
Abstract
Lithium intercalation of MoS2 is generally believed to introduce a phase transition from H phase (semiconducting) to T phase (metallic). However, during the intercalation process, a spatially sharp boundary is usually formed between the fully intercalated T phase MoS2 and non-intercalated H phase MoS2. The intermediate state, i.e., lightly intercalated H phase MoS2 without a phase transition, is difficult to investigate by optical-microscope-based spectroscopy due to the narrow size. Here, we report the stabilization of the intermediate state across the whole flake of twisted bilayer MoS2. The twisted bilayer system allows the lithium to intercalate from the top surface and enables fast Li-ion diffusion by the reduced interlayer interaction. The E2g Raman mode of the intermediate state shows a peak splitting behavior. Our simulation results indicate that the intermediate state is stabilized by lithium-induced symmetry breaking of the H phase MoS2. Our results provide an insight into the non-uniform intercalation during battery charging and discharging, and also open a new opportunity to modulate the properties of twisted 2D systems with guest species doping in the Moiré structures.
Similar content being viewed by others
Introduction
Molybdenum disulfide (MoS2), as one of the most well-known layered transition metal dichalcogenides (TMDs) with stable semiconducting H phase and metastable metallic T phase1,2,3, has attracted intensive research interest4. Considerable effort has been devoted to studying the H phase MoS2 in the fields of electronics, photonics, etc5,6. The T phase has a wide variety of applications such as catalysis, energy storage, superconductivity, etc7,8,9,10, but the metastable nature makes it difficult to synthesis and store1. Therefore, various methods have been developed to induce the H to T phase transition in MoS211,12, and these methods have flourished with the advances in atomically-thin 2D materials in recent decades13,14,15,16,17. Among these methods, lithium intercalation has been one of the most popular approaches18,19. Since 1983, it was found that the phase transition occurs in LixMoS2 when 0.2 < x < 111,12. Until now, both electrochemical and chemical methods have been developed for lithium intercalation of MoS220,21. Electrochemical methods are widely used to provide dynamical and reversible control of the lithium intercalation of MoS211. Chemical intercalation provides a simple approach, by soaking the MoS2 into highly reducible lithium solution (e.g., n-Butyllithium), where the lithium atoms can spontaneously move into the van der Waals gaps of the MoS222. It was generally believed that lithium diffuses into MoS2 through the edge13,14. Recently, it was discovered that the lithium can also can go through across the layers via defects in thin flakes15.
In recent years, a number of investigations have been performed with state-of-art characterization techniques (spectroscopic methods in particular) to study lithium intercalation of MoS213,14,23,24,25. The in-situ observation of dynamic lithium intercalation into MoS2 under potential control by using differential optical microscopy, discovered that the intercalation starts from the edge, and distinct phase separation of lithiated and delithiated regions is observed14. Similar with the electrochemical intercalation, chemical intercalation also shows a distinct phase separation with a very narrow boundary region (~few hundreds of nanometers) (Fig. 1a and Supplementary Fig. 1)13,14,23. The intermediate state, with lithium intercalation in H phase MoS2 before phase transformation, is only a few hundreds of nanometers wide, which is barely studied by optical-microscopy-based techniques due to the narrow size.

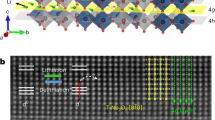
a Lithium intercalation in bilayer 2H-MoS2 through the edge, resulting a phase separation (left side: top view (view along the c-axis), right side: cross-section view). b Lithium intercalation in twisted bilayer MoS2 (left side: top view (view along the c-axis), right side: cross-section view).
The lithium diffusion in MoS2 is an interplay between charge-transfer process at the step and the force that pushes a row of Li ions into the interior of the basal plane of the crystal14. Thus, the sharp domain boundaries never show a gradual decrease in contrast with time14. The bilayer system provides a platform for uniform intercalation from the top surface, and the reduced interlayer interaction in the Moiré structure allows a fast diffusion of the lithium ions. The fast ion diffusion in Moiré structure originates from the reduced interlayer binding forces26. Therefore, instead of the formation of distinct phase boundary, the intermediate stated can be introduced in a whole flake (Fig. 1b). Here, by using twisted bilayer MoS2, the intermediate state can be induced in an entire flake and was signaled by a splitting of the E2g mode in Raman spectroscopy. Our discovery offers new insight into the diffusion problem in batteries27,28, and also offers new possibilities to twistronics by introducing guest species in the Moiré superlattice29,30.
Results and discussion
The twisted bilayer MoS2 was fabricated by transferring one chemical vapor deposition (CVD) grown monolayer MoS2 onto another one. The transfer process was performed by PMMA- and PDMS- assisted target transfer (Supplementary Fig. 2)31. The twisting angle can be identified by the edge of the triangular CVD grown MoS2. Before intercalation, Raman spectroscopy was conducted on three different positions (Fig. 2a). Position 1 and 2 are monolayer regions on the bottom and top monolayer flakes, respectively, and position 3 is a twisted bilayer region (negligible variations of the Raman spectra in the same region were observed). As shown in the Raman spectra (Fig. 2b), before intercalation, the E2g peak was located at ~386.1 cm−1 and the A1g peak was at ~404.3 cm−1 in both of the monolayer regions. The ~18 cm−1 distance between the two modes reflected the monolayer H phase32. In the bilayer region, the E2g peak showed a slight blueshift to ~385.5 cm−1 and the position of the A1g peak showed negligible change. The twisting angle of 32° implies a relatively narrow E2g and A1g mode frequency difference in the bilayer region33. Twisted bilayer MoS2 flakes with various angles can be obtained in one transfer process (Supplementary Fig. 3).

a Optical microscope image of a twisted bilayer MoS2 (scale bar 10 μm). b Raman spectra of the positions 1, 2, and 3 in a before intercalation. c Raman spectra of the same positions after intercalation. d Zoom-in image of the E2g and A1g peaks after intercalation.
Chemical intercalation was then conducted on the bilayer system by soaking the samples in dilute n-Butyllithium solution. After intercalation, no strong color change was observed under the optical microscope. Raman spectroscopy was performed again on positions 1, 2, and 3. It can be seen that after exposure to n-Butyllithium, both of the E2g modes in monolayer region positions 1 and 2 shifted to smaller wavenumber. The shift of the E2g mode resulted from the charge transfer between lithium and monolayer MoS2. The E2g mode in the top layer showed a larger shift (~384 cm−1) than that in the bottom layer (~385 cm−1), which is probably due to more defects in the transferred top layer which induced more lithium absorption. The difference of the E2g peak shift in the top and bottom monolayer region can be reduced by transferring two monolayers to ensure that every layer experiences the same transfer process (Supplementary Fig. 4). The frequencies of the A1g mode in all three regions show negligible change after intercalation due to the low concentration of lithium during intercalation. Remarkably, the E2g mode in the twisted bilayer region showed a striking splitting behavior (intermediate state): the original E2g peak at ~385.5 cm−1 was split into two peaks at ~383 cm−1 and 386.6 cm−1 respectively. Although the E2g modes showed slightly different shifts in positions 1 and 2, it is clear that the two split peaks in region 3 were not a simple combination of the two monolayer peaks due to the differences in frequency.
To reveal the Raman spectra evolution during intercalation, we immersed a twisted bilayer MoS2 in the n-butyllithium solution for different lengths of time (Fig. 3) and examined the Raman spectra variations. During the whole intercalation process, both the E2g and A1g peak were softened. As we began the lithium intercalation, the E2g peak is slightly broadened in the first 5 min. After 10 min, the relative intensity of the E2g mode was softened and the onset of the peak splitting was observed. More intercalation time resulted in a clear splitting and broadening of the E2g mode. After 20 min of intercalation, we started to see the J modes (J1, J2, and J3) in T phase MoS2 and the peak splitting of the E2g mode disappeared. Finally, after 30 min, the J modes were clear, indicating a complete phase transition from H to T of the intercalated MoS2.

a Raman spectra of one twisted bilayer MoS2 for different intercalation times. The variation of the frequency (b) and FWHM (c) of E2g and split E2g’ modes as a function of the intercalation time.
The dependence of the frequencies of the E2g peak and the split peak (which we designate as E2g’ here) on intercalation time were plotted in Fig. 3b. The peak splitting was observed after 5 min of intercalation and both the E2g and E2g’ peak shifted to low frequency with the increase of intercalation time. The split E2g’ peak suddenly vanished when the phase transited to 1 T, suggesting that the peak splitting was only in the H phase MoS2. After the formation of 1 T phase MoS2, the E2g peak started to shift to a higher frequency with a higher concentration of intercalated lithium. The value of the full-width-at-half-maximum (FWHM) of E2g and E2g’ were extracted from the spectra (Fig. 3c). In general, intercalation induced a broad shear mode, which could be caused by the increased electron–phonon coupling. Around the phase transition time, the FWHM of E2g peak showed some variations, likely caused by disorder during the phase transition.
To further characterize the samples in the intermediate state, photoluminescence (PL) was conducted on a twisted bilayer MoS2 in intrinsic, intermediate, and 1 T states (Fig. 4a). The PL spectra beyond 750 nm of another sample are shown in Supplementary Fig. 5, where an indirect bandgap peak can be observed34. The ratio of A and B excitons and the position of the indirect bandgap peak of the twisted bilayer MoS2 are angle-dependent, which is related to the interlayer coupling31,34. The location of A excitons was at ~670 nm, indicating the bandgap of the twisted bilayer MoS2 was ~1.85 eV. For the intermediate state, the intensity of the PL became much weaker with a redshift of the A exciton, signifying a narrower bandgap, but also indicating that the MoS2 still maintained the semiconducting H phase. It should be noted that some lithium residue on the surface of MoS2 could affect the PL intensity, but the H phase was not affected by a degree of surface doping35. Another twisted bilayer MoS2 sample was fabricated as a field-effect transistor to study the variation of conductivity (Fig. 4b insert). By sweeping the gate voltage from −60 V to 60 V, the source-drain current was measured with a constant bias voltage of 1 V. Before intercalation, the device showed typical n-type semiconductor behavior. For the intermediate state, the sample still maintained n-type behavior with one order of magnitude enhancement of the conductivity at zero gate voltage. The fully intercalated T phase twisted bilayer MoS2 showed a constant conductivity regardless of the gate voltage (additional electrical characterization at different source-drain voltages can be seen in Supplementary Fig. 6). Cryogenic transmission electron microscopy (TEM) was used to characterize the twisted bilayer samples after 15 min of intercalation. The Moiré pattern was observed under high-resolution TEM and confirmed by the two series of MoS2 lattices with a twist angle in the electron diffraction pattern. However, given both the low concentration of intercalated lithium and relatively small atomic number of lithium, the lithium was not directly observable in the TEM. Nevertheless, electron energy-loss spectroscopy (EELS) shows that lithium is incorporated in the bilayer system (Supplementary Fig. 7).

a Photoluminescence of a twisted bilayer MoS2 sample before intercalation, in the intermediate state, and after transformation to the 1 T phase. b Room-temperature transfer characteristics for a twisted bilayer MoS2 FET before intercalation, in the intermediate state, and after transformed to 1 T phase with 1 V applied bias voltage (the width of the contact is 10 μm). c High resolution cryo-TEM image (scale bar: 2 nm) of a twisted bilayer MoS2 after 15 min lithium intercalation and d the corresponding electron diffraction pattern (scale bar: 10 nm−1).
The splitting of the shear mode was observed in lithium intercalation of graphite but never seen in MoS236,37. In graphite, the intercalants shift the frequency of the shear mode, and incomplete intercalation induced a mixture of bare and intercalated graphite, which results in the observed double peak30. However, the situation is different with our twisted bilayer MoS2 case since we have uniform lithium intercalation in the bilayer system without a staging effect. Recent calculations showed that in twisted bilayer systems, some high-symmetry points exist in the Moiré superlattice that is energy favorable for lithium38,39. When lithium goes into the twist bilayer MoS2, it tends to fill these specific regions of the Moiré superlattice and then distribute to other equivalent locations. To understand the physics behind these phenomenon, first-principles calculations were conducted based on density function theory (DFT) to simulate the phonon spectrum.
Since the intermediate state was observed at different angles, we started from a twisted bilayer Moiré supercell with a random twisting angle ((3,4)-configuration with a twist angle of 9.43°, Supplementary Fig. 8a). To study the effect of Li intercalation in twisted bilayer MoS2, we constructed two additional models where one Li atom is placed in between the twisted bilayers and the stacking order locally is approximately MX (Supplementary Fig. 8b) and MM (Supplementary Fig. 8c) respectively. MX (MM) refers to the stacking order where the metal atom in the top MoS2 layer is directly on top of the chalcogenide (metal) atom in the bottom MoS2 layer40. The MX and MM sites locally possess approximate hexagonal symmetry as in the untwisted MoS2 bilayer. The Li atom is placed at a tetrahedrally coordinated site bonded with four S atoms. The phonon density of states (PDOS) of the pristine twisted bilayer MoS2 and the two Li-intercalated configurations were calculated and plotted in Supplementary Fig. 8d–f. The PDOS for the Li-intercalated twisted bilayer MoS2 showed a significant broadening across the frequency spectrum compared with that of the pristine twisted bilayer MoS2. This indicates that the presence of the Li atom reduces the symmetry of the lattice locally, increasing the anharmonicity of vibrational eigenmodes of the whole lattice. On the other hand, due to the very low Li concentration in these configurations (<2%), the frequencies of the eigenmodes of MoS2 vibrations in the Li-intercalated models do not differ noticeably compared to the pristine MoS2 case.
However, it is challenging to predict the phonon dispersion with more lithium in the Moiré supercell, because the variations of lithium positions and phonon DOSs are exponentially sensitive to the amount of lithium in the system. Therefore, we simulate an extreme case: 180° rotation, i.e., 2H bilayer MoS2 (Fig. 5a). Usually, when the H phase MoS2 is fully lithiated, a phase transition from H to T is unavoidable, yet here we consider the structure of the H phase so that the effect of lithium intercalation in the H phase MoS2 can be examined. The phonon DOS of pure bilayer 2H-MoS2 was calculated and shown by the blue curve in Fig. 5c. The calculated E2g and A1g modes were located at ~374 cm−1 and 406 cm−1 respectively. The A1g mode is singly degenerate and the E2g mode is doubly degenerate due to the lattice symmetry. For fully intercalated 2H MoS2, each lithium atom occupies a tetrahedrally coordinated site bonded with four S atoms, where the local stacking order is MX (Fig. 5b). In the DOS of the fully lithiated 2H MoS2, the E2g mode was split into two peaks located at 341 cm−1 and 353 cm−1 with a large shift and the broadening. In fact, the curves in Fig. 5c were smoothed from the raw data in Supplementary Fig. 9, and the two split peaks originated from a combination of multiple modes. The symmetry-breaking caused by lithium in the H phase MoS2 induced the formation of multiple modes. The simulated atomic vibrational movements associated with the E2g mode before and after intercalation is shown in the Supplementary Movie 1. In Li-MoS2, the lower frequency in-plane modes correspond to the vibration of the upper layer MoS2, and higher frequency in-plane modes corresponds to the bottom layer vibration. The A1g mode was extinguished in the fully lithiated 2H-MoS2 due to the dissociation of interlayer bonding due to lithium, which is similar with the 1 T MoS2 with lithium13. Consequently, we believe that the splitting of the E2g mode was caused by the lithium-induced symmetry breaking in MoS2. The twist structure is not thermodynamically essential for the peaking splitting behavior. Hence, we should expect the peaking splitting behavior in non-twisted bilayer MoS2.

Schematic illustration of bilayer 2H-MoS2 (a) and fully intercalated bilayer 2H-MoS2 (b). c The calculated phonon DOS of the structure in a and b. d CVD grown bilayer 2H and 3R MoS2, and the corresponding Raman spectra in the intrinsic and intermediate state (scale bar: 10 μm). e The intercalation time required for phase transition of CVD grown 2H/3R bilayer MoS2 and transferred twisted bilayer MoS2. The error bars are standard deviations based on three samples for each condition.
The intermediate state was observed in both 2H and 3R MoS2 (Fig. 5d). However, since the Li ion diffusion in the CVD grown bilayers is much slower than that in the twisted bilayer MoS2, the time required for the observation of CVD bilayers shows large variations, and a sharp 2H/1T boundary can be observed in some samples due to the large amount of lithium intercalation through the edges. To qualitatively evaluate the ion diffusivity in MoS2, we summarized the time required for observing the phase transition in untwisted bilayers and twisted bilayers as shown in Fig. 5e. The intercalation time for the phase transition of the untwisted bilayer is over one order of magnitude longer than that of the twisted bilayer, suggesting a faster ion diffusion in the twisted bilayers. The peak splitting behavior can also be induced in the exfoliated bilayer MoS2 (see Supplementary Fig. 10). Therefore, although the twist structure is not thermodynamically necessary for the formation of peaking splitting, it facilities the fast kinetics of lithium diffusion, enabling an accessible observation of the phenomenon.
The intercalation time of the twisted bilayer MoS2 showed some variations, which is probably due to the variations of the twist angle. The twist angle not only defines the density of the localized energy favorable position38, but also controls the interlayer binding energy. A recent study shows that the ion diffusion in van der Waals interlayer is strongly related to the van der Waals interlayer expandability, and a higher interlayer expandability usually leads to a higher ion diffusivity26. Previous reports have demonstrated the angle dependence of the interlayer coupling of twisted bilayer MoS231,34, suggesting an angle dependence of the ion diffusion in twisted bilayer MoS2. However, due to the low controllability of chemical intercalation and the uncertainty of the measurement of ion diffusivity by optical methods, further efforts are needed to accurately investigate the dependence of twist angle and the kinetics of ion diffusion.
In conclusion, we have observed an intermediate state during lithium intercalation of twisted bilayer MoS2 by Raman spectroscopy. The bilayer structure allows lithium to intercalate from the top surface and the Moiré pattern facilitates the spread of lithium in MoS2. The splitting of the shear mode was caused by the lithium-induced symmetry breaking in MoS2. Our discovery is meaningful for understanding nonuniform intercalation in battery materials during fast charge and discharge41, and presents a potential spatially distributed doping technique for twistronics42.
Methods
Synthesis of Monolayer of MoS2
Monolayer MoS2 was synthesized by chemical evaporation deposition (CVD) on 300 nm SiO2/Si substrate with molybdenum oxide (MoO3) and sulfur as precursors, and argon as the carrier gas, at a growth temperature of 750 °C43. Sodium chloride (NaCl) can be used in the growth to improve the yield of monolayer growth44. The detailed transfer process is described in the Supplementary Note 1.
Chemical intercalation
The chemical intercalation was conducted by immersing the samples into 1.6 M n-Bbutyllithium hexane solution at room temperature in an argon-filled glovebox with oxygen and water concentration less than 0.1 ppm. After intercalation, samples were washed by anhydrous hexane for several times and dried by argon gas flow to remove organic residues. The intercalated samples were sealed in the argon glove box before transferring out for other characterizations.
Characterization
The Raman data was obtained by using a Horiba Labram HR Evolution Raman System with 532 nm laser wavelength and 1800 l/mm grading. The ideal resolution is ~1/3 cm−1 in the measurement range. The ideal laser spot size was 564 nm in diameter under a 100× objective. The FET devices were fabricated by e-beam lithography and e-beam evaporation of 3/50 nm Cr/Au to form contacts. The electrical tests were conducted on an Agilent B1500A Semiconductor Device Parameter Analyzer. Cryo-TEM experiments were carried out using a FEI Titan 80-300 aberration-corrected environmental TEM operated at 300 kV with Gatan 626 cryo-transfer holder. The TEM sample was prepared by first transferring the bilayers onto TEM grid, and then conducting intercalation. The TEM grid was transferred from glovebox to the TEM holder by using the cryo-transfer method45.
Density Functional Theory Calculations
The calculations were performed with the Density Functional Theory (DFT) code Quantum ESPRESSO46. The non-local functional vdW-df-cx47 is used as the exchange-correlation functional to describe the interlayer van der Waals interaction. The Projected Augmented Wave (PAW) pseudopotentials of PSlibrary1.048 are used to describe the effective core electron wavefunctions of Mo, S, and Li. The 7 × 7 × 1 k-point grid and the Gamma k-point are used for the (3,4) twisted MoS2 bilayer supercells and the 2H-MoS2 unit cells, respectively. The energy cutoff is chosen to be 680 eV. The energy and forces are converged to within 10−4 eV and 10−3 eV/Å respectively. The length of the vacuum is 15 Å to minimize artificial interlayer interaction due to periodicity of supercell. The phonon calculations are performed using Density Functional Perturbation Theory (DFPT)49 for the 2H-MoS2 unit cells, and finite displacement method for the (3,4) twisted MoS2 bilayer supercells. The 5 × 5 × 1 q-point grid is used for the DFPT calculations; the length of the displacement is 0.02 Å.
Data availability
The data that support the findings of this study are available from the authors upon reasonable request.
Code availability
Codes related to the DFT calculations are available from the authors upon reasonable request.
References
Wypych, F. & Schollhorn, R. 1T-MoS2, a New Metallic Modification of Molybdenum-Disulfide. J. Chem. Soc. Chem. Comm., 1386-1388 (1992).
Yang, D., Sandoval, S. J., Divigalpitiya, W. M. R., Irwin, J. C. & Frindt, R. F. Structure of single-molecular-layer MoS2. Phys. Rev. B 43, 12053–12056 (1991).
Edmondson, D. R. Electronic band structure of the layer-type crystal 2H-MoS2. Solid State Commun. 10, 1085–1088 (1972).
Manzeli, S., Ovchinnikov, D., Pasquier, D., Yazyev, O. V. & Kis, A. 2D transition metal dichalcogenides. Nat. Rev. Mater. 2, 17033 (2017).
Wang, Q. H., Kalantar-Zadeh, K., Kis, A., Coleman, J. N. & Strano, M. S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 7, 699–712 (2012).
Mak, K. F., Lee, C., Hone, J., Shan, J. & Heinz, T. F. Atomically thin MoS2: A new direct-gap semiconductor. Phys. Rev. Lett. 105, 136805 (2010).
Acerce, M., Voiry, D. & Chhowalla, M. Metallic 1T phase MoS2 nanosheets as supercapacitor electrode materials. Nat. Nanotechnol. 10, 313–318 (2015).
Wang, H. T. et al. Electrochemical tuning of vertically aligned MoS2 nanofilms and its application in improving hydrogen evolution reaction. P. Natl Acad. Sci. USA 110, 19701–19706 (2013).
Peng, J. et al. High phase purity of large-sized 1T’-MoS2 monolayers with 2D superconductivity. Adv. Mater. 31, 1900568 (2019).
Shi, S. L., Sun, Z. X. & Hu, Y. H. Synthesis, stabilization and applications of 2-dimensional 1T metallic MoS2. J. Mater. Chem. A 6, 23932–23977 (2018).
Py, M. A. & Haering, R. R. Structural destabilization induced by lithium intercalation in MoS2 and related-compounds. Can. J. Phys. 61, 76–84 (1983).
Imanishi, N., Toyoda, M., Takeda, Y. & Yamamoto, O. Study on lithium intercalation into MoS2. Solid State Ion. 58, 333–338 (1992).
Xiong, F. et al. Li intercalation in MoS2: In situ observation of its dynamics and tuning optical and electrical properties. Nano Lett. 15, 6777–6784 (2015).
Azhagurajan, M., Kajita, T., Itoh, T., Kim, Y. G. & Itaya, K. In situ visualization of lithium ion intercalation into MoS2 single crystals using differential optical microscopy with atomic layer resolution. J. Am. Chem. Soc. 138, 3355–3361 (2016).
Zhang, J. S. et al. Reversible and selective ion intercalation through the top surface of few-layer MoS2. Nat. Commun. 9, 5289 (2018).
Pandey, M., Bothra, P. & Pati, S. K. Phase transition of MoS2 bilayer structures. J. Phys. Chem. C. 120, 3776–3780 (2016).
Zhu, J. Q. et al. Argon plasma induced phase transition in monolayer MoS2. J. Am. Chem. Soc. 139, 10216–10219 (2017).
Benavente, E., Santa Ana, M. A., Mendizabal, F. & Gonzalez, G. Intercalation chemistry of molybdenum disulfide. Coord. Chem. Rev. 224, 87–109 (2002).
Woollam, J. A. & Somoano, R. B. Physics and chemistry of Mos2 intercalation compounds. Mater. Sci. Eng. 31, 289–295 (1977).
Jung, Y., Zhou, Y. & Cha, J. J. Intercalation in two-dimensional transition metal chalcogenides. Inorg. Chem. Front. 3, 452–463 (2016).
Wan, J. Y. et al. Tuning two-dimensional nanomaterials by intercalation: materials, properties and applications. Chem. Soc. Rev. 45, 6742–6765 (2016).
Dines, M. B. Lithium intercalation via n-Butyllithium of the layered transition metal dichalcogenides. Mater. Res. Bull. 10, 287–291 (1975).
Yoshida, K., Kawasaki, T., Kuwabara, A., Ukyo, Y. & Ikuhara, Y. In situ electron microscopic observation of electrochemical Li-intercalation into MoS2. Solid State Ion. 357, 115488 (2020).
Wang, L. F., Xu, Z., Wang, W. L. & Bai, X. D. Atomic mechanism of dynamic electrochemical lithiation processes of MoS2 nanosheets. J. Am. Chem. Soc. 136, 6693–6697 (2014).
Zou, J. L., Li, F. X., Bissett, M. A., Kim, F. & Hardwick, L. J. Intercalation behaviour of Li and Na into 3-layer and multilayer MoS2 flakes. Electrochim. Acta 331, 135284 (2020).
Zou, Y. C. et al. Ion exchange in atomically thin clays and micas. Nat. Mater. 20, 1677–1682 (2021).
Goodenough, J. B. & Kim, Y. Challenges for rechargeable Li batteries. Chem. Mater. 22, 587–603 (2010).
Hu, Z., Liu, Q., Chou, S.-L. & Dou, S.-X. Two-dimensional material-based heterostructures for rechargeable batteries. Cell Rep. Phys. Sci. 2, 100286 (2021).
Carr, S. et al. Twistronics: Manipulating the electronic properties of two-dimensional layered structures through their twist angle. Phys. Rev. B 95, 075420 (2017).
Cao, Y. et al. Unconventional superconductivity in magic-angle graphene superlattices. Nature 556, 43–50 (2018).
Huang, S. X. et al. Probing the interlayer coupling of twisted bilayer MoS2 using photoluminescence spectroscopy. Nano Lett. 14, 5500–5508 (2014).
Li, H. et al. From bulk to monolayer MoS2: Evolution of Raman scattering. Adv. Funct. Mater. 22, 1385–1390 (2012).
Lin, M.-L. et al. Moiré phonons in twisted bilayer MoS2. ACS Nano 12, 8770–8780 (2018).
Liu, K. H. et al. Evolution of interlayer coupling in twisted molybdenum disulfide bilayers. Nat. Commun. 5, 4966 (2014).
Li, Y., Duerloo, K. A. N., Wauson, K. & Reed, E. J. Structural semiconductor-to-semimetal phase transition in two-dimensional materials induced by electrostatic gating. Nat. Commun. 7, 10671 (2016).
Sole, C., Drewett, N. E. & Hardwick, L. J. In situ Raman study of lithium-ion intercalation into microcrystalline graphite. Faraday Discuss. 172, 223–237 (2014).
Zou, J. L., Sole, C., Drewett, N. E., Velicky, M. & Hardwick, L. J. In situ study of Li Intercalation into highly crystalline graphitic flakes of varying thicknesses. J. Phys. Chem. Lett. 7, 4291–4296 (2016).
Lu, Z. Y., Carr, S., Larson, D. T. & Kaxiras, E. Lithium intercalation in MoS2 bilayers and implications for moire flat bands. Phys. Rev. B 102, 125424 (2020).
Larson, D. T., Carr, S., Tritsaris, G. A. & Kaxiras, E. Effects of lithium intercalation in twisted bilayer graphene. Phys. Rev. B 101, 075407 (2020).
Weston, A. et al. Atomic reconstruction in twisted bilayers of transition metal dichalcogenides. Nat. Nanotechnol. 15, 592–597 (2020).
Liu, Y. Y., Zhu, Y. Y. & Cui, Y. Challenges and opportunities towards fast-charging battery materials. Nat. Energy 4, 540–550 (2019).
Andrei, E. Y. & MacDonald, A. H. Graphene bilayers with a twist. Nat. Mater. 19, 1265–1275 (2020).
Wu, Y. et al. A two-dimensional MoS2 catalysis transistor by Solid-State Ion Gating Manipulation and Adjustment (SIGMA). Nano Lett. 19, 7293–7300 (2019).
Zhou, J. D. et al. A library of atomically thin metal chalcogenides. Nature 556, 355–359 (2018).
Li, Y. Z. et al. Atomic structure of sensitive battery materials and Interfaces revealed by cryo-electron microscopy. Science 358, 506–510 (2017).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Berland, K. & Hyldgaard, P. Exchange functional that tests the robustness of the plasmon description of the van der Waals density functional. Phys. Rev. B 89, 035412 (2014).
Dal Corso, A. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci. 95, 337–350 (2014).
Baroni, S., de Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001).
Acknowledgements
This work was supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Materials Sciences and Engineering (Contract No. DE-AC02-76SF00515). Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152.
Author information
Authors and Affiliations
Contributions
Y.W., H.Y.H., and Y.C. conceived the project. Y.W. carried out the materials synthesis, optical microscopy, Raman spectroscopy, and diffusion model calculation. J.W. and L.W.W. conducted the DFT simulation and calculation. Y.L. performed TEM measurements. B.Y.W. assisted the electrical tests. J.Z. assisted the Raman characterization. A.Y. assisted the chemical intercalation. Y.W., J.W., H.Y.H., and Y.C. cowrote the manuscript, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Penghong Ci, and the other, anonymous, reviewer for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Y., Wang, J., Li, Y. et al. Observation of an intermediate state during lithium intercalation of twisted bilayer MoS2. Nat Commun 13, 3008 (2022). https://doi.org/10.1038/s41467-022-30516-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-30516-z
This article is cited by
-
Direct visualization of stacking-selective self-intercalation in epitaxial Nb1+xSe2 films
Nature Communications (2024)
-
Moiré superlattice engineering of two-dimensional materials for electrocatalytic hydrogen evolution reaction
Nano Research (2023)
-
Chemical vapor deposition synthesis of V-doped MoS2
Rare Metals (2023)
-
Regulating the Electrical and Mechanical Properties of TaS2 Films via van der Waals and Electrostatic Interaction for High Performance Electromagnetic Interference Shielding
Nano-Micro Letters (2023)
-
Electrostatic gating and intercalation in 2D materials
Nature Reviews Materials (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
