
Abstract
Colloidal chemistry grants access to a wealth of materials through simple and mild reactions. However, even few elements can combine in a variety of stoichiometries and structures, potentially resulting in impurities or even wrong products. Similar issues have been long addressed in organic chemistry by using reaction-directing groups, that are added to a substrate to promote a specific product and are later removed. Inspired by such approach, we demonstrate the use of CsPbCl3 perovskite nanocrystals to drive the phase-selective synthesis of two yet unexplored lead sulfochlorides: Pb3S2Cl2 and Pb4S3Cl2. When homogeneously nucleated in solution, lead sulfochlorides form Pb3S2Cl2 nanocrystals. Conversely, the presence of CsPbCl3 triggers the formation of Pb4S3Cl2/CsPbCl3 epitaxial heterostructures. The phase selectivity is guaranteed by the continuity of the cationic subnetwork across the interface, a condition not met in a hypothetical Pb3S2Cl2/CsPbCl3 heterostructure. The perovskite domain is then etched, delivering phase-pure Pb4S3Cl2 nanocrystals that could not be synthesized directly.
Similar content being viewed by others
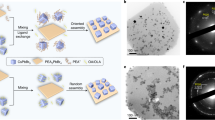


Introduction
Since its early days1, the research on colloidal nanocrystals (NCs) has provided access to a variety of inorganic compounds, spanning from metals and alloys2 to oxides3,4, phosphides5, chalcogenides6,7,8,9,10,11, halides12,13, and more. Nowadays, NCs are exploited in many different fields, as NCs-based products are steadily finding their way to commercial applications14,15,16,17,18. A colloidal approach to the synthesis of inorganic materials offers several advantages over more traditional solid-state chemistry techniques, such as milder reaction conditions, ease of tunability deriving from a wider choice of precursors, better processability of the products, and straightforward access to nanostructured materials. However, both this flexibility and the push toward materials combining a higher number of elements come at the cost of increased complexity. One example is the field of colloidal semiconductors, where lead-based NCs are among the most explored compounds due to the appealing optoelectronic properties demonstrated by lead halide perovskites in the UV-VIS and by lead chalcogenides in the IR spectral ranges6,7,12,13,19. There, the combination of as little as four elements (Cs, Pb, X and E, where X = F, Cl, Br or I, and E = S, Se or Te) can yield NCs of a variety of phases: the binaries CsX, PbX2, and PbE6,7,20,21,22, the well-known cesium lead halides (Cs4PbX6, CsPbX3, and CsPb2X5)12,19,23,24, and the still little explored lead chalcohalides (Pb4S3Br2 and Pb4S3I2)25,26. All these compounds are often obtained under similar reaction conditions, and can therefore compete during the synthesis, requiring a careful tuning of the synthetic protocols to achieve an impurity-free synthesis of the desired product.
This condition is not dissimilar from that of many organic chemistry reactions, where two molecules can react following different pathways and resulting in different products. Similar issues are often addressed by exploiting reaction-directing groups, that are moieties which are attached to one of the reagents in order to direct the reaction pathway toward the desired product, and are removed at a later stage27. This requires a multi-step rational design of the synthetic procedure, that is common for organic chemistry but is missing in the colloidal synthesis of NCs, as most procedures consist of a one-step reaction.
In this work, we exploit the formation of epitaxial heterostructures as a reaction-directing step for the phase-selective synthesis of colloidal NCs, and we take advantage of this approach to further expand the family of lead chalcohalides. These materials share the general formula PbaEbXc, and only four stable compositions are known in bulk (PbTeF6, Pb7S2Br10, Pb5S2I6, Pb4SeBr6)28,29, together with two recently reported high-pressure metastable phases (Pb4S3I2, Pb3Se2Br2)30,31. Our group pioneered the investigation of lead chalcohalides at the nanoscale, discovering two additional compounds (Pb4S3Br2 and a yet not identified Pb-S-Cl phase) in the form of NCs25. More recently, we found that Pb4S3Br2 can match epitaxially the CsPbX3 perovskite to form colloidal heterostructures, thus demonstrating a remarkable synthetic and structural compatibility26. Here we focus our attention on the lead sulfochlorides, demonstrating that the yet unknown Pb-S-Cl phase obtained through direct synthesis is Pb3S2Cl2, whose structure, here identified as monoclinic, is a distorted version of the cubic Pb3Se2Br2 prototype (Fig. 1, left reaction route)31. Conversely, the introduction of CsPbCl3 nanoclusters in the reaction medium, under comparable reaction conditions, produces Pb4S3Cl2/CsPbCl3 heterostructures while suppressing the formation of Pb3S2Cl2. Once the heterostructures are formed, the perovskite domain can be etched by exploiting the solubility of CsPbCl3 in polar solvents, while leaving the Pb4S3Cl2 domains intact. Hence, the use of CsPbCl3 as a disposable template yields colloidally stable Pb4S3Cl2 NCs that could not be obtained by direct synthesis due to the competitive nucleation of Pb3S2Cl2 (Fig. 1, top-right reaction route).

A solution of PbCl2 reacts with sulfur dissolved in 1-octadecene (S-ODE) to form Pb3S2Cl2 NCs. When reacted with Cs-oleate (R-COO-Cs+) it produces instead CsPbCl3 nanoclusters, that can be further reacted with Pb-oleate, S-ODE, and an alkyl thiol (R-SH) to form Pb4S3Cl2/CsPbCl3 heterostructures. The CsPbCl3 domain can be then selectively etched by the sequential addition of oleylamine (R-NH2), dimethyl formamide (DMF), and oleic acid (R-COOH), yielding colloidally stable Pb4S3Cl2 NCs that could not be obtained by direct synthesis. Atoms color code: Cs = cyan; Pb = gray; S = yellow; Cl = green.
The reason why CsPbCl3 induces such phase selectivity lies in the fact that the structures of Pb3S2Cl2 and Pb4S3Cl2 are remarkably different25,30, to the point that only Pb4S3Cl2 can match epitaxially with the CsPbCl3 perovskite. Indeed, Pb4S3Cl2 meets two strict structural constraints: i) the presence of a perovskite-like atomic plane26, and ii) the continuity of the cationic subnetwork across the chalcohalide/perovskite interface32. Such conditions are not met for Pb3S2Cl2, whose homogeneous nucleation is, therefore, less favorable than the heterogeneous nucleation of Pb4S3Cl2 templated by the CsPbCl3 perovskite.
Overall, our method effectively combines the concept of sacrificial substrates, such as the alkali halide single crystals used for the large-area synthesis of films and ultrathin materials33,34,35,36, with the phase-selection capabilities ensured by the epitaxial templating, all in the liquid phase.
Results
Structure solution of Pb3S2Cl2 NCs
We start by discussing the synthesis and structure solution of lead sulfochloride NCs prepared by homogenous nucleation, that is, in the absence of halide perovskites. We recently reported the synthesis of the same NCs by a different procedure25, but in our previous work the small size of the particles and the heavy PbS contamination prevented us from determining the composition and crystal structure, which had therefore remained unknown to date. To solve the structure in the present work, we prepared large-size NCs via a two-step procedure, consisting of nucleation and seeded growth (Fig. 2a, b, see Supplementary Discussion, section 1 for details).

a Pb3S2Cl2 NCs as synthesized (left) and after the accretion process (right). b HAADF-STEM image of a Pb3S2Cl2 NC. c [010] projection of the Pb3S2Cl2 reciprocal lattice measured by 3D-ED, together with a representation of the reciprocal lattice axes and unit cell. d XRPD (top) and PDF (bottom) fits obtained by refining the Pb3S2Cl2 structure in the Cc space group (experimental data = blue, fit curves = orange, residuals = gray). The XRPD profiles are plotted in the Cu-Kα 2θ scale to ease the comparison with lab-grade diffraction patterns. e Two views of the Cc monoclinic Pb3S2Cl2 structure are shown along the high symmetry directions of the corresponding pseudocubic cell ([100] top, [111] bottom). A projection of the monoclinic cell is overlaid in blue, and the directions of the corresponding lattice vectors are indicated by labeled arrows. Atoms color code: Pb = gray; S = yellow; Cl = green. Source data for d are provided as a Source Data file.
The synthesis yielded particles with a diameter of 29.5 ± 2.0 nm, as estimated by Transmission Electron Microscopy (TEM). Beyond this size the NCs became insoluble in the reaction medium, and the accretion process halted. The composition measured by SEM-EDX was Pb:S:Cl = 3.2:1.8:2.0, which is compatible with our previously advanced hypothesis of a Pb3S2Cl2 compound25. However, the X-Ray Powder Diffraction (XRPD) pattern could not be fitted based on the supposedly related Pb3Se2Br2 cubic prototype25,31, suggesting a different structure. Therefore, we exploited a combination of techniques to solve it. First, we performed a single-NC 3D Electron Diffraction (3D-ED)37 experiment to identify the unit cell parameters and a list of possible space groups (Fig. 2c). For some of them we also produced ab-initio solutions; however, a definitive structural model could not be obtained due to the small size of the NCs, which made the integrated intensity of the diffraction spots inaccurate. Thus, we exploited the information from 3D-ED to repeat the ab-initio structure solution from XRPD data with the software EXPO201438, eventually obtaining a model close to the cubic Pb3Se2Br2 in terms of connectivity, but with a lower symmetry. Finally, we refined this model in both reciprocal and direct spaces on data collected at the Brookhaven National Laboratory synchrotron, on setups optimized for XRPD and Pair Distribution Function (PDF) experiments respectively (Fig. 2d).
The solution was found in the monoclinic Cc space group (Fig. 2e, see Supplementary Discussion, section 2 for details), capturing deviations from the Pb3Se2Br2 cubic prototype that are likely due to the small ionic radii of S2− and Cl−. Density Functional Theory calculations (DFT, see Supplementary Discussion, section 3) confirmed that as a bulk crystal the monoclinic Pb3S2Cl2 would be more stable than a hypothetical cubic polymorph by 164 kcal/mol. The ionic radii are also likely to play a major role in favoring the Pb3S2Cl2 phase over Pb4S3Cl2 for free-standing lead sulfochloride NCs. Indeed, Pb3E2X2 appears to be favored if \({r}_{{E}^{2-}}/{r}_{{X}^{-}}\approx 1\) (Se2−/Br− = 1.01; S2−/Cl− = 1.02)31, while \({r}_{{E}^{2-}}/{r}_{{X}^{-}} < 1\) favors Pb4E3X2 (S2−/Br− = 0.94; S2−/I− = 0.84)25,30,39. Indeed, in both structures Pb2+ is surrounded by eight anions, and S2− features a distorted octahedral coordination. Conversely, the coordination of the halide changes: in Pb3S2Cl2 the smaller Cl− ions share the same octahedral coordination as S2−, while the larger Br− and I− ions in Pb4S3X2 are surrounded by seven Pb2+ ions in a pentagonal bipyramidal configuration. The reason is likely that bulkier anions can accommodate an enlarged coordination environment. Interestingly, Pb3S2Cl2 is the only lead chalcohalide where Cl− is coordinated by an octahedron of Pb2+ ions. Such coordination has been recently proposed on the surface of PbS NCs synthesized in excess of PbCl2 to account for the formation of a Cl-rich shell that improves the optical properties40,41,42,43, suggesting that these NCs might be passivated by a layer of some lead sulfochloride compound. For further comparisons between the structures of Pb3S2Cl2 and Pb3S2X2 compounds see Supplementary Discussion, section 4, which includes the structure refinements of Pb4S3Br2 and Pb4S3I2 NCs and a comparison of PDF profiles by RootProf44.
Compatibility of lead sulfochlorides with CsPbX3
Our prior studies demonstrated that the lead sulfobromide Pb4S3Br2 can grow epitaxially on lead halide perovskite domains26, suggesting that other isostructural compounds, i.e., Pb4S3I2 and a hypothetical Pb4S3Cl2, would do the same. Conversely, the Pb3S2Cl2 structure we hereby solved is remarkably different from that of the Pb4S3X2 compounds, to the point that the epitaxial compatibility with CsPbX3 would be lost. This prompted us to think that the synthesis of a sulfochloride/perovskite heterostructure would produce domains of the yet unknown compound Pb4S3Cl2 instead. Indeed, the Pb4S3X2/CsPbX3 match, regardless of the halide, is strictly structure specific. First, both the chalcohalide and the perovskite share a plane of Pb2+ ions arranged in a square grid, which serves as an interface between the two domains (in red in Fig. 3a, right)26. No such plane can be found in Pb3S2Cl2. Moreover, Pb4S3X2 shares deep similarities with some of the Cs-Pb-X phases and the heterostructures they form with CsPbX3, which are easily overlooked at first sight. Indeed, the Cs-Pb-X compounds share a common cationic subnetwork of Cs+ ions, which in Cs-Pb-X/Cs-Pb-X heterostructures is preserved across the interface and ensures the lattice compatibility32. Remarkably, similar connectivity is found across the Pb4S3X2/CsPbX3 interface. Relevant examples for our discussion are the CsPb2X5/CsPbX3 heterostructures45,46. In CsPb2X5, the Cs+ subnetwork encloses layers of [Pb2X5]− bidimensional polyanions32. Remarkably, Pb4S3X2 is almost isostructural to CsPb2X5, being formed by a Pb2+ subnetwork enclosing [Pb2S3X2]− polyanions of identical geometry. The only difference is the presence of an extra layer of Pb2+ ions in between each polyanion, that is needed to maintain the charge balance (Fig. 3a, left). This makes the Pb2+ subnetwork of Pb4S3X2 resemble that of another Cs-Pb-X phase, namely Cs2PbX4. Consequently, the cationic subnetwork of Pb4S3X2 is similar to that found in Cs-Pb-X compounds and can find its natural prosecution in the CsPbX3 perovskite (Fig. 3a, right), ensuring the stability of the interface and remarking the pivotal role of ionic subnetworks in the formation of colloidal heterostructures. Deeper insights into the heterostructure interface connectivity and the similarities with Cs-Pb-X/Cs-Pb-X heterostructures can be found in the Supplementary Discussion, section 5.

a On the left, models of the Cs2PbX4-like Pb2+ subnetwork (orange Pb2+) and of the CsPb2X5-like [Pb2S3Cl2]− polyanion (white Pb2+, yellow S2-, green Cl−) found in Pb4S3Cl2. On the right, model of the Pb4S3Cl2/CsPbCl3 epitaxial interface superimposed on a close-up view of c, highlighting the continuity of the Pb2+/Cs+ cationic subnetwork. The Pb4S3Cl2 structure has been represented by adapting the Pb4S3Br2 structure. Atoms color code: Cs = cyan; Pb = orange/white; S = yellow; Cl = green. b Low resolution TEM image of as-synthesized Pb4S3Cl2/CsPbCl3 heterostructures. c HAADF-STEM image of a single Pb4S3Cl2/CsPbCl3 heterostructure. Insets: FFTs of the Pb4S3Cl2 (top) and CsPbCl3 (bottom) domains. The spots circled in a solid line correspond to planes parallel to the heterostructure interface and to each other; those circled in a dashed line are instead perpendicular to the interface and share similar periodicities, ensuring the match of the two lattices. d Column intensity map of the Pb-containing columns in the perovskite phase and of the Pb columns in the Cs2PbX4-like subnetwork of the Pb4S3Cl2 domain. The color in the intensity map correlates with the total intensity scattered from the corresponding atomic column (red = higher intensity; blue = lower intensity). The map is compatible with the interface model depicted in Fig. 3a, and highlights the preservation and merging of the cationic subnetwork of the two phases across the interface. Source data for d are provided as a Source Data file.
Synthesis of Pb4S3Cl2/CsPbCl3 heterostructures
To test whether the natural affinity of the Pb4S3X2 phases for lead halide perovskites would lead to the formation of Pb4S3Cl2, we adapted our previously reported method to attempt the synthesis of Pb4S3Cl2/CsPbCl3 heterostructures26. In the first step of the procedure, CsPbCl3 nanoclusters, prepared separately, were reacted with elemental sulfur in the presence of lead oleate and dodecanethiol at 200 °C, indeed producing heterostructures. The resulting particles were recovered through antisolvent precipitation, and then treated with a solution of PbCl2, oleylamine, and oleic acid to restore their colloidal stability (see Supplementary Discussion, section 6). The heterostructures were homogeneous in terms of size and shape (Fig. 3b), and no single-phase perovskite or sulfochloride NCs were observed.
High-Angle Annular Dark-Field Scanning TEM (HAADF-STEM) showed that the heterostructures were composed of two highly crystalline domains jointed along a flat interface spanning across the entire nanoparticle (Fig. 3a, c). As expected, the fast Fourier transform (FFT) of the HAADF-STEM image revealed that the CsPbCl3 domain matches with the perovskite structure, while that of the lead sulfochloride domain is incompatible with Pb3S2Cl2. Instead, the unit cell parameters and the overall FFT symmetry match with what is expected for a Pb4S3X2 domain based on our previous observations on Pb4S3Br2/CsPbX3 heterostructures (insets of Fig. 3c)26. Such phase selectivity was granted by the transformation of CsPbCl3 nanoclusters into NCs during the early stages of the reaction26. The perovskite NCs then acted as phase-selective heterogeneous nucleation seeds for Pb4S3Cl2, providing a significant advantage over the homogeneous nucleation of Pb3S2Cl2, which was indeed suppressed.
The predicted structure of the Pb4S3Cl2/CsPbCl3 interface, superimposed in Fig. 3a to a high-resolution HAADF-STEM image of the heterostructure, was further confirmed by highlighting the Pb-containing atomic columns involved in the Pb2+ cationic subnetwork through the quantitative analysis of HAADF-STEM images (Fig. 3d). Indeed, it is worth noting that a prosecution of the chalcohalide-Pb2+ subnetwork, identified in Fig. 3a with the perovskite-Cs+ subnetwork in analogy with the CsPb2X5/CsPbX3 heterostructures, can also be found in the Pb2+ subnetwork of the perovskite domain, as it shares the same symmetry as the Cs+ one (see Supplementary Discussion, section 5 for details).
Selective etching of the CsPbCl3 domain
Our templated synthesis approach could be pushed a step further as we took advantage of the solubility of CsPbCl3 in polar solvents to selectively etch the perovskite domains and recover phase-pure Pb4S3Cl2 NCs (Fig. 4). Briefly, the heterostructures dispersed in hexane were first treated with oleylamine to improve the colloidal stability of the etched NCs in nonpolar media. Then, we added to the dispersion an equal volume of dimethylformamide (DMF), and the resulting mixture was vortexed for 30 s, forming an emulsion. After its complete separation, DMF was removed, and predried oleic acid was introduced in the NCs hexane dispersion. Subsequently, the particles were recovered through antisolvent precipitation followed by a centrifugation, and redispersed in toluene for further use. The procedure yielded remarkably uniform Pb4S3Cl2 NCs, whose structure was confirmed via high-resolution HAADF-STEM imaging and analysis of the lattice by means of FFTs. Interestingly, the NCs appeared to be sphere-shaped despite the original chalcohalide domains in the heterostructures being hemispherical. We attribute this change of morphology to the need of lowering the surface energy of the extended and non-passivated facet left after the dissolution of the perovskite. It is likely that the excess ions released by the etching process will recrystallize on the chalcohalide domains, thus reshaping them into a more stable spherical morphology. See Supplementary Discussion, section 7 for details on the etching of heterostructures.

a Scheme of the DMF-assisted etching procedure developed to transform the Pb4S3Cl2/CsPbCl3 heterostructures into Pb4S3Cl2 NCs in the presence of surfactants (R-NH2 = oleylamine, R-COOH = oleic acid). The cyan cubes and the orange spheres represent the CsPbCl3 and the Pb4S3Cl2 domains of the heterostructures, respectively. b TEM image of the starting heterostructures. The top inset shows an HAADF-STEM image of one heterostructure prior to the etching, while the bottom inset shows the FFT of the chalcohalide domain of the same image. c TEM image of the etched NCs. The top inset shows an HAADF-STEM image of one NC after the etching, while the bottom inset shows the FFT of the same image. The two FFTs demonstrate that the Pb4S3Cl2 domains retain the same crystal structure before and after the etching procedure.
Optoelectronic properties of lead sulfochloride NCs
Obtaining both Pb3S2Cl2 and Pb4S3Cl2 NCs, and the latter both in the form of free-standing NCs and heterostructures with CsPbCl3, gave us the opportunity to investigate the impact of stoichiometry, structure, and presence of a heterojunction on the optoelectronic properties of lead sulfochlorides. Figure 5 summarizes the experimental results, along with data from DFT calculations. The free-standing Pb3S2Cl2 and Pb4S3Cl2 NCs exhibited basically overlapping and featureless absorption spectra, with onset at ∼1.8 eV (∼690 nm, Fig. 5a, solid blue and orange plots). The Tauc plot analysis suggested an indirect band gap of ∼1.8 eV in both cases, in agreement with the DFT predictions (see Supplementary Discussion, sections 3 and 8). This is in stark contrast with the direct bandgap observed for lead chalcogenides (PbE) and many lead halides (e.g., CsPbX3), remarking that the electronic properties of lead chalcohalides are not straightforwardly related to those of chemically similar compounds. A comparable absorption profile was found in the heterostructures spectrum, combined with the blunt absorption edge of the direct-gap CsPbCl3 domain (∼3.1 eV = 400 nm24,47, Fig. 5a, solid black plot). Such edge was instead sharp for free-standing CsPbCl3 NCs (Fig. 5a, solid cyan plot), suggesting an intimate electronic connection between the two domains in the heterostructures. This was further supported by the almost complete suppression of the CsPbCl3 photoluminescence (PL) when the perovskite domain was involved in the heterojunction (Fig. 5b, inset).

a Absorption (ABS, solid lines) and photoluminescence (PL, dashed lines) spectra of free-standing Pb3S2Cl2, Pb4S3Cl2, CsPbCl3 NCs and of Pb4S3Cl2/CsPbCl3 heterostructures. The CsPbCl3 NCs were prepared for comparison purposes by heating the CsPbCl3 precursor nanoclusters (see Supplementary Discussion, section 6). b Decay curves of the ∼410 nm PL from CsPbCl3 as free-standing NCs and in the Pb4S3Cl2/CsPbCl3 heterostructures. The dashed gray line is the time response of the experimental setup. Inset: PL spectra of CsPbCl3 as free-standing NCs and in the heterostructures, showing a drastic quenching in the latter case. The PL intensity of the heterostructures is multiplied ×10 to make it more visible. The slight redshift of the heterostructure PL maximum is likely due to the relaxation of quantum confinement of the perovskite domain caused by the partial delocalization of the exciton in the chalcohalide domain. c Decay curves of the ∼1.8 eV (∼690 nm) emission of the three samples at room temperature and at 77 K (inset). Color code for a–c: cyan = CsPbCl3, blue = Pb4S3Cl2, orange = Pb3S2Cl2, black = Pb4S3Cl2/CsPbCl3 heterostructures. d Electronic structure of free-standing Pb3S2Cl2 (left) and Pb4S3Cl2 (right) NC models computed at the DFT/PBE level of theory. The color code indicates the elemental contribution to each molecular orbital (Pb = gray; S = yellow; Cl = green). e Electronic structure of the Pb4S3Cl2/CsPbCl3 heterostructure model computed at the DFT/PBE level of theory, color coded by domain (Pb4S3Cl2 = orange; CsPbCl3 = cyan). The Inverse Participation Ratio (IPR, left panel) quantifies the degree of localization of a state (1/Natoms = completely delocalized; 1 = localized on one single atom), and indicates that most of the states surrounding the band gap are localized trap states. f Representation of molecular orbitals corresponding to band edge delocalized states (1,4) and to band edge trap states (2,3). In both cases, the band edge states are localized on the Pb4S3Cl2 domain. Source data for a–e are provided as a Source Data file.
DFT calculations suggested that the cause for the PL quenching was the nearly type-I band alignment between Pb4S3Cl2 and CsPbCl3 (Fig. 5e, see also Supplementary Discussion, section 3). Interestingly, this contrasts with the structurally equivalent Pb4S3Br2/CsPbBr3 heterostructures investigated in our previous work, which were instead identified as quasi-type-II junctions26. Indeed, the molecular orbitals corresponding to the band edge states of the heterostructures are strongly localized on the sulfochloride domain (Fig. 5f). It is worth noting that the predicted type-I alignment should promote the migration of photocarriers into the sulfochloride domain of the heterostructure. Indeed, the decay of the barely detectable PL of CsPbCl3 in the heterostructures was strongly accelerated if compared with that of free-standing CsPbCl3 NCs (τavg ~8 ps vs ~18 ps, Fig. 5b).
Owing to the relaxation of the momentum conservation at the nanoscale, both the free-standing sulfochlorides and the heterostructures exhibited a broad PL peak at ∼1.8 eV (∼690 nm, see Fig. 5a, dashed lines), indicating in all the cases a sulfochloride band edge emission. Such emission was negligible at room temperature (PLQY < 1%) due to the efficient thermal quenching. However, lowering the temperature to 77 K resulted in a ~100-fold intensification of the PL, accompanied in the case of heterostructures by a brightening of the residual, yet still negligible, CsPbCl3 emission. In all three cases, the PL kinetics of the lead chalcohalides was markedly non-exponential, with a dominant sub-microsecond drop followed by a slower decay (Fig. 5c). The process was slightly slower in the heterostructures with respect to both free-standing Pb3S2Cl2 and Pb4S3Cl2 NCs, suggesting that the CsPbCl3 domain might suppress some trapping losses by passivating part of the sulfochloride domain surface. This is also consistent with our observation of a more intense PL. At low temperatures, all the PL decays became substantially longer. Fitting the dataset with an Arrhenius function yielded for all the samples an activation energy of ∼15 meV. Remarkably, the spectral shape of all samples followed a nearly identical trend with the temperature, showing a progressive shift to ∼1.5 eV (∼827 nm) and a band narrowing, further corroborating the strong similarities between the optoelectronic properties of these three systems. See Supplementary Discussion, section 8 for details on the spectroscopic characterizations.
In general, our analysis indicates that the properties of lead sulfochlorides are largely independent of both stoichiometry and structure. This completes our preliminary observations on Pb4S3Br2 and Pb4S3I2 NCs, that shared nearly identical absorption spectra despite containing different halides25. We rationalized such behavior as a consequence of two factors. First, in these materials the band edge states feature a prominent participation of Pb2+ and S2−, while the halides mostly contribute to states buried deep in the valence band, making the electronic properties almost halide-insensitive. Second, regardless of the structure of the specific chalcohalide, the geometry and connectivity of the coordination polyhedra surrounding both Pb2+ and S2− is the same, making the electronic properties basically structure-insensitive.
Discussion
In this work, we have exploited the epitaxial templating effect of CsPbCl3 to control the synthesis of lead sulfohalide NCs through the formation of Pb4S3Cl2/CsPbCl3 heterostructures. Indeed, while a direct synthesis approach (i.e., in the absence of CsPbCl3 acting as a template) yielded Pb3S2Cl2 NCs, only the selective etching of the heterostructures perovskite domain allowed us to recover free-standing Pb4S3Cl2 NCs. This ultimately means that we could select the reaction product between two competing phases, a problem of great relevance in the synthesis of colloidal inorganic nanomaterials. Both the obtained lead sulfochlorides shared remarkably similar optoelectronic properties. This, combined with our previous observation on bromine- and iodine-based sulfohalides25, suggests that lead sulfohalide compounds are mostly stoichiometry- and structure-insensitive. Interestingly, the discovery of the Pb3S2Cl2 structure demonstrates that Cl− in lead chalcohalides can be coordinated by an octahedron of Pb2+ ions, differently from Br− and I−. This supports the recent observations of a PbClx-rich shell on the surface of PbS NCs synthesized using PbCl2 as a precursor, a condition that is remarkably similar to our synthetic protocol and might indeed lead to the formation of a lead sulfochloride surface layer40,41,42,43.
We remark that our use of perovskite NCs as disposable and phase-selective epitaxial templates parallels that of reaction-directing groups in traditional organic chemistry and catalysis27, and allowed us to design the phase-selective synthesis of two yet unknown materials (Pb3S2Cl2 and Pb4S3Cl2) based on structural considerations only. Such an approach to a deterministic synthesis of NCs may be extended to other phases with known or predictable epitaxial relations, taking advantage of the vast library of already reported nanomaterials as starting templates. For example, in the field of chalcohalides it might lead to yet unknown selenium- and tellurium-based materials, or even to compounds where the halide is replaced by pseudohalide ions (e.g., CN− and SCN−). In conclusion, we envision that our phase-selective templating approach will open new routes for the colloidal syntheses of nanomaterials which are now hindered by an excessive activation energy for the homogeneous nucleation, or by the competitive formation of undesired phases.
Methods
Chemicals
1-octadecene (ODE, technical grade, 90%), oleic acid (OA, technical grade, 90%), oleylamine (OLA, technical grade, 70%), lead(II) chloride (PbCl2, 98%), lead(II) bromide (PbBr2, 98%), lead(II) iodide (PbI2, 98%), lead(II) thiocyanate (Pb(SCN)2, 99.5%), lead(II) acetate trihydrate (Pb(OAc)2·3H2O, 99.99%), cesium(I) carbonate (Cs2CO3, 99%), dodecanethiol (DDT, 99.9%), and sulfur powder (S, 99.99%) were purchased from Sigma‐Aldrich. All reagents were used as received without any further purification.
Synthesis: S-ODE stock solution for Pb3S2Cl2 NCs
0.064 g (2 mmol) of S powder were mixed with 10 mL of ODE (pre-degassed at 120 °C under vacuum for an hour) in a 20 mL glass vial inside a glove box filled with N2. The resulting mixture was sonicated until the complete dissolution of the sulfur powder.
Synthesis: Pb3S2Cl2 nanocrystals (NCs)
0.21 g of PbCl2 (0.8 mmol) were solubilized at 120 °C in a mixture of 2 mL of OLA, 2 mL of OA and 20 mL of ODE in a 100 mL flask. The solution was then heated up to 170 °C, and 2 mL of the S-ODE stock solution, pre-heated at 150 °C, were swiftly injected. The NCs formed ∼10–30 s after the injection, as revealed by the color change of the solution from pale yellow to red. After 1 min, the reaction was quenched by cooling the flask with a room-temperature water bath. The NCs were recovered by adding 40 mL of ethyl acetate, followed by a centrifugation at 6000 rpm (∼2940 × g) for 5 min. The precipitate was discarded, and the supernatant was suspended in hexane or toluene. If needed, the sample could be washed multiple times by adding ethyl acetate and repeating the centrifugation. The maximum diameter for Pb3S2Cl2 NCs obtained by direct synthesis was 7–8 nm (see Supplementary Discussion, section 1).
Synthesis: accretion of larger Pb3S2Cl2 NCs
Larger NCs were obtained through a seeded growth approach, performed by using as starting seeds Pb3S2Cl2 NCs prepared as detailed above. A stock solution of reagents was prepared by dissolving 0.2966 g of PbCl2 (1.06 mmol) and 0.6898 g of Pb(SCN)2 (2.13 mmol) in a mixture of 4 mL of OLA, 4 mL of OA, and 40 mL of ODE in a 100 mL flask at 120 °C. During its preparation, the stock solution must not exceed this temperature to avoid the thermal decomposition of the thiocyanate, which would cause the nucleation of PbS NCs25. Once the two solids were completely solubilized, the stock solution was cooled to room temperature, filtered with a 0.2 μm PTFE syringe filter, and loaded on a syringe pump. Then, a crude Pb3S2Cl2 reaction mixture in ODE was obtained by following the procedure detailed in the previous paragraph up to before the addition of ethyl acetate. 12 ml of such solution were heated to 170 °C, and the stock solution of PbCl2 and Pb(SCN)2 was added dropwise at a controlled rate of 10 mL/h with the help of a syringe pump. Finally, the reaction mixture was washed with ethyl acetate as described in the previous paragraph, and the NCs were suspended in hexane. A last step of filtration performed with a 0.2 μm PTFE syringe filter allowed to remove any impurity of PbS that may have formed during the accretion process. The so-obtained Pb3S2Cl2 NCs measured 29.5 ± 2.0 nm on average, and were used to solve the structure of the material by a combination of 3D-ED, XRPD, and PDF experiments. See Supplementary Discussion, section 1 for additional details on the accretion process.
Synthesis: Pb4S3Br2 (or Pb4S3I2) NCs
NCs of Pb4S3Br2 (and Pb4S3I2) were prepared following our previously published method25. In short, 0.2 mmol of PbBr2 (or PbI2) and 0.2 mmol of Pb(SCN)2 where dissolved in a mixture of 10 mL ODE and 250 μL of OLA and OA at 120 °C in a 25 mL three-necked flask. Then, the solution was quickly heated (∼20 °C/min) and started turning from light-yellow to bloody red above 150 °C while the NCs nucleated and grew. The reaction was quenched by cooling the flask in a water bath. The NCs were recovered by simple centrifugation or by ethyl-acetate assisted precipitation followed by centrifugation (6000 rpm for 5 min in both cases, ∼2940 × g). The so-obtained Pb4S3Br2 and Pb4S3I2 NCs were used to refine the structure of both materials by XRPD and PDF experiments (see Supplementary Discussion, section 4).
Synthesis: Cs-oleate precursor for CsPbCl3 nanoclusters
The Cs-oleate precursor was prepared by increasing the concentration of oleic acid in comparison to our previously reported method26. In a typical synthesis, Cs2CO3 (0.652 g, 2 mmol), OA (5 mL, 15 mmol) and ODE (17.5 mL) were loaded into a 50 mL 3-neck flask, dried for 1 h at 110 °C and then heated under N2 to 150 °C until the solution turned clear. The resulting mixture was transferred into a N2 filled glass vial, which was stored inside a glovebox for further use.
Synthesis: PbCl2 stock solution for heterostructures
PbCl2 powder (0.556 g, 2 mmol), OA (5 mL), and OLA (5 mL) were mixed with ODE (30 mL) in a 100 mL three neck round bottom flask. The reaction mixture was dried/degassed under vacuum for 15 min at 110 °C. Then, the flask was filled with N2 and the temperature was raised to 150 °C. After complete dissolution of PbCl2 salt, the solution was cooled down to room temperature (25 °C) and transferred into a N2-filled glass vial.
Synthesis: Pb(OA)2 stock solution for heterostructures
The lead oleate stock solution was prepared following our previously reported method26. Briefly, Pb(OAc)2·3H2O powder (0.76 g, 2 mmol) and OA (1.3 mL), were mixed with ODE (18.7 mL) in a 50 ml three neck round bottom flask. The reaction mixture was degassed under vacuum for 1 h at 110 °C and then heated under N2 to 150 °C until all Pb(OAc)2·3H2O reacted with OA. Thereafter, the solution was cooled down to room temperature (25 °C) and transferred into N2-filled glass vials.
Synthesis: CsPbCl3 nanoclusters
CsPbCl3 nanoclusters were synthesized following a previously reported method26. Briefly, 4 mL of the above-mentioned PbCl2 stock solution were transferred into the N2 filled 20 ml glass vial. Thereafter, 0.25 mL of the Cs-oleate precursor were injected into the PbCl2 stock solution at 40 °C and the resulting mixture was kept under stirring for 20 min. After about 20 min, the mixture (turbid white) was centrifuged at 8000 rpm (∼3935 × g) for 5 min, the supernatant was discarded, and the precipitate was redispersed in 0.9 mL of degassed ODE.
Synthesis: Pb4S3Cl2/CsPbCl3 heterostructures
4.0 mL of degassed ODE were added to a 20 mL glass vial under N2. The vial was heated to 200 °C, and 100 μL of the above-mentioned Pb(OA)2 solution and 20 μL DDT (diluted in 200 μL predried ODE) were added into the reaction system. Then, the mixture of S (0.1 mL) and CsPbCl3 nanoclusters (0.9 mL) was rapidly injected into the reaction mixture. The reaction was allowed to proceed for 5 min and was subsequently quenched by immersing the vial in an ice and water bath. The crude solution was then centrifuged by adding methyl acetate (with volume ratio of 1 to 1) and the precipitate was redispersed in toluene. See Supplementary Discussion, section 6 for details.
Synthesis: surface treatment of heterostructures
A PbCl2 solution, separately prepared by dissolving PbCl2 salt (1 mmol) in ODE (15 mL) in the presence of oleic acid (2.5 mL) and oleylamine (2.5 mL), was added to the toluene dispersion containing the heterostructures, and the mixture was vortexed for 1 min. Then, the solution was centrifuged by adding methyl acetate and the precipitate was redispersed in hexane or toluene for further use. The role of this treatment is to reconstruct the surface of the CsPbCl3 perovskite domains after the antisolvent-assisted precipitation, and to ensure the long-term colloidal stability of the sample.
Synthesis: CsPbCl3 NCs
4.0 mL of ODE were added to a 20 mL glass vial under air and heated to 150 °C, then 1.0 mL of the above-mentioned seed cluster solution (CsPbCl3, without S-ODE) was swiftly injected. The mixture was annealed for 5 min and subsequently cooled down by using an ice water bath. The resulting mixture was centrifuged at 8000 rpm (∼3935 × g) for 5 min, the supernatant was discarded, and the precipitate was dispersed in 4 mL of toluene.
Synthesis: etching of Pb4S3Cl2/CsPbCl3 heterostructures
1 mL of heterostructures dispersed in hexane (corresponding to one entire batch of heterostructures) was treated first with oleylamine (60 μL). Then, an equal volume ratio of dimethylformamide (DMF, 1 mL) was added to the heterostructures dispersion and the resulting mixture was vortexed for 30 s. After the complete phase separation of solvents (2–3 min), DMF was removed by using a syringe and pre-dried oleic acid (60 μL) was introduced in the hexane dispersion. Subsequently, the now etched NCs were centrifuged by adding methyl acetate (with 1 to 1 volume ratio) and were redispersed in toluene for further use. See Supplementary Discussion, section 7 for details.
Characterization: TEM
Bright-field TEM images of the samples were acquired with a JEOL-1100 transmission electron microscope operating at an acceleration voltage of 100 kV. The samples were prepared by drop casting diluted solutions of NCs or heterostructures onto carbon film-coated 200 mesh copper grids for low-resolution TEM.
Characterization: Lab-grade XRPD
Lab-grade XRPD analyses were performed in θ:2θ scan mode on a Panalytical Empyrean diffractometer, equipped with a 1.8 kW Cu-Kα ceramic anode working at 45 kV–40 mA and a PIXcel3D detector. XRPD data were acquired on samples in the form of dry powders or drop-casted solutions; the measurements were carried out in air at room temperature using a zero-diffraction silicon substrate.
Characterization: HAADF-STEM
High-resolution HAADF-STEM images were acquired with a probe-corrected cubed Thermo Fisher Scientific Themis Z Microscope operating at 300 kV with a probe semi-convergence angle of 20.5 mrad. For a quantitative analysis of the HAADF-STEM image, the intensities of the individual atomic columns in a single heterostructure were analyzed by using the StatSTEM software48. The color code in the intensity map correlates with the total intensity scattered from each atomic column. The intensity is calculated by fitting a Gaussian function to each atomic column: the intensity value of a column equals the volume of its Gaussian peak.
Characterization: 3D-ED
3D-ED data were collected on a Zeiss Libra TEM operating at 120 kV and equipped with a LaB6 source. Data were acquired in STEM mode after defocusing the beam to achieve a parallel illumination of the sample. A beam size of about 150 nm in diameter was obtained by inserting a 5 μm C2 condenser aperture. A mild illumination was adopted to avoid any alteration or amorphization of the sample, and to slow down the accumulation of organic contaminants. 3D-ED data were recorded with an ASI Timepix detector49, which is able to detect single electrons and deliver a pattern that is virtually background-free. The camera length was 180 mm, with a theoretical resolution limit of 0.75 Å.
Characterization: synchrotron XRPD and PDF data collection
Synchrotron diffraction data were collected at the 28ID-2 beamline of the National Synchrotron Light Source (NSLS-II) of the Brookhaven National Laboratory with an X-ray energy of 67.17 keV (0.1846 Å) and a 0.5 mm × 0.5 mm beam size. A Perkin Elmer XRD 1621 digital imaging detector (2048 × 2048 pixels and 200 × 200 µm pixel size) was mounted orthogonal to the beam path in two positions: 228 mm and 1365 mm downstream of the sample. Samples of Pb3S2Cl2, Pb4S3Br2, and Pb4S3I2 were measured in both setups, optimized for PDF and XRPD measurements, respectively. Lanthanum hexaboride (LaB6) was measured as a standard material to calibrate the wavelength and the detector geometry, including the sample-to-detector distance. An empty capillary was measured for background estimation. Diffraction images were azimuthally integrated and converted into intensity profiles vs 2θ and vs momentum transfer \(Q=4{{\pi }}{{\sin }}\vartheta /{\lambda }_{X-{ray}}\) by using the FIT2D program50. PDF profiles were calculated up to interatomic distances r of 50 Å from the Q profiles with the program PDFGetX351. The parameters for PDF calculation (background subtraction, scale factor, minimum and maximum values of Q, degree of data-correction polynomial) were optimized on individual PDF profiles, to avoid large termination effects and preserve the signal to noise ratio. The Qmax values were 23.7 Å−1 for Pb3S2Cl2 and 22.0 Å−1 for Pb4S3Br2 and Pb4S3I2.
Characterization: NCs structure solution and refinement
The solution and refinement of the Pb3S2Cl2 NCs structure was performed through a combination of 3D-ED, XRPD, and PDF analyses. 3D-ED37,52 was used to determine the starting unit cell parameters and a set of possible space groups. In the process, the software ADT3D53 was used. XRPD was exploited to select the most promising space groups for the structure solution, extract the integrated intensities of reflections and perform the ab-initio structure solution by Direct Methods. In the process, the software EXPO201438 was used. Both XRPD and DFT data were exploited to refine the structure of NCs, by alternating the refinement in the direct (PDF) and reciprocal (XRPD) spaces. To analyze the PDF profiles, the PDFGUI44,54,55 and DiffPy-CMI16 software packages were used. The same refinement strategy was applied to Pb4S3Br2 and Pb4S3I2 NCs, while the structure solution step was not needed because the starting structure was already known25,30. All the crystal structure illustrations shown in this work were produced by using the software VESTA56. Additional details are provided in the Supplementary Discussion, section 4.
Characterization: optical spectroscopies
Absorbance and photoluminescence (PL) spectra of solution NCs and heterostructures were recorded on a Cary 500 spectrophotometer and Cary Eclipse spectrofluorometer, respectively. The spectra were measured in 10 × 10 mm quartz cuvettes. A wavelength of 350 nm was used for excitation during the collection of PL spectra. Regarding the extended spectroscopy: for PL and transient PL measurements, the samples were excited using a Laser-export Co. Ltd., frequency tripled, pulsed Nd:YAG laser at 3.49 eV (355 nm) and at 2.33 eV (532 nm) with 1 kHz repetition rate and detected respectively using an Hamamatsu Mini-Spectrometer and an Oriel Instrument Cornerstone 1/4 m monochromator coupled with a Hamamatsu UV–VIS photomultiplier tube. Cryogenic Measurements were performed in a liquid nitrogen-cooled Oxford Instruments Limited cryostat.
DFT: comparison of monoclinic and cubic Pb3S2Cl2 structures
To assess the impact of the cubic symmetry breaking on the stability of Pb3S2Cl2 phase, we compared by DFT calculations the stability of the monoclinic Pb3S2Cl2 structure that we solved with that of a hypothetic cubic Pb3S2Cl2 structure. We performed this investigation on the corresponding 2 × 2 × 2 supercells at the gamma point. In particular, we relaxed both atomic positions and cell parameters at the DFT level using the PBE exchange−correlation functional57, and a double-ζ basis set plus polarization functions (DZVP) on all atoms as implemented in the CP2K 8.1 package58. Scalar relativistic effects were incorporated as effective core potentials in the basis set. With this approach, we determined that the monoclinic Pb3S2Cl2 2 × 2 × 2 supercell is more stable than the prototypal cubic one by 164 kcal/mol.
DFT: calculations on NCs models
Both the structural relaxation and the electronic structure calculations of atomistic NCs models were carried out at the DFT/PBE/DZVP level of theory with CP2K. In order to identify any surface localized states that could trap charge carriers, we also computed the Inverse Participation Ratio (IPR) for chalcohalide and perovskite, both in the form of free-standing NCs and in the heterostructure. The IPR, as also demonstrated for other NCs26,59, quantifies the orbital localization of a given molecular orbital and it is defined as:
Here, Pα,i represents the weight of molecular orbital i on a given atom α expanded in an atomic orbital basis. For finite systems, the IPR provides an estimate of the number of atoms that contribute to a given electronic state i. It can range from the inverse of the number of atoms in the system (when the wave function is distributed equally over all atoms in the system) to 1 in the case of states localized on single atoms. In other terms, IPR values very close to 0 identify delocalized states.
Data availability
The data obtained from 3D-ED, X-ray diffraction, HRSTEM column intensity analysis, optical spectroscopy, particle size distribution analysis, the results of the DFT calculations, and the structure models generated in this study have been deposited in the Zenodo database under accession code https://doi.org/10.5281/zenodo.6430001. The data obtained from X-ray diffraction, HRSTEM column intensity analysis, optical spectroscopy, particle size distribution analysis, and the results of the DFT calculations generated in this study are provided in the Source Data file. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CSD 2181723 (Pb3S2Cl2), 2181721 (Pb4S3Br2) and 2181722 (Pb4S3I2). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. An atomistic model of the Pb4S3Cl2/CsPbCl3 epitaxial interface is provided as a Supplementary Data file (Supplementary Data 1.xyz). Source data are provided with this paper.
References
Antonii, F. Panacea Aurea-Auro Potabile. (Bibliopolio Frobeniano, 1618).
Xia, Y., Xiong, Y., Lim, B. & Skrabalak, S. E. Shape-controlled synthesis of metal nanocrystals: simple chemistry meets complex physics? Angew. Chem. - Int. Ed. 48, 60–103 (2009).
Lee, J., Zhang, S. & Sun, S. High-temperature solution-phase syntheses of metal-oxide nanocrystals. Chem. Mater. 25, 1293–1304 (2013).
Cargnello, M., Gordon, T. R. & Murray, C. B. Solution-phase synthesis of titanium dioxide nanoparticles and nanocrystals. Chem. Rev. 114, 9319–9345 (2014).
Tamang, S., Lincheneau, C., Hermans, Y., Jeong, S. & Reiss, P. Chemistry of InP nanocrystal syntheses. Chem. Mater. 28, 2491–2506 (2016).
Lu, H., Carroll, G. M., Neale, N. R. & Beard, M. C. Infrared quantum dots: progress, challenges, and opportunities. ACS Nano 13, 939–953 (2019).
Mcdonald, S. A. et al. Solution-processed PbS quantum dot infrared photodetectors and photovoltaics. Nat. Mater. 4, 138–142 (2005).
Kolny-Olesiak, J. & Weller, H. Synthesis and application of colloidal CuInS2 semiconductor nanocrystals. ACS Appl. Mater. Interfaces 5, 12221–12237 (2013).
Gréboval, C. et al. Mercury chalcogenide quantum dots: material perspective for device integration. Chem. Rev. 121, 3627–3700 (2021).
Murray, C. B., Norris, D. J. & Bawendi, M. G. Synthesis and characterization of nearly monodisperse CdE (E = S, Se, Te) semiconductor nanocrystallites. J. Am. Chem. Soc. 115, 8706–8715 (1993).
Kim, S., Fisher, B., Eisler, H. J. & Bawendi, M. Type-II quantum dots: CdTe/CdSe(core/shell) and CdSe/ZnTe(core/shell) heterostructures. J. Am. Chem. Soc. 125, 11466–11467 (2003).
Dey, A. et al. State of the art and prospects for halide perovskite nanocrystals. ACS Nano 15, 10775–10981 (2021).
Akkerman, Q. A., Rainò, G., Kovalenko, M. V. & Manna, L. Genesis, challenges and opportunities for colloidal lead halide perovskite nanocrystals. Nat. Mater. 17, 394–405 (2018).
Talapin, D. V., Lee, J. S., Kovalenko, M. V. & Shevchenko, E. V. Prospects of colloidal nanocrystals for electronic and optoelectronic applications. Chem. Rev. 110, 389–458 (2010).
Losch, P. et al. Colloidal nanocrystals for heterogeneous catalysis. Nano Today 24, 15–47 (2019).
Carey, G. H. et al. Colloidal quantum dot solar cells. Chem. Rev. 115, 12732–12763 (2015).
Mitchell, M. J. et al. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 20, 101–124 (2020).
Park, Y. S., Roh, J., Diroll, B. T., Schaller, R. D. & Klimov, V. I. Colloidal quantum dot lasers. Nat. Rev. Mater. 6, 382–401 (2021).
Toso, S., Baranov, D. & Manna, L. Metamorphoses of cesium lead halide nanocrystals. Acc. Chem. Res. 54, 498–508 (2021).
Shamsi, J. et al. Colloidal CsX (X = Cl, Br, I) nanocrystals and their transformation to CsPbX3 nanocrystals by cation exchange. Chem. Mater. 30, 79–83 (2018).
Xu, J. et al. Ultrasonically-prepared copper-doped cesium halide nanocrystals with bright and stable emission. Nanoscale 13, 9659–9667 (2021).
Klein, E., Lesyuk, R. & Klinke, C. Insights into the formation mechanism of two-dimensional lead halide nanostructures. Nanoscale 10, 4442–4451 (2018).
Akkerman, Q. A. et al. Nearly monodisperse insulator Cs4PbX6 (X = Cl, Br, I) nanocrystals, their mixed halide compositions, and their transformation into CsPbX3 Nanocrystals. Nano Lett. 17, 1924–1930 (2017).
Protesescu, L. et al. Nanocrystals of cesium lead halide perovskites (CsPbX3, X = Cl, Br, and I): novel optoelectronic materials showing bright emission with wide color gamut. Nano Lett. 15, 3692–3696 (2015).
Toso, S. et al. Nanocrystals of lead chalcohalides: a series of kinetically trapped metastable nanostructures. J. Am. Chem. Soc. 142, 10198–10211 (2020).
Imran, M. et al. Halide perovskite-lead chalcohalide nanocrystal heterostructures. J. Am. Chem. Soc. 143, 1435–1446 (2021).
Rousseau, G. & Breit, B. Removable directing groups in organic synthesis and catalysis. Angew. Chem. - Int. Ed. 50, 2450–2494 (2011).
Ider, A., Laval, J. P., Frit, B., Carré, J. & Bastide, J. P. Crystal structure of PbTeF6. J. Fluor. Chem. 78, 15–20 (1996).
Krebs, B. Die Kristallstrukturen von Pb4SeBr6, Pb5S2J6 und Pb7S2Br10. ZAAC ‐. J. Inorg. Gen. Chem. 396, 137–151 (1973).
Ni, D., Guo, S., Yang, Z. S., Powderly, K. M. & Cava, R. J. Pb4S3I2 –a high-pressure phase in the PbS-PbI2 system. Solid State Sci. 91, 49–53 (2019).
Ni, D., Guo, S., Powderly, K. M., Zhong, R. & Cava, R. J. A high-pressure phase with a non-centrosymmetric crystal structure in the PbSe–PbBr2 system. J. Solid State Chem. 280, 106–111 (2019).
Toso, S., Baranov, D. & Manna, L. Hidden in plain sight: the overlooked influence of the Cs+ substructure on transformations in cesium lead halide nanocrystals. ACS Energy Lett. 5, 3409–3414 (2020).
Graham, C. et al. NaCl substrates for high-temperature processing and transfer of ultrathin materials. Sci. Rep. 10, 7253 (2020).
Dish, N., Behera, R., Satyaprasad, A. & Gautam, A. Epitaxial growth of copper, gold, and silver on vicinal NaCl surfaces. Appl. Surf. Sci. 574, 151643 (2022).
Dong, W. J., Kim, S., Park, J. Y., Yu, H. K. & Lee, J.-L. Ultrafast and chemically stable transfer of au nanomembrane using a water-soluble NaCl sacrificial layer for flexible solar cells. ACS Appl. Mater. Interfaces 11, 30477–30483 (2019).
Lee, D. K. et al. Water-soluble epitaxial NaCl thin film for fabrication of flexible devices. Sci. Rep. 7, 1–7 (2017).
Gemmi, M. et al. 3D electron diffraction: the nanocrystallography revolution. ACS Cent. Sci. 5, 1315–1329 (2019).
Altomare, A. et al. EXPO2013: a kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 46, 1231–1235 (2013).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 32, 751–767 (1976).
Winslow, S. W., Liu, Y., Swan, J. W. & Tisdale, W. A. Quantification of a PbClx shell on the surface of PbS nanocrystals. ACS Mater. Lett. 1, 209–216 (2019).
Green, P. B. et al. PbS nanocrystals made using excess lead chloride have a halide-perovskite-like surface. Chem. Mater. 10, 27 (2021).
Green, P. B., Li, Z. & Wilson, M. W. B. PbS nanocrystals made with excess PbCl2 have an intrinsic shell that reduces their Stokes shift. J. Phys. Chem. Lett. 10, 5897–5901 (2019).
Brittman, S. et al. Effects of a lead chloride shell on lead sulfide quantum dots. J. Phys. Chem. Lett. 10, 1914–1918 (2019).
Caliandro, R. & Belviso, D. B. RootProf: software for multivariate analysis of unidimensional profiles. J. Appl. Crystallogr. 47, 1087–1096 (2014).
Zheng, Y. et al. All-inorganic dual-phase halide perovskite nanorings. Nano Res 13, 2994–3000 (2020).
Huang, Z. P. et al. In situ growth of 3D/2D (CsPbBr3/CsPb2Br5) perovskite heterojunctions toward optoelectronic devices. J. Phys. Chem. Lett. 11, 6007–6015 (2020).
Akkerman, Q. A. et al. Tuning the optical properties of cesium lead halide perovskite nanocrystals by anion exchange reactions. J. Am. Chem. Soc. 137, 10276–10281 (2015).
De Backer, A., van den Bos, K. H. W., Van den Broek, W., Sijbers, J. & Van Aert, S. StatSTEM: an efficient approach for accurate and precise model-based quantification of atomic resolution electron microscopy images. Ultramicroscopy 171, 104–116 (2016).
Gemmi, M. & Lanza, A. E. 3D electron diffraction techniques. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 75, 495–504 (2019).
Hammersley, A. P., Svensson, S. O., Hanfland, M., Fitch, A. N. & Häusermann, D. Two-dimensional detector software: from real detector to idealised image or two-theta scan. High. Press. Res. 14, 235–248 (1996).
Juhás, P., Davis, T., Farrow, C. L. & Billinge, S. J. L. PDFgetX3: a rapid and highly automatable program for processing powder diffraction data into total scattering pair distribution functions. J. Appl. Crystallogr. 46, 560–566 (2013).
Yun, Y., Zou, X., Hovmöller, S. & Wan, W. Three-dimensional electron diffraction as a complementary technique to powder X-ray diffraction for phase identification and structure solution of powders. IUCrJ 2, 267–282 (2015).
Kolb, U., Krysiak, Y. & Plana-Ruiz, S. Automated electron diffraction tomography—development and applications. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 75, 463–474 (2019).
Farrow, C. L. et al. PDFfit2 and PDFgui: Computer Programs for Studying Nanostructure in Crystals. J. Phys. Condens. Matter 19, 335219 (2007).
Juhás, P., Farrow, C. L., Yang, X., Knox, K. R. & Billinge, S. J. L. Complex modeling: a strategy and software program for combining multiple information sources to solve Ill posed structure and nanostructure inverse problems. Acta Crystallogr. Sect. A Found. Adv. 71, 562–568 (2015).
Momma, K. & Izumi, F. VESTA: a three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 41, 653–658 (2008).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kühne, T. D. et al. CP2K: An electronic structure and molecular dynamics software package - Quickstep: efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
Houtepen, A. J., Hens, Z., Owen, J. S. & Infante, I. On the origin of surface traps in colloidal II-VI semiconductor nanocrystals. Chem. Mater. 29, 752–761 (2017).
Acknowledgements
The authors would like to acknowledge Dr. Joka Buha for the help with preliminary tests preceding this project, and Dr. B. M. Aresta and Dr. L. Cassano for their administrative support. The authors acknowledge financial support from the Research Foundation - Flanders (FWO) through a postdoctoral fellowship to N.J.S. (FWO Grant No. 1238622N, N.J.S). S.B. acknowledges financial support from the European Commission by ERC Consolidator grant REALNANO (No. 815128, S.B.). L.M. acknowledges financial support from the Italian Ministry of University and Research (MIUR) through the Flag-Era JTC2019 project “Solution-Processed Perovskite/Graphene Nanocomposites for Self-Powered Gas Sensors” (PeroGaS, L.M.). The access to the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886 (NSLS-II Proposal Number 307441).
Author information
Authors and Affiliations
Contributions
S.T. synthesized and characterized Pb3S2Cl2, Pb4S3Br2, and Pb4S3I2 NCs. M.I. and Y.W. synthesized and characterized Pb4S3Cl2/CsPbCl3 heterostructures and Pb4S3Cl2 NCs. S.T., E.M., A.M, R.C., and C.G. performed the structures solution and refinement. J.Z., F.Z., and I. I. performed the DFT simulations. N.S. performed the HRSTEM and the HAADF-STEM characterizations. A.P performed the spectroscopic characterizations. M.G., C.G., S. Brovelli, I.I, S. Bals, and L.M. coordinated the research work. S.T., L.M., and M.I. wrote the manuscript. All the authors contributed to reviewing and editing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rajesh Sardar and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Toso, S., Imran, M., Mugnaioli, E. et al. Halide perovskites as disposable epitaxial templates for the phase-selective synthesis of lead sulfochloride nanocrystals. Nat Commun 13, 3976 (2022). https://doi.org/10.1038/s41467-022-31699-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-31699-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
