res. 43, 1461–1475 (2010).
Sundararaju, B., Achard, M. & Bruneau, C. Transition metal catalyzed nucleophilic allylic substitution: activation of allylic alcohols via π-allylic species. Chem. Soc. Rev. 41, 4467–4483 (2012).
Tosatti, P., Nelson, A. & Marsden, S. P. Recent advances and applications of iridium-catalysed asymmetric allylic substitution. Org. Biomol. Chem. 10, 3147–3163 (2012).
Butt, N. A. & Zhang, W. Transition metal-catalyzed allylic substitution reactions with unactivated allylic substrates. Chem. Soc. Rev. 44, 7929–7967 (2015).
Hethcox, J. C., Shockley, S. E. & Stoltz, B. M. Iridium-Catalyzed Diastereo-, Enantio-, and Regioselective Allylic Alkylation with Prochiral Enolates. ACS Catal. 6, 6207–6213 (2016).
Shintani, R. Recent Progress in Copper-Catalyzed Asymmetric Allylic Substitution Reactions Using Organoboron Nucleophiles. Synthesis 48, 1087–1100 (2016).
Bayeh, L. & Tambar, U. K. Catalytic Asymmetric Intermolecular Allylic Functionalization of Unactivated Internal Alkenes. ACS Catal. 7, 8533–8543 (2017).
Qu, J. & Helmchen, G. Applications of Iridium-Catalyzed Asymmetric Allylic Substitution Reactions in Target-Oriented Synthesis. Acc. Chem. Res. 50, 2539–2555 (2017).
Cheng, Q. et al. Iridium-Catalyzed Asymmetric Allylic Substitution Reactions. Chem. Rev. 119, 1855–1969 (2019).
Rössler, S. L., Petrone, D. A. & Carreira, E. M. Iridium-Catalyzed Asymmetric Synthesis of Functionally Rich Molecules Enabled by (Phosphoramidite,Olefin) Ligands. Acc. Chem. Res. 52, 2657–2672 (2019).
Pàmies, O. et al. Recent Advances in Enantioselective Pd-Catalyzed Allylic Substitution: From Design to Applications. Chem. Rev. 121, 4373–4505 (2021).
Zhang, W. et al. Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 353, 1014–1018 (2016).
Ge, L. et al. Iron-catalysed asymmetric carboazidation of styrenes. Nat. Catal. 4, 28–35 (2021).
Zhang, C., Li, Z.-L., Gu, Q.-S. & Liu, X.-Y. Catalytic enantioselective C(sp3)–H functionalization involving radical intermediates. Nat. Commun. 12, 475 (2021).
Zhou, Q., Chin, M., Fu, Y., Liu, P. & Yang, Y. Stereodivergent atom-transfer radical cyclization by engineered cytochromes P450. Science 374, 1612–1616 (2021).
Jiao, K.-J. et al. Palladium-catalyzed reductive electrocarboxylation of allyl esters with carbon dioxide. Org. Chem. Front. 5, 2244–2248 (2018).
Zhang, H.-H., Zhao, J.-J. & Yu, S. Enantioselective Allylic Alkylation with 4-Alkyl-1,4-dihydro-pyridines Enabled by Photoredox/Palladium Cocatalysis. J. Am. Chem. Soc. 140, 16914–16919 (2018).
Dhawa, U. et al. Enantioselective Pallada-Electrocatalyzed C−H Activation by Transient Directing Groups: Expedient Access to Helicenes. Angew. Chem. Int. Ed. 59, 13451–13457 (2020).
Dhawa, U. et al. Enantioselective palladaelectro-catalyzed C–H olefinations and allylations for N–C axial chirality. Chem. Sci. 12, 14182–14188 (2021).
Zhang, H.-H., Tang, M., Zhao, J.-J., Song, C. & Yu, S. Enantioselective Reductive Homocoupling of Allylic Acetates Enabled by Dual Photoredox/Palladium Catalysis: Access to C2-Symmetrical 1,5-Dienes. J. Am. Chem. Soc. 143, 12836–12846 (2021).
Alvaro, G., Cardullo, F., D’Adamo, L., Piga, E. & Seri, C. N-benzyl-3-phenyl-3-heterocyclyl-propionamide compounds as tachykinin/serotonin reuptake inhibitors. Assignee: Glaxo Group Limited, UK, international patent WO2004005255A1. p. 105 (2004).
Jary, H., Temal, T. & Deprez, P. New thiazole- or benzothiazole-substituted urea derivatives, their for preparation, pharmaceutical compositions containing them and their use as ion inorganic, particularly, calcium receptor modulators. Assignee: Proskelia Sas, Fr., french patent FR2885129A1. p. 107 (2006).
Zeng, L. et al. Electrochemical oxidative aminocarbonylation of terminal alkynes. Nat. Catal. 3, 438–445 (2020).
Fu, N. et al. New Bisoxazoline Ligands Enable Enantioselective Electrocatalytic Cyanofunctionalization of Vinylarenes. J. Am. Chem. Soc. 141, 14480–14485 (2019).
Song, L. et al. Dual electrocatalysis enables enantioselective hydrocyanation of conjugated alkenes. Nat. Chem. 12, 747–754 (2020).
Xiong, P., Hemming, M., Ivlev, S. I. & Meggers, E. Electrochemical Enantioselective Nucleophilic α-C(sp3)–H Alkenylation of 2-Acyl Imidazoles. J. Am. Chem. Soc. 144, 6964–6971 (2022).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (nos. 22071105 and 22031008, for CX) and by the QingLan Project of Jiangsu Education Department (CX).
Author information
Authors and Affiliations
Contributions
Experimental and computational investigation: W.D. Methodology: W.D., and M.L. Substrate synthesis: M.L., and J.F. Writing–original draft: W.D., and X.C. Review & editing, X.C. Supervision X.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ding, W., Li, M., Fan, J. et al. Palladium-catalyzed asymmetric allylic 4-pyridinylation via electroreductive substitution reaction. Nat Commun 13, 5642 (2022). https://doi.org/10.1038/s41467-022-33452-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-33452-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
