
Abstract
Organochalcogen heterocycles are ubiquitously present and widely utilized in various fields. Among them, oxirane has been extensively studied, and all of the stereoisomeric forms are readily available. In contrast, synthetic studies on thiirane were rarely reported, and thus the useful sulfur-congener of oxirane has been difficult to access in a stereodefined form. In this research, a general stereoselective synthesis of cis-thiiranes is accomplished by taking advantage of stereospecific electrocyclization of trans-thiocarbonyl ylides, which are generated in situ from readily available E,E-aldazine N-oxides upon treatment with Lawesson’s reagent. This newly developed practical method provides a variety of cis−1,2-diarylthiiranes as essentially single diastereomers in high yields under mild reaction conditions. The intermediacy of trans-thiocarbonyl yilde is confirmed by mechanistic experiments, and the excellent stereocontrol is rationalized by DFT calculation.
Similar content being viewed by others
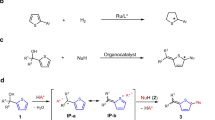

Introduction
The chalcogens, elements in group 16 of the periodic table, are widely utilized in various fields (Fig. 1a). In particular, non-covalent interaction between chalcogen atoms and electron donors, so-called chalcogen bonding plays important roles in materials chemistry1,2,3 as well as noncovalent organocatalysis1,4,5. Moreover, the combination with other electropositive elements composes chalcogenides, which have numerous applications such as chalcogenide glasses6,7,8, polymer solar cells9, wide band gap semiconductors10, and electrochemical sensors11. Furthermore, chalcogens are often found in natural products and pharmacologically active compounds. Among them, three-membered heterocycles containing a chalcogen are ubiquitous subunits, and significant efforts have been dedicated to the development of synthetic methods for oxiranes12,13. The sulfur congeners, thiiranes are also useful synthetic precursors that can be transformed into an array of functional groups via ring-opening or desulfurization14,15,16,17,18,19,20. In addition, versatile sulfur-containing polymers can be produced via ring-expansion polymerizations or copolymerizations21,22,23,24,25. Moreover, thiiranes have been examined as analogs of biologically active oxiranes26,27,28, and several derivatives showed inhibitory activity against gelatinases29,30. However, despite the apparent utility, synthetic methods for thiiranes are not well established compared to oxiranes (Fig. 1b). Indeed, the stereoselective construction of thiiranes remains a great challenge. For example, whereas all of the stereoisomers of stilbene oxide can be easily prepared and are even commercially available, the related stilbene sulfides are typically produced through stereospecific conversion of the corresponding stilbene oxides31. Clearly, it is highly desirable to develop a general and practical stereoselective synthesis of thiiranes.

a Importance of organochalcogens. b Comparison between oxiranes and thiiranes.
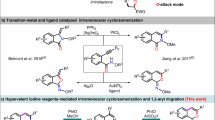
Thiirane synthesis has been generally accomplished via the conversion from oxirane analogs with thiourea or thiocyanate as a sulfurating reagent (Fig. 2a)32,33,34,35,36,37,38. Consequently, the stereostructure of thiiranes necessarily relies on the configuration of oxiranes. In a special case, it is possible to introduce a stereochemical element during the chalcogen substitution step via a kinetic resolution process as recently demonstrated by the List group utilizing the chiral phosphoric acid catalysis39,40. However, only terminal thiiranes can be prepared by this strategy. Alternatively, the episulfidation of alkenes has been investigated15, but this seemingly straightforward epoxidation-like approach is applicable only to a limited range of cyclic alkenes. Otherwise, stereoselective synthesis of thiiranes has rarely been developed41,42,43,44,45, and most of the reported examples have serious drawbacks such as low stereoselectivity, narrow substrate scope, and/or the requirement of highly toxic reagents. Among these precedents, a unique synthetic sequence of the Barton–Kellogg reaction is noteworthy (Fig. 2b)46,47,48,49,50,51,52,53. The Kellogg group developed a sequential process involving non-stereoselective sulfurization of azines followed by dehydrogenation to furnish thiadiazolines (1) as a mixture of diastereomers48. Interestingly, the isolated trans- and cis-1 were stereospecifically transformed to the corresponding cis- and trans-thiiranes, respectively. The stereochemical outcome was rationalized by 4π-electrocyclization of the putative thiocarbonyl ylides (2), which were derived from 1 via thermal extrusion of dinitrogen gas. Unfortunately, despite the intriguing stereochemical properties, this method has not received much attention because of the use of a highly toxic and inconvenient gaseous sulfurating reagent, H2S. Moreover, the formation of 1 requires harsh and toxic oxidants such as Pb(OAc)2 or diethyl azodicarboxylate (DEAD). The most critical drawback is the lack of stereoselectivity in the sulfurization step, which diminishes the practicality of this method quite substantially. Our group has been interested in the unusual reactivity of electrophilically activated 1,2-diazines and related organonitrogen compounds54,55. While investigating the reactions of aldazine N-oxides (5) with various electrophiles, we serendipitously observed the highly diastereoselective formation of cis-disubstituted thiiranes (6) upon treatment with Lawesson’s reagent (LR) (Fig. 2c).

a Common method for thiirane synthesis. b Previous approach to thiiranes via the intermediacy of thiocarbonyl ylides. c This work: Diastereoselective synthesis of cis-thiiranes from aldazine N-oxides. LR Lawesson’s reagent, d.r. diastereomeric ratio.
Herein, we report a general synthetic method for cis-diarylthiiranes through the stereospecific sulfurization of aldazine N-oxides. Our newly discovered process resembles the Barton–Kellogg reaction but exhibits several distinct advantages. Because azine is pre-oxidized under relatively mild conditions56,57,58 prior to the sulfurization, harsh oxidation conditions are avoided. In addition, a more convenient and less toxic sulfurating reagent, LR, is utilized. Most importantly, cis-thiiranes are produced with an excellent level of diastereoselectivity.
Results and discussion
Aldazine N-oxides (5) were readily prepared by a two-step sequence involving double condensation of aldehydes with hydrazine59,60 and the following N-oxidation (see Section 2.1 of the Supplementary Information for details).56,57,58 All of 5 were obtained with E,E-configuration59,60 and are bench-stable solids that can be stored for months without decomposition. On the other hand, it was challenging to access unsymmetrical substrates because of the disproportionation during both C = N condensation and N-oxidation, and aliphatic aldazines are unstable under the N-oxidation conditions. At the outset, the reaction conditions for thiirane formation were optimized with benzaldazine N-oxide (5a) and LR (Table 1). In the presence of 1.0 equiv of LR in CH2Cl2 at −10 °C, 2,3-diphenylthiirane (6a) was produced in 13% yield with a reasonably high 84:16 cis/trans selectivity (entry 1). Although the yield was low, the dominant formation of cis-isomer was encouraging. When the reaction was conducted in polar MeCN, both yield (23%) and cis-selectivity (94:6) were increased (entry 2). Thus, a few other polar solvents were examined. DMSO was unsuitable because it reacted vigorously with LR. Gratifyingly, the use of DMF resulted in a synthetically useful 62% yield with an enhanced 97:3 cis-selectivity (entry 3). The analysis of the crude mixture revealed that a small amount of DMTF was generated by thionation of DMF. Hence, DMTF was employed as a solvent in order to evaluate the potentially promoting activity, but a drastically decreased 25% yield was afforded (entry 4). Subsequently, upon a brief survey of other amide solvents, an improved 67% yield and an excellent 99:1 cis-selectivity were obtained in DMPU (entry 5), whereas the use of DMA was less effective (entry 6). In these experiments, a noticeable exotherm was detected during the addition of LR. It was hypothesized that the elevated internal temperature could have caused unproductive side reactions, leading to moderate chemical yields. Therefore, the reaction was carried out at a lower temperature. For this purpose, DMF was employed as a solvent because of the high melting point of DMPU. At −50 °C, an increased 71% yield was obtained with an essentially exclusive cis-selectivity (entry 7). To our delight, when LR was added slowly as a solution for finer temperature control, the yield was even further improved to 78% (entry 8). In this case, DMPU was used as a solvent for LR because DMF reacts with LR at room temperature61. With this protocol, higher and lower loadings of LR were examined, but the yields were slightly diminished in both cases (entries 9 and 10).
With the optimal reaction conditions in hand, a wide range of 1,2-diarylaldazine N-oxides was surveyed (Fig. 3). Electron-withdrawing groups were well tolerated. Not only p-fluorine but also sterically hindering o-chlorine/bromine substituents could be introduced, producing 6b–6d in 59–73% yields. Moreover, highly electron-deficient, p-trifluoromethyl-substituted thiirane 6e was successfully afforded in 63% yield. It was possible to install electron-donating alkyl groups at any positions on the aryl rings, too. Thiiranes with p-methyl (6f), p-tert-butyl (6g), o-methyl (6h), or m-methyl (6i) groups were formed in 71–91% yields. Strongly electron-donating alkoxy groups could be present at the o-positions, again, without steric encumbrance problem, providing 6j in still high yield and diastereoselectivity. The presence of m-methoxy groups was allowed to give 6k in 71% yield, as well. Unfortunately, p-alkoxy substitution resulted in an unstable product. Even though the predominant formation of 6l was observed by 1H NMR analysis of the crude mixture, the thiirane decomposed slowly to the corresponding alkene upon purification on silica gel. Thus, 6l was obtained as a mixture with alkene (86:14). Moreover, the loss of sulfur atom took place spontaneously over time to increase the amount of alkene upon storage even in the fridge (Fig. 4).14 This desulfurization process is not stereospecific, and thus a mixture of Z- and E-alkene isomers 7 was produced from an essentially diastereopure thiirane. Such decomposition was suppressed by attenuating the electron-donating ability of the alkoxy group. Hence, 6m containing p-trifluoromethoxy groups could be isolated in 69% yield. Finally, heteroaromatic substrates were examined, and both 3-thienyl and 3-furyl moieties were successfully employed to give 6n and 6o in 64% and 57% yields, respectively. On the other hand, the isomeric 2-thienyl and 2-furyl substrates could not be employed because the corresponding thiirane products decomposed rapidly on silica gel. Overall, cis-diarylthiiranes were consistently obtained in good yields with excellent diastereoselectivity regardless of the electronic and steric properties of the substituents. The cis-configuration of thiirane products was unambiguously established by X-ray crystallographic analysis of 6d (Supplementary Fig. 4).

a Reaction conditions: 5 (1.0 mmol) in DMF (5.0 mL) and LR (1.0 mmol) in DMPU (5.0 mL). Isolated yields after column chromatography are given. Diastereomeric ratio was determined by 1H NMR analysis of the isolated material. Data after recrystallization are given in the parenthesis. b Contaminated by alkene (86:14).

Non-stereospecific, spontaneous desulfurization of p-alkoxy-cis-thiirane.
Mechanistic experiments were carried out for the newly developed cis-selective thiirane synthesis (Fig. 5). On the basis of Kellogg’s study48, it was hypothesized that the observed cis-configuration of thiiranes would be originated from the intermediacy of a trans-thiocarbonyl ylide via stereospecific conrotatory 4π-electrocyclization. Therefore, trapping of a putative trans-thiocarbonyl ylide 8 was pursued, and the 1,3-dipolar cycloadducts 9a and 9b were successfully obtained employing N-phenylmaleimide and maleic anhydride (Fig. 5a)62. The 1,3-trans-configurations of these compounds were determined by single-crystal X-ray diffraction analysis (Supplementary Figs. 2 and 3), highly supporting the presence of trans-8. In addition, a crossover experiment was conducted with a 1:1 mixture of 5a and 5f (Fig. 5b). Under the standard reaction conditions, only 6a and 6f were afforded, and the scrambled product 6af was not detected. Therefore, the intermolecular reaction pathway was excluded.

a Trapping the trans-thiocarbonyl ylide intermediate with dipolarophiles. b Crossover experiment with two different aldazine N-oxides.
To gain insight into the detailed reaction mechanism, a computational study was performed by the density functional theory calculation at the M06-2X/6-311 + G(d,p)/PCM(DMF) level of theory (Fig. 6, Supplementary Data 1)63,64,65,66. Whereas typical thionation with LR requires heating, our thiirane formation proceeds at a subzero temperature. Thus, dissociation of LR is less likely under our reaction conditions, and the dimeric form was employed in the computational analysis. The process is initiated by the coordination of aldazine N-oxide 5a onto dimeric LR to generate Int I67, and the subsequent cyclization affords oxathiazaphospholidine sulfide Int II, completing a stepwise formal [3 + 2] cycloaddition68. Then, the exergonic production of zwitterion Int III is driven by the formation of a stable P = O bond67. Until here, even after cleavage of covalent bonds in the LR moiety, the electron-deficient phosphorus centers and the electron-rich sulfur anions still appear to associate through electrostatic interaction. Notably, a few calculated structures were located only when the solvent effect of DMF was considered in the computation. This result is consistent with the experimental observation. During the reaction conditions optimization, the yield of thiirane was substantially improved upon the use of DMF (Table 1, entry 3), whereas the reactions in less polar solvents gave inferior results (Table 1, entries 1, 2, and 4). It is presumed that a highly polar solvent is needed for the stabilization of multiply charge-separated transition structures and intermediates. Subsequently, a facile intramolecular nucleophilic attack of the reactive sulfur anion onto the neighboring diazenium moiety furnishes trans-thiadiazoline Int IV with concomitant release of an oxygenated LR analog 10. The calculated pathway up to this point from I is composed of a series of stereospecific transformations, all of which take place at one side of 5a. In consequence, the E,E-geometry of aldazine N-oxide leads to the trans-configuration of thiadiazoline. Then, upon a concerted extrusion of dinitrogen gas, the experimentally confirmed trans-thiocarbonyl ylide Int V is produced69. Alternatively, Int IV may split into a diazo compound and a thial (Int V’) via a [3 + 2] cycloreversion (red line)50. This potential side reaction is endergonic and reversible. Hence, the stereochemical integrity of Int IV may be compromised through unselective recombination. Fortunately, the activation energy of the desired pathway is 4.3 kcal/mol lower. Moreover, the intermolecular mechanism has been ruled out by the crossover experiment (Fig. 5b). Finally, Int V undergoes thermal conrotatory 4π-electrocyclization to give the product cis-thiirane II.

Gibbs free energy profile at the M06-2X/6-311 + G(d,p)/PCM(DMF) level of theory.
In summary, a generally applicable, highly stereoselective synthesis of cis-thiiranes has been realized under mild reaction conditions utilizing Lawesson’s reagent as a convenient sulfur source. The newly developed method provides access to a wide variety of cis-1,2-diarylthiiranes from E,E-aldazine N-oxides in high yields with almost exclusive diastereoselectivity. Mechanistic experiments, as well as DFT calculation, suggest a reaction mechanism composed of stereospecific transformations involving 4π-electrocyclization of a trans-thiocarbonyl ylide as the origin of the exquisite diastereocontrol. Through this research, the useful sulfur congener of oxirane becomes available in a stereodefined form. Further expansion of this unique reactivity is currently investigated in our laboratories.
Methods
A representative procedure for LR-mediated synthesis of cis-thiiranes from aldazine N-oxides
To a mixture of benzaldazine N-oxide (5a, 224 mg, 1.00 mmol) in DMF (5.0 mL) was added LR (404 mg, 1.00 mmol) as a solution in DMPU (5.0 mL) at –50 °C dropwise under Ar. The change in internal temperature should not exceed 1 °C when the LR solution was added. After 4 h, the reaction mixture was transferred to a 100 mL separatory funnel and diluted with CH2Cl2 (4 mL). The mixture was washed with water (50 mL × 3) and brine (50 mL). The combined aqueous layers were extracted with CH2Cl2 (3 mL × 6). The combined organic layers were dried over anhydrous MgSO4 (2 g), filtered through a glass frit, and concentrated in vacuo. The residue was purified immediately by flash column chromatography (SiO2, ø = 3.5 cm, l = 5.0 cm, hexanes, Rf = 0.20, KMnO4) to give cis-2,3-diphenylthiirane (6a, 165 mg, 78%, cis:trans = 99:1) as white solid. Data for 6a: 1H NMR (400 MHz, CDCl3): δ 7.14–7.10 (m, 10H), 4.38 (s, 2H); 13C NMR (100 MHz, CDCl3): δ 135.2, 129.5, 127.8, 127.3, 44.2; HRMS (ESI): [M–H]– calcd for C14H11S: 211.0587; found: 211.0586.
Data availability
The data supporting the findings of this study are available within this article and its Supplementary Information, which contains experimental details, characterization data, copies of NMR spectra for all new compounds, and DFT calculation data. Crystallographic data for 6d, 9a, and 9b have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 2119467, 2119466, and 2119465, respectively. Copies of the data can be accessed free of charge via https://www.ccdc.cam.ac.uk/structures/.
References
Vogel, L., Wonner, P. & Huber, S. M. Chalcogen bonding: an overview. Angew. Chem. Int. Ed. 58, 1880–1891 (2019).
Ho, P. C., Wang, J. Z., Meloni, F. & Vargas-Baca, I. Chalcogen bonding in materials chemistry. Coord. Chem. Rev. 422, 213464 (2020).
Mahmudov, K. T., Kopylovich, M. N., Guedes da Silva, M. F. C. & Pombeiro, A. J. L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 46, 10121–10138 (2017).
Young, C. M. et al. The importance of 1,5-oxygen···chalcogen interactions in enantioselective isochalcogenourea catalysis. Angew. Chem. Int. Ed. 59, 3705–3710 (2020).
Wang, W. et al. Chalcogen–chalcogen bonding catalysis enables assembly of discrete molecules. J. Am. Chem. Soc. 141, 9175–9179 (2019).
Eggleton, B. J., Luther-Davies, B. & Richardson, K. Chalcogenide photonics. Nat. Photonics 5, 141–148 (2011).
Zakery, A. & Elliott, S. R. Optical properties and applications of chalcogenide glasses: a review. J. Non Cryst. Solids 330, 1–12 (2003).
Sanghera, J. S., Shaw, L. B. & Aggarwal, I. D. Applications of chalcogenide glass optical fibers. C. R. Chim. 5, 873–883 (2002).
Freitas, J. N., Gonçalves, A. S. & Nogueira, A. F. A comprehensive review of the application of chalcogenide nanoparticles in polymer solar cells. Nanoscale 6, 6371–6397 (2014).
Woods-Robinson, R. et al. Wide band gap chalcogenide semiconductors. Chem. Rev. 120, 4007–4055 (2020).
Suresh, R., Pandiaraj, M., Sankaralingam, M. & Giribabu, K. Graphene–metal chalcogenide modified electrochemical sensor. In Graphene-Based Electrochemical Sensors for Biomolecules (eds Pandikumar, A. & Rameshkumar, P.) Ch. 6, 139–153 (Elsevier, 2019).
Faveri, G. D., Ilyashenko, G. & Watkinson, M. Recent advances in catalytic asymmetric epoxidation using the environmentally benign oxidant hydrogen peroxide and its derivatives. Chem. Soc. Rev. 40, 1722–1760 (2011).
Zhu, Y., Wang, Q., Cornwall, R. G. & Shi, Y. Organocatalytic asymmetric epoxidation and aziridination of olefins and their synthetic applications. Chem. Rev. 114, 8199–8256 (2014).
Sander, M. Thiiranes. Chem. Rev. 66, 297–339 (1966).
Adam, W. & Bargon, R. M. Synthesis of thiiranes by direct sulfur transfer: the challenge of developing effective sulfur donors and metal catalysts. Chem. Rev. 104, 251–261 (2004).
Murphree, S. S. Three-membered heterocycles. Structure and reactivity. In Modern Heterocyclic Chemistry Vol. 4 (eds Alvarez-Builla, J., Vaquero, J. J. & Barluenga, J.), Ch. 2, 11–162 (Wiley, 2011).
Rogers, E., Araki, H., Batory, L. A., Mclnnis, C. E. & Njardarson, J. T. Highly selective copper-catalyzed ring expansion of vinyl thiiranes: application to synthesis of biotin and the heterocyclic core of plavix. J. Am. Chem. Soc. 129, 2768–2769 (2007).
Iranpoor, N., Firouzabadi, H. & Jafari, A. A. Conversion of thiiranes to β-chlorothioacetates catalyzed with CoCl2. Synth. Commun. 33, 2321–2327 (2003).
Sauve, A. A. & Groves, J. T. Synthesis of trithiolanes and tetrathianes from thiiranes catalyzed by ruthenium salen nitrosyl complexes. J. Am. Chem. Soc. 124, 4770–4778 (2002).
Xu, J. Recent synthesis of thietanes. Beilstein J. Org. Chem. 16, 1357–1410 (2020).
Chao, J.-Y. et al. Controlled disassembly of elemental sulfur: an approach to the precise synthesis of polydisulfides. Angew. Chem. Int. Ed. 61, e202115950 (2022).
Kudo, H. et al. Living ring-expansion polymerization of thiirane with cyclic monocarbamothioates. Macromolecules 53, 4733–4740 (2020).
Takahashi, A., Tsunoda, S., Yuzaki, R. & Kameyama, A. Thioacyl-transfer ring-expansion polymerization of thiiranes based on a cyclic dithiocarbamate initiator. Macromolecules 53, 5227–5236 (2020).
Takahashi, A., Yuzaki, R., Ishida, Y. & Kameyama, A. Controlled ring-expansion polymerization of thiiranes based on cyclic aromatic thiourethane initiator. J. Polym. Sci. Part A: Polym. Chem. 57, 2442–2449 (2019).
Schuetz, J.-H., Sandbrink, L. & Vana, P. Insights into the ring-expansion polymerization of thiiranes with 2,4-thiazolidinedione. Macromol. Chem. Phys. 214, 1484–1495 (2013).
Schramm, F., Müller, A., Hammer, H., Paschke, A. & Schüürmann, G. Epoxide and thiirane toxicity in vitro with the ciliates tetrahymena puriformis: structural alerts indicating excess toxicity. Environ. Sci. Technol. 45, 5812–5819 (2011).
Gao, M. et al. Acceleration of diabetic wound healing using a novel protease–anti-protease combination therapy. Proc. Natl Acad. Sci. USA 112, 15226–15231 (2015).
Lee, M. et al. Synthesis of chiral 2-(4-phenoxyphenylsulfonylmethyl)thiiranes as selective gelatinase inhibitors. Org. Lett. 7, 4463–4465 (2005).
Fabre, B. et al. New clicked thiirane derivatives as gelatinase inhibitors: the relevance of the P1’ segment. RSC Adv. 4, 17726–17735 (2014).
Lee, M. et al. Structure–activity relationship for thiirane-based gelatinase inhibitors. ACS Med. Chem. Lett. 3, 490–495 (2012).
Ketcham, R. & Shah, V. P. cis- and trans-stilbene sulfides. J. Org. Chem. 28, 229–230 (1963).
Akhlaghinia, B., Rahimizadeh, M., Eshghi, H., Zhaleh, S. & Rezazadeh, S. Green synthesis of thiiranes from oxiranes under solvent- and catalyst-free conditions. J. Sulfur Chem. 33, 351–361 (2012).
Zeynizadeh, B., Baradarani, M. M. & Eisavi, R. A practical and eco-friendly method for conversion of epoxides to thiiranes with immobilized thiourea on CaCo3. Phosphorus Sulfur Silicon Relat. Elem. 186, 2208–2215 (2011).
Wu, L., Wang, Y., Yan, F. & Yang, C. Facile conversion of epoxides to thiiranes with ammonium thiocyanate catalyzed with etidronic acid. Bull. Korean Chem. Soc. 31, 1419–1420 (2010).
Zeynizadeh, B. & Yeghaneh, S. A green protocol for solvent-free conversion of epoxides to thiiranes with dowex-50WX8–supported thiourea. Phosphorous Sulfur Silicon Relat Elem 184, 362–368 (2009).
Yadav, J. S. et al. Iodine as a mild, efficient, and cost-effective catalyst for the synthesis of thiiranes from oxiranes. Monatch. Chem. 139, 1363–1367 (2008).
Bandgar, B. P., Patil, A. V., Kamble, V. T. & Totre, J. V. An efficient synthesis of thiiranes from oxiranes using fluoroboric acid adsorbed on silica gel (HBF4–SiO2) as a catalyst under mild conditions in the absence of solvent. J. Mol. Catal. A Chem. 273, 114–117 (2007).
Yadav, J. S., Reddy, B. V. S. & Baishya, G. Indium tribromide: a novel and highly efficient reagent for the conversion of oxiranes to thiiranes. Synlett 3, 396–398 (2003).
Liao, S., Leutzsch, M., Monaco, M. R. & List, B. Catalytic enantioselective conversion of epoxides to thiiranes. J. Am. Chem. Soc. 138, 5230–5233 (2016).
Duan, M. et al. Chiral phosphoric acid catalyzed conversion of epoxides into thiiranes: mechanism, stereochemical model, and new catalyst design. Angew. Chem. Int. Ed. 61, e202113204 (2022).
Lin, X. et al. Asymmetric catalytic (2+1) cycloaddition of thioketones to synthesize tetrasubstituted thiiranes. Angew. Chem. Int. Ed. 61, e202201151 (2022).
Schmidt, T. A. & Sparr, C. Catalyst-controlled stereoselective Barton–Kellogg olefination. Angew. Chem. Int. Ed. 60, 23911–23916 (2021).
Cano, I. et al. N-(Diazoacetyl)oxazolidin-2-thiones as sulfur-donor reagents: asymmetric synthesis of thiiranes from aldehydes. Angew. Chem. Int. Ed. 51, 10856–10860 (2012).
Zhou, C., Fu, C. & Ma, S. Highly selective thiiranation of 1,2-allenyl sulfones with Br2 and Na2S2O3: mechanism and asymmetric synthesis of alkylidenethiiranes. Angew. Chem. Int. Ed. 46, 4379–4381 (2007).
Collazo, L. R. & Guziec, F. S. Jr. Stereoselective synthesis of unhindered olefins by 2-fold extrusion reactions. J. Org. Chem. 58, 43–46 (1993).
Kellogg, R. M. The molecules R2CXCR2 including azomethine, carbonyl and thiocarbonyl ylides. their syntheses, properties and reactions. Tetrahedron 32, 2165–2184 (1976).
Barton, D. H. R. & Willis, B. J. Olefin synthesis by two-fold extrusion processes. Part I Preliminary experiments. J. Chem. Soc. Perkin Trans. 1, 305–310 (1972).
Buter, J., Wassenaar, S. & Kellogg, R. M. Thiocarbonyl ylides. Generation, properties, and reactions. J. Org. Chem. 37, 4045–4060 (1972).
Mlostoń, G., Pipiak, P. & Heimgartner, H. Diradical reaction mechanisms in [3+2]-cycloadditions of hetaryl thioketones with alkyl- or trimethylsilyl-substituted diazomethanes. Beilstein J. Org. Chem. 12, 716–724 (2016).
Mlostoń, G., Jasiński, R., Kula, K. & Heimgartner, H. A DFT study on the Barton–Kellogg reaction—the molecular mechanism of the formation of thiiranes in the reaction between diphenyldiazomethane and diaryl thioketones. Eur. J. Org. Chem. 2020, 176–182 (2020).
Kowalski, M. K., Obijalska, E., Mlostoń, G. & Heimgartner, H. Generation and reactions of thiocarbonyl S-(2,2,2-trifluoroethanides). Synthesis of trifluoromethylated 1,3-dithiolanes, thiiranes and alkenes. J. Fluor. Chem. 200, 102–108 (2017).
Shermolovich, Y. G. et al. Reaction of N,N-disubstituted polyfluoroalkanethioamides with diazomethane: entry to new thiirane derivatives. Eur. J. Org. Chem. 2021, 6524–6529 (2021).
Mlostoń, G. & Heimgartner, H. Reactions for thiocarbonyl compounds with electrophilic and nucleophilic carbenes as well as with their metal complexes. J. Sulfur Chem. 41, 672–700 (2020).
Im, J. K. et al. N‐Chlorinative ring contraction of 1,4‐dimethoxyphthalazines via a bicyclization/ring opening mechanism. Synthesis 53, 1760–1770 (2021).
Im, J. K., Yang, B., Jeong, I., Choi, J.-H. & Chung, W.-j N-Chlorination-Induced, oxidative ring contraction of 1,4-dimethoxyphthalazines. Tetrahedron Lett. 61, 152048 (2020).
Williams, W. M. & Dolbier, W. R. Jr. Thermal and photochemical rearrangements of azine oxides I. A novel pyrolytic decomposition to nitrile. J. Org. Chem. 34, 155–157 (1969).
Chuang, K. V., Xu, C. & Reisman, S. E. A 15-Step synthesis of (+)-ryanodol. Science 353, 912–915 (2016).
Soldaini, G., Cardona, F. & Goti, A. Catalytic oxidation of imines based on methyltrioxorhenium/urea hydrogen peroxide: a mild and easy chemo- and regioselective entry to nitrones. Org. Lett. 9, 473–476 (2007).
Safari, J. & Gandomi-Ravandi, S. Structure, synthesis and application of azines: a historical perspective. RSC Adv. 4, 46224–46249 (2014).
Karmakar, R., Choudhury, C. R., Batten, S. R. & Mitra, S. Two new copper(II) complexes with the shortest (N–N) diazine based rigid ligand: example of unusual tridentate coordination mode. J. Mol. Struct. 826, 75–81 (2007).
Iashin, V. et al. Metal-free C–H borylation of N-heteroarenes by boron trifluoride. Chem. Eur. J. 26, 13873–13879 (2020).
Huisgen, R. et al. Recent developments of the chemistry of thiocarbonyl ylides. Bull. Soc. Chim. Belg. 93, 511–532 (1984).
Zhao, Y. & Truhlar, D. G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41, 157–167 (2008).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Frisch, M. J. et al. Gaussian 16, Revision C.01 (Gaussian, Inc., Wallingford, CT, 2016).
Legault, C. Y. CYLview20 (Université de Sherbrooke, 2020).
Legnani, L. et al. Computational mechanistic study of thionation of carbonyl compounds with Lawesson’s reagent. J. Org. Chem. 81, 7733–7740 (2016).
Dubau-Assibat, N., Baceiredo, A. & Bertrand, G. Lawesson’s reagent: an efficient 1,3-dipole trapping agent. J. Org. Chem. 60, 3904–3906 (1995).
Burns, J. M., Clark, T. & Williams, C. M. Comprehensive computational investigation of the Barton–Kellogg reaction for both alkyl and aryl systems. J. Org. Chem. 86, 7515–7528 (2021).
Acknowledgements
This research was supported by Samsung Science and Technology Foundation (Project Number SSTF‐BA1501‐10, W.-J.C.). We thank Prof. Junseong Lee at Chonnam National University for the X-ray crystallographic analysis of 6d, 9a, and 9b.
Author information
Authors and Affiliations
Contributions
W.-J.C. conceived the research concept. W.-J.C. and S.-M.S. designed the synthetic strategy. S.-M.S. and J.J. performed the synthetic work. J.-H.C. directed the computational study. S.-M.S. conducted the mechanistic analysis including the DFT calculations. S.-M.S. and W.-J.C. wrote the manuscript. All authors discussed the results and contributed to editing the manuscript and preparing the Supplementary Information.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, Sm., Jin, J., Choi, JH. et al. Synthesis of cis-thiiranes as diastereoselective access to epoxide congeners via 4π-electrocyclization of thiocarbonyl ylides. Nat Commun 13, 4818 (2022). https://doi.org/10.1038/s41467-022-32499-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32499-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
