
Abstract
The C-type lectin receptor Mincle is known for its important role in innate immune cells in recognizing pathogen and damage associated molecular patterns. Here we report a T cell–intrinsic role for Mincle in the pathogenesis of experimental autoimmune encephalomyelitis (EAE). Genomic deletion of Mincle in T cells impairs TH17, but not TH1 cell-mediated EAE, in alignment with significantly higher expression of Mincle in TH17 cells than in TH1 cells. Mechanistically, dying cells release β-glucosylceramide during inflammation, which serves as natural ligand for Mincle. Ligand engagement induces activation of the ASC-NLRP3 inflammasome, which leads to Caspase8-dependent IL-1β production and consequentially TH17 cell proliferation via an autocrine regulatory loop. Chemical inhibition of β-glucosylceramide synthesis greatly reduces inflammatory CD4+ T cells in the central nervous system and inhibits EAE progression in mice. Taken together, this study indicates that sensing of danger signals by Mincle on TH17 cells plays a critical role in promoting CNS inflammation.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system (CNS)1,2,3. Numerous studies indicate that the inflammatory process in MS and experimental autoimmune encephalomyelitis (EAE) is initiated by autoreactive CD4+ T cells that are reactive against myelin4,5. During the initiation stage of EAE, antigen-presenting cells (APCs) produce cytokines that regulate the differentiation of effector CD4+ T cells, polarizing these cells to TH1 (producing IFNγ) and TH17 (producing IL-17) T-cell lineages6,7. Previous studies have reported the critical roles of pattern recognition receptors on APCs in autoimmune inflammatory responses8,9. While Toll-like receptors (TLRs) are well known for their ability in modulating TH1 and TH17 responses10,11; C-type lectin receptors (CLRs) have also begun to take a center stage in T-cell-mediated autoimmune diseases, including MS and EAE12,13,14. Notably, type II transmembrane CLRs carry a carbohydrate-recognition domain; this family of CLRs included Dectin-1, Dectin-2, and macrophage-inducible C-type lectin (Mincle). These CLRs are important immune modulators through the recognition of pathogen-associated molecular patterns and damage-associated molecular patterns (DAMPs). The activation of CLR signaling activates APCs, enabling the differentiation of CD4+ IL-17-producing effector T cells (TH17 cells) during host defense against fungal infection and pathogenesis of autoimmune diseases such as MS and EAE15,16,17,18. In addition to the indirect roles of TLRs and CLRs in promoting T-cell differentiation through DC maturation and production of regulatory cytokines, emerging evidence indicates that TLR signaling via TLR2 and TLR4 is activated in CD4+ T cells to promote cytokine secretion or modulate their function19,20. Considering the robust impact of CLRs on TH17 responses, it is critical to examine the possible expression of CLRs on CD4+ T cells and determine whether they have any direct role in promoting TH17 cells.
Here we report a T cell-intrinsic Mincle-mediated inflammasome activation that results in IL-1β production critical for TH17-mediated EAE pathogenesis. Unexpectedly, we observed that Mincle was highly expressed in polarized TH17 cells, but not TH1 cells. TH17 polarizing cytokines IL-1 and IL-6 induced the expression of Mincle in CD4+ cells. We observed that T-cell-intrinsic Mincle was required for the effector stage of EAE, and Mincle deficiency in T cells impaired TH17, but not TH1, cell-mediated EAE. Mechanistically, Mincle activation (by endogenous and exogenous ligands) drove the activated TH17 cells to produce IL-1β via ASC-NLRP3–dependent caspase-8 activation. While IL-1 receptor (IL-1R) was specifically expressed on TH17 cells but not on TH1 cells, Mincle-activated TH17 cells exhibited enhanced cell proliferation in an IL-1R-dependent manner, suggesting autocrine action of Mincle-promoted TH17-derived IL-1β. Interestingly, Mincle endogenous ligand, β-glucosylceramide released by dying cells, promoted TH17 cell proliferation in a Mincle-dependent manner; blockade of β-glucosylceramide synthesis rescued the mice from EAE. Lipids extracted from the spinal cord of EAE mice promoted TH17 cell expansion, whereas lipid extracts from the spinal cord of mice treated with glucosylceramide synthase inhibitor-AMP-DNM failed to promote TH17 cells expansion.
Results
Mincle is highly expressed in TH17 cells
Mincle is well known as an inducible receptor on innate immune cells, including macrophages21. However, little is known regarding the expression and function of Mincle in T cells. Interestingly, we found that Mincle mRNA and protein were highly elevated in TH17 cells, but not in TH0, TH1, TH2 or Treg cells (Fig. 1a, b). In the presence of anti-CD3 and anti-CD28, Mincle mRNA expression was highly induced in CD4 T cells by IL-6, and to a lesser extent by IL-1β and IL-23 (Fig. 1c). Notably, Mincle deficiency had no impact on the polarization of TH1, TH2, TH17, or Treg cells (Fig. 1d). Furthermore, we reanalyzed three RNA seq datasets of CD4 or TH17 cells from EAE mice22,23,24 and found that Mincle is among the highly expressed CLRs genes in splenic CD4 T cells and as well as in CNS TH17 cells (Supplemental Fig. 1a–c). Similar to the increased expression of Mincle in TH17, other members of CLR family, including Dectin-1 (Clec7a), Dectin-2 (Clec4n), and Dectin-3 (Clec4d, MCL), were also induced in TH17. However, this response was not affected by Mincle deficiency (Supplemental Fig. 1d). Dectin-3/MCL is required for induction of Mincle in response to stimulation by TDM (trehalose-6,6-dimycolate)13. Further, our data showed that Mincle is only abundantly expressed at the late stages of in vitro polarization and in vivo priming (Supplemental Fig. 1e). Therefore, we tested MCL was also essential for the induction of Mincle in TH17 cells. However, expression of Clec4d mRNA in TH17 was much lower compared to other CLRs and Dectin-3/MCL protein was not detectable by western blot (Supplemental Fig. 1d, f). Taken together, these data indicate that Mincle is likely induced in TH17 cells in an MCL-independent manner.

a Real-time PCR analysis for mRNA levels of Mincle in TH0, TH1, TH2, TH17, and Treg cells after 3 days polarization. The expression levels were normalized to the expression of β-actin, n = 3 biological replicates. b Western analysis of Mincle protein in polarized TH1, TH2, TH17, and Treg cells from WT and Mincle-deficient mice, β-actin as a loading control, data are representative of three independent experiments, density values measured using Image J for the representative blot shown, ND not detected. c Real-time PCR analysis for Mincle mRNA levels in CD4+ cells stimulated with anti-CD3/CD28 in the presence of indicated cytokines for 72 h, n = 3 biological replicates. d Flow cytometry analysis of wild-type and Mincle-deficient TH1, TH1, TH17, and Treg cells with the indicated antibodies, n = 4 biological replicates. ***P < 0.001 (Two sided student’s t test for a and d, Two-way ANOVA for c) Data are represented as mean ± SD. Exact P values for asterisks (from left to right): a 0.00004, c 0.0015, 0.0007, 0.0089, 0.0056, 0.0086.
T-cell-specific deficiency of Mincle delays and reduces EAE
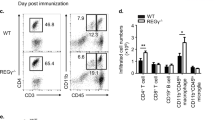
To investigate whether Mincle has a T cell-intrinsic role, we crossed a mouse strain in which exon 3–5 of the gene Clec4e (which encodes Mincle) (Mincle f/f mice) is flanked by loxP sites onto the Lck-Cre transgenic mouse strain, which expresses Cre under the control of the Lck proximal promoter, generating Mincle f/fLck-Cre and Mincle f/+Lck-Cre mice (Fig. 2a and Supplementary Fig. 2a, b). Mincle expression was efficiently and specifically deleted on T cells isolated from Mincle f/fLck-cre mice (Fig. 2a). We then tested the impact of T-cell-specific Mincle deletion on neuroinflammation and demyelination by immunizing Mincle f/fLck-Cre and littermate control Mincle f/+Lck-Cre with the neuroantigen MOG35-55 peptide. Mice with T-cell-specific Mincle deficiency had attenuated disease severity compared with controls (Fig. 2b). As a control, we showed that Mincle+/+ and Mincle f/f mice developed comparable EAE disease, indicating that the floxed allele did not affect the development of EAE (Supplementary Fig. 2c). Inflammatory mononuclear cell infiltration in the brain, including CD4+ T cells, neutrophils, and macrophages, was substantially reduced in mice with T-cell-specific Mincle deletion compared with controls (Fig. 2c, d), and the expression of inflammatory cytokines and chemokines in the spinal cord was also significantly decreased (Fig. 2e). Histopathological analysis showed substantially reduced accumulation of infiltrating immune cells and demyelination in spinal cords of Mincle f/fLck-Cre mice than that in littermate control Mincle f/+Lck-Cre mice (Fig. 2f). Further analysis of infiltrating CD4+ T cells showed that T-cell-specific Mincle deficiency resulted in a reduction of pathogenic TH17 cells (IL-17A+, GM-CSF+), but not IL-10 and IFN-γ producing CD4+ T cells (Fig. 2g). Likewise, Mincle f/fCD4-cre exhibited substantially reduced EAE disease severity compared to the littermate control Mincle f/+CD4-Cre mice (Supplementary Fig. 2d). Infiltrating immune cells and demyelination were also dramatically reduced in the spinal cord of Mincle f/fCD4-cre mice (Supplementary Fig. 2e, f). Unexpectedly, deletion of Mincle in myeloid cells or microglia had little impact on EAE pathogenesis (Supplementary Fig. 2g, h). Taken together, these data suggest that T-cell-intrinsic Mincle plays a critical role in the pathogenesis of EAE.

a Targeting vector design for the generation of a mouse strain with flanking Clec4e exon 3-5 by loxP sites and western analysis of Mincle protein expression in TH17 cells and bone marrow macrophages (1 μg/ml LPS, 6 h) from Mincle f/+Lck-Cre and Mincle f/fLck-Cre mice, n = 2 for each genotype, density values measured using Image J for the representative blot shown, ND not detected. b Mean clinical score of EAE in Mincle f/+Lck-Cre and Mincle f/fLck-Cre mice (n = 6 mice in each group) induced by active immunization with MOG35-55. c, d Absolute cell numbers (c) and gating strategy (d) of CNS-infiltrating cells were measured at the peak of disease by analyzing brain mononuclear infiltrating cells through flow cytometry with indicated antibodies, n = 4 biological replicates. e Real-time PCR analysis of relative mRNA expression of inflammatory genes in the spinal cord from Mincle f/+Lck-Cre and Mincle f/fLck-Cre mice at the peak of disease. Expression was normalized to β-actin mRNA, n = 4 biological replicates. f Hematoxylin and eosin (H&E) staining (upper panels) and Luxol fast blue staining (lower panels) of lumbar spinal cords from Mincle f/+Lck-Cre and Mincle f/fLck-Cre mice harvested at the peak of disease, Scale bars represent 100 μm. Arrows in the upper panel indicate inflammatory cells infiltration, and arrows in the lower panel indicate demyelination area. Representative data are shown for n = 4. g Flow cytometry analysis of infiltrated cytokine-producing CD4 T cells in CNS at the peak of disease, n = 4 biological replicates. *P < 0.05, **P < 0.01 (Two-sided student’s t test, c, e). *P < 0.05 (Two-way ANOVA for b). Data are represented as mean ± SD. Exact P values for asterisks (from left to right): b 0.0002 c 0.0032 0.0012 0.0017 0.0068 d 0.0446 0.0053 0.0062 0.0341 0.0410 0.0021 0.0023 0.0003 0.0005 0.0001 g 0.0014 0.0030.
T-cell-specific Mincle deficiency did not affect T-cell priming
Since the EAE phenotype was greatly reduced in Mincle f/fLck-Cre and Mincle f/fCD4-Cre mice after immunization with MOG35-55 peptide, we next examined the importance of Mincle for the priming of MOG35-55-reactive effector T-cell populations in secondary lymphoid organs. Notably, IL-17A, IFN-γ and GM-CSF cytokine production from the culture of MOG restimulated lymph node cells were similar from Mincle f/fLck-Cre mice to those in controls (Fig. 3a–c and Supplementary Fig. 3a). Further characterization of draining lymph nodes on day 9 post MOG immunization revealed similar CD4+ T-cell activation, proliferation and cytokine production, suggesting that peripheral T-cell priming was not affected in Mincle f/fLck-cre mice (Supplementary Fig. 3b–e). These results are consistent with the ex vivo polarization experiments, which showed that Mincle deficiency had no impact on the polarization of TH1, TH2, TH17 or Treg cells. Taken together, these results indicate that T-cell-specific Mincle deficiency had no impact on ex vivo T-cell differentiation or primary MOG35-55-specific T-cell priming in vivo.

a–c Lymph nodes were harvested from MOG immunized mice on Day 9 post immunization, and cells were cultured with increasing concentrations of MOG for 72 h. Cytokine concentrations in the culture media were measured by ELISA, n = 4 biological replicates. d, e Mean clinical score of EAE mice (n = 5) induced by adoptive transfer of MOG-reactive d TH1 or e TH17 cells. f Brain lymphocytes from TH17 recipient mice were harvested at the peak of disease and analyzed by flow cytometry with indicated antibodies by flow cytometry, n = 5 biological replicates. g H&E staining and Luxol fast blue staining of lumbar spinal cords at peak of the disease in recipient mice. Scale bar, 100 μM. Arrows in the upper panel indicate inflammatory cells infiltration, and arrows in the lower panel indicate demyelination area. Representative data are shown for n = 4. h Percentage of CD45+ CD4+ cells of total CNS infiltrated cells from Rag1−/− mice that received MOG-reactive TH17 cells, n = 4 biological replicates. i Cell numbers of CNS-infiltrating CD4+ T cells from (h, j). Percent of Ki67/CD4 double-positive cells from spinal cords from adoptive transfer experiments 9 days after transfer (representative images shown in Supplemental Fig. 3f.). n = 5, biological replicates. **P < 0.01 (Two-sided student’s t test for a–c, f, h–j). ***P < 0.001 (two-way ANOVA for d, e). Data are represented as mean ± SD. Exact P values for asterisks (from left to right): e <0.0001 f 0.0009 0.0033 0.0037 h 0.0025 i 0.0069 j 0.0015.
T-cell-intrinsic Mincle is required for TH17, but not TH1, -mediated EAE
The fact that T-cell-specific Mincle deficiency attenuated EAE pathogenesis but not T-cell priming, promoted us to examine the pathogenic role of Mincle-deficient TH1 and TH17 cells via adoptive transfer into naive recipients. Wild-type recipients receiving TH1 cells from Mincle f/fLck-Cre mice developed a similar diseases as those receiving TH1 cells from Mincle f/+Lck-Cre mice (Fig. 3d). But wild-type mice receiving TH17 cells from Mincle f/fLck-Cre mice developed disease with reduced severity compared with that in mice receiving TH17 cells from Mincle f/+Lck-Cre mice (Fig. 3e). Flow cytometry analysis of the infiltrating mononuclear cells in the brains showed that the numbers of CD4+ T cells, macrophages, and neutrophils were also reduced in mice receiving Mincle-deficient TH17 cells compared with controls (Fig. 3f). Likewise, histopathological analysis revealed reduced inflammatory-cell infiltration, accompanied by reduced demyelination in mice receiving Mincle-deficient TH17 cells (Fig. 3g). Together, these findings indicate that T-cell-intrinsic Mincle is required for TH17, but not TH1, cell-mediated EAE.
To test the effect of Mincle signal on TH17 migration and survival, we transferred MOG-reactivated TH17 cells from Mincle f/fLck-Cre and Mincle f/+Lck-Cre into Rag1−/− mice. Following TH17 cell transfer, we examined the CD4+ cells in the peripheral (spleen) and the CNS (brain) on days 3, 6, and day 10. TH17 cells from mice with T-cell-specific Mincle deficiency egressed from the spleen and migrated to the CNS normally on days 3 and 6 post-transfer (Fig. 3h, i). However, by day 10 after adoptive transfer, mice with TH17 cells from Mincle f/fLck-Cre mice had fewer CD4+ T cells in the CNS than did mice with TH17 cells from Mincle f/+Lck-Cre mice. Taken together, these results suggest that Mincle is required for TH17 cell survival and/or expansion in the CNS. Consistent with this hypothesis, staining of the proliferation marker Ki67 in the spinal cord revealed more proliferating/infiltrating CD4+ T cells after adoptive transfer of wild-type TH17 cells compared to mice receiving Mincle-deficient TH17 cells (Fig. 3j and Supplementary 3f).
β-glucosylceramide, a Mincle ligand, promotes TH17 cell proliferation
Since our data indicated a critical role of Mincle for in vivo TH17 cell function, we next investigated whether and how Mincle activation may impact TH17 cells ex vivo. Mincle senses divergent ligands released by non-self-pathogenic microbiome and self-ligands released from dying cells21. Recent studies have shown that the intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity25. To test the potential impact of β-glucosylceramide on TH17 cells, we cultured the TH17 cells with soluble anti-CD3 and anti-CD28 on plates coated with β-glucosylceramide in the presence of IL-6 and TGFβ. Polarizing TH17 cells in the presence of β-glucosylceramide resulted in a higher percentage of IL-17A+ cells (Fig. 4a). Next, the polarized TH17 cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) to examine the potential impact of β-glucosylceramide on TH17 cell division. We indeed found that Mincle activation with β-glucosylceramide promoted TH17 cell proliferation (Fig. 4b). Importantly, the impact of β-glucosylceramide on TH17 cells was impaired in Mincle-deficient TH17 cells (Fig. 4a, b). Similarly, challenge with another Mincle ligand, trehalose-6,6-dihehenate (TDB), a synthetic analog of mycobacterial tuberculosis cord factor- trehalose-6,6-dimycolate (TDM), to the TH17 cells under polarizing conditions also resulted in a higher percentage of TH17 cells in a Mincle-dependent manner (Supplementary Fig. 4a). In addition to enhanced cell proliferation, Mincle activation promoted inflammatory gene expression in TH17 cells, including increased expression of Csf2, Tnf, and Ifng mRNA (Fig. 4c). β-glucosylceramide stimulation also induced robust induction of Gm-csf+ TH17 cells; this response was substantially reduced by Mincle deficiency (Supplementary Fig. 4b). Heat killed, Mycobacterium Tuberculosis (HK-Mtb), an adjuvant commonly used for the induction of EAE, contains abundant immunostimulatory ligands, including the Mincle ligand trehalose-6,6’dimycolate (TDM). To evaluate whether there could be a direct effect of HK-Mtb on TH17 cells, we polarized TH17 cells in the presence or absence of HK-Mtb. Surprisingly, HK-Mtb increased TH17 polarization in both Mincle/+fLck-Cre and Mincle f/fLck-Cre cells (Supplementary Fig. 4c), indicating that additional Mincle-independent mechanisms are likely involved in the recognition of HK-Mtb by TH17 cells. Taken together, these findings suggest that β-glucosylceramide might activate TH17 cells via Mincle signaling to promote inflammatory TH17 cells, but HK-Mtb has little impact on the activation of Mincle signaling in TH17 cells.

a Flow cytometric analysis of IL-17A and IFNγ from Mincle f/+Lck-Cre and Mincle f/fLck-Cre TH17 polarized with or without β-glucosylceramide (5 μg/ml) stimulation, n = 4 biological replicates. b CFSE staining of TH17 cells polarized with or without β-glucosylceramide (5 μg/ml). Data are presented as mean fluorescent intensity, n = 4 biological replicates. c Real-time PCR of mRNA of inflammatory genes in TH17 cells polarized with or without β-glucosylceramide (1 μg/ml), n = 3 biological replicates. Expression was normalized to expression of β-actin. *p < 0.05, **p < 0.01 ***p < 0.001 (Two sided student’s t test). Data are represented as mean ± SD. Exact P values for asterisks (from left to right): a <0.0001 0.00016 b <0.0001 < 0.0001 c 0.00005 0.00088 0.0036 0.0172 0.0225 0.0341 0.0207 0.0178 0.0101 0.0128.
Mincle activation promotes TH17 cell proliferation via the production of mature IL-1β
Mincle activation in myeloid cells results in the secretion of mature IL-1β in LPS-primed condition26,27. Furthermore, we previously reported that IL-1β produced by TH17 acts in an autocrine manner on TH17 cells to promote inflammation in CNS28. Thus, we next asked whether activation of Mincle on TH17 cells would also promote IL-1β processing and secretion. β-glucosylceramide stimulation indeed induced IL-1β production in TH17 cells; this response was reduced in Mincle-deficient TH17 cells (Fig. 5a). β-glucosylceramide-induced IL-1β production was dependent on ASC-NLRP3 inflammasome (Fig. 5b–d). Interestingly, we noted that β-glucosylceramide induced the cleavage and activation of caspase 8, in TH17 cells (Fig. 5b–f). Furthermore, β-glucosylceramide-induced IL-1β production was blocked by caspase 8 inhibition (Fig. 5g) and formation of an ASC-NLRP3-caspase 8 complex was detected upon the β-glucosylceramide stimulation (Fig. 5h).

a–d Polarized TH17 cells from indicated murine strains were stimulated with β-glucosylceramide (0, 5, 50 μg/ml) for 12 h, followed by western blot analysis of supernatants and cell lysates with the indicated antibodies, density values measured using Image J for the representative blot shown, ND not detected. e Supernatants from a–d were harvested and IL-1β concentrations were determined by ELISA, n = 3 biological replicates. f TH17 cells were stimulated with β-glucosylceramide (50 μg/ml) for 12 h, caspase 8 Glo Assay reagent was added to the media for another 1 h, followed by analysis by luminescence, n = 3 biological replicates. g IL-1β concentrations were analyzed from the supernatant of polarized TH17 cells pretreated with caspase inhibitors (YVAD-fmk/caspase1 inhibitor, IETD-fmk/caspase 8 inhibitor) and stimulated with β-glucosylceramide, n = 3 biological replicates. h Cell lysates from TH17 treated with β-glucosylceramide (50 μg/ml) were subjected to immunoprecipitation with anti-ASC, followed by western analysis with the indicated antibodies, data is representative of three independent experiments. *p < 0.05, ***p < 0.001 (two-sided student’s t test for e–g). Data are represented as mean ± SD. Exact P values for asterisks (from left to right): e 0.00015 0.00024 0.0015 f 0.0011 0.00002 0.0039 g <0.0001 0.0002.
We next investigated the critical question as to the potential link between Mincle-dependent IL-1β production and TH17 cell proliferation. Using TH17 cells from wild-type and IL-1β-deficient mice, we noted that IL-1β deficiency abolished β-glucosylceramide-induced expansion of inflammatory TH17 cells (Supplementary Fig. 5a). Stimulation of TH17 cells with β-glucosylceramide increased the association of two key downstream mediators of Mincle signaling, FcRγ and Syk kinase, with Mincle (Supplementary Fig. 5b). Although canonical caspase 8 activation leads to cell death, activation of Mincle signaling did not affect the viability of TH17 cells (Supplementary Fig. 5c). Taken together, these data demonstrate that β-glucosylceramide activated a Mincle-FcRγ-Syk and ASC-NLRP3-caspase 8-dependent IL-1β production and enhanced the proliferation of TH17 cells.
β-glucosylceramide accumulation during EAE development aggravates the disease
Sensing of DAMPs released by damaged or dying cells elicits and promotes sterile inflammatory response in the CNS in models of MS and EAE29. β-glucosylceramide is released by dying cells25, which may activate Mincle expressing cells in the CNS. Similar to other studies showing increased plasma concentrations of β-glucosylceramide in inflammatory and neurological diseases25,30, plasma concentrations of β-glucosylceramides were increased in plasma from EAE mice compared to naive mice (Fig. 6a). We then investigated the influence of the administration of exogenous β-glucosylceramide on inflammation in the CNS. IV injection of β-glucosylceramide at the onset of disease worsened EAE (Fig. 6b), associated with increased immune cell infiltration and demyelination (Fig. 6c). We also detected more leukocyte infiltration in the CNS of β-glucosylceramide-treated mice (Fig. 6d). Notably, IL-17A+ TH17 cells were substantially increased in β-glucosylceramide-treated mice compared to control mice (Fig. 6e). These data suggest that β-glucosylceramide administration at the onset of disease might directly promote the function of infiltrated TH17 cells in the CNS. As an important control, treatment of Mincle f/fCD4-Cre mice with β-glucosylceramide did not affect the development of CNS inflammation (Supplementary Fig. 6a). On the other hand, daily administration of glucosylceramide synthase inhibitor-AMP-DNM protected mice from EAE progression and reduced immune cell infiltration to the CNS (Fig. 6f–h); however, treatment of Mincle f/fCD4-Cre mice with glucosylceramide synthase inhibitor-AMP-DNM failed to further reduce EAE disease (Supplementary Fig. 6b). Notably, blood glucose and serum cholesterol remained at similar concentrations in untreated and AMP-DNM-treated mice (Supplementary Fig. 6c, d). Taken together, these data suggest that the accumulation of β-glucosylceramide during the development of EAE exacerbates the EAE symptoms via activation of Mincle.

a Quantification of β-glucosylceramide derivatives obtained from the serum of naive and EAE mice at the peak of the disease, n = 4 biological replicates. b EAE clinical scores of wild-type mice treated with synthetic β-glucosylceramide (150 μg/mice) or vehicle (PBS) on day 10 after EAE induction, n = 5 mice. c Hematoxylin and eosin (H&E) staining (upper panels) and Luxol fast blue staining (lower panels) of lumbar spinal cords EAE mice harvested at the peak of disease. Scale bar, 100 μm. Arrows in the upper panel indicate inflammatory cells infiltration, and arrows in the lower panel indicate demyelination area. Representative data are shown for n = 4. d Absolute numbers of CNS-infiltrating cells were measured at the peak of disease by flow cytometry with indicated antibodies, n = 3 biological replicates. e Flow cytometry analysis of CD4+ lymphocytes from the brain of EAE mice at the peak of the disease, n = 4 biological replicates. f EAE clinical score of wild-type mice treated with AMP-DNM (25 mg/kg) or vehicle (EtOH) since the start of EAE symptom, n = 5 mice. g Hematoxylin and eosin (H&E) staining (upper panels) and Luxol fast blue staining (lower panels) of lumbar spinal cords from EAE mice harvested at the peak of disease. Scale bar, 100 μm. Arrows in the upper panel indicate inflammatory cells infiltration, and arrows in the lower panel indicate demyelination area. Representative data are shown for n = 4. h Absolute numbers of CNS-infiltrating cells were measured at the peak of disease by flow cytometry with the indicated antibodies, n = 4 biological replicates. *P < 0.05, **P < 0.01 (Two-sided student’s t test for a, d, e, h). **P < 0.01 (two-way ANOVA, b, d). Data are represented as mean ± SD. Exact P values for asterisks (from left to right): a 0.0016 0.0061 0.0178 0.0103 0.0129 b < 0.0001 d 0.0024 0.0144 0.0007 e 0.00056 0.01513 f 0.0033 h 0.0023 0.0323 0.0177 0.0057.
To test whether there is an accumulation of β-glucosylceramide in the CNS of EAE mice, we stimulated TH17 cells with lipids extracted from the spinal cord of EAE mice. Lipids extracted from the spinal cord of EAE mice promoted TH17 cell expansion, whereas lipids extracted from the spinal cord of mice treated with glucosylceramide synthase inhibitor-AMP-DNM failed to promote TH17 cells (Fig. 7a). Further, β-glucosylceramide concentration was much higher in the spinal cord of EAE mice compared to mice treated with glucosylceramide synthase inhibitor-AMP-DNM (Fig. 7b). Oligodendrocyte death contributes to the pathogenesis of EAE31,32. Since TNF is known to drive apoptosis of oligodendrocytes during EAE, we used ex vivo cultures to model TNF-mediated cell death and release of β-glucosylceramide. Lipids extracted from the supernatant of TNF-treated oligodendrocytes promoted TH17 cell expansion (Fig. 7c); this response was blocked by incubation with glucosylceramide synthase inhibitor-AMP-DNM. β-glucosylceramide was indeed accumulated in the supernatant of TNF-treated oligodendrocytes, but decreased in the supernatant of cells treated by the glucosylceramide synthase inhibitor-AMP-DNM (Fig. 7d).

a Flow cytometric analysis of IL-17A and IFNγ following TH17 polarization with lipids extracted from spinal cord of naive, EAE or AMP-DNM treated EAE mice, n = 4 biological replicates. b Measurement of β-glucosylceramide from the spinal cord of naive, EAE and AMP-DNM treated EAE mice, n = 3, 5, 4 biological replicates. c Flow cytometric analysis of IL-17A and IFNγ following TH17 polarization with lipids extracted from supernatants of oligodendrocytes after the indicated treatments, n = 4 biological replicates. d Measurement of β-glucosylceramide from the oligodendrocyte culture medium under indicated stimulations for 12 h, n = 3 biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 (Two sided student’s t test). Data are represented as mean ± SD. Exact P values for asterisks (from left to right): a < 0.0001 0.00004 0.0091 b 0.0376 0.0289 0.00047 0.0065 0.01178 0.0313 0.0193 0.00043 0.0030 0.0056 c 0.00013 0.00038 0.00366 d 0.0131 0.0055 0.0005 0.0005 0.0001 0.0393 0.0220 0.0190.
Discussion
This study reports that sensing of danger signals by Mincle on TH17 cells plays a critical role in promoting CNS inflammation. We demonstrated a T-cell-intrinsic role of Mincle in mediating TH17 cell expansion in the CNS for EAE pathogenesis. Mincle was highly expressed in polarized TH17 cells, but not TH1 cells and T-cell-specific deletion of Mincle substantially reduced TH17, but not TH1, cell-mediated EAE. Consistently, β-glucosylceramide, an endogenous Mincle ligand, promoted TH17 cell proliferation and blockade of β-glucosylceramide synthesis reduced EAE. Mechanistically, Mincle ligands stimulated the production of IL-1β via ASC-NLRP3-dependent caspase-8 mechanism in activated TH17 cells.
Notably, T-cell-specific Mincle deficiency did not affect antigen-dependent priming of TH17 cell or TH1 cell polarization in the spleen, suggesting that IL-1β produced by macrophages and dendritic cells in the peripheral environment is sufficient to support initial TH17 cell priming and expansion. Through TH17 adoptive transfer experiments, while wild-type and Mincle-deficient TH17 cells had similar first-wave infiltration into the CNS, Mincle-deficient TH17 cells in the CNS failed to proliferate or recruit the second-wave infiltration of inflammatory cells with reduced production of pro-inflammatory cytokines compared with wild-type TH17 cells. In support of this, previous studies utilizing the mouse EAE model have suggested that IL-1β stimulation actually induces the secretion of IL-17A, IFN-γ, GM-CSF, and TNF from TH17 cell–polarized brain-infiltrating cells33. Notably, IL-1R is robustly induced during TH17 cell differentiation34. Mice deficient in IL-1R have shown significant reductions in EAE disease severity29,35,36, whereas mice deficient in IL-1Ra, the endogenous soluble IL-1R antagonist, were shown to have a worse disease than wild-type controls. IL-1β stimulation of TH17 cells leads to strong and prolonged activation of the mammalian target of the rapamycin (mTOR) pathway, which has a critical role in cell proliferation and survival and is required for TH17 cell-dependent EAE pathogenesis37,38. In support of this, we have previously shown that Rag1−/− mice reconstituted with CD4+ T cells from IL1-β−/− mice were protected from the development of EAE28.
The cord factor (TDM) from HK-Mtb, used as an adjuvant in models of EAE, is a known ligand of Mincle39. However, our data suggest that HK-Mtb signaling via Mincle has a limited role in EAE priming or TH17 differentiation. One possible explanation for this discrepancy could be the presence of other T-cell ligands in the HK-Mtb, as HK-Mtb also contains TLR2, TLR4, and TLR9 ligands, such as lipoproteins and lipomannan40,41,42. Intriguingly, both TLR2 and TLR4 signals have been reported to promote TH17 responses and pathogenesis of autoimmune diseases19,20. Another possible explanation might be the differences in sensitivity of Mincle compared to other receptors for ligands in HK-Mtb. For example, compared with TLR2, Mincle requires 100–1000× higher concentration of HK-Mtb for activation; TLR2-elicited responses are stronger than Mincle-dependent responses when stimulated with same concentration of HK-Mtb43,44,45. Further study will be required to clarify which signals are involved in the HK-Mtb triggered TH17 responses.
Our previous work reported that pro-IL-1β in TH17 cells could be processed and secreted in response to stimulation with extracellular ATP in a manner dependent on both ASC and NLRP328. Consistently, the T-cell-intrinsic NLRP3 adaptor ASC was required for the effector stage of EAE. However, that study left one question unresolved: what other danger signals can re-activate TH17 cells in the CNS to stimulate their expansion and conversion towards inflammatory TH17 cells. In this study, we found that β-glucosylceramide, an endogenous Mincle ligand released by dying cells, promoted TH17 cell proliferation in a Mincle-dependent manner; blockade of β-glucosylceramide synthesis rescued the mice from EAE. Lipids extracted from the spinal cord of EAE mice promoted TH17 cell expansion, whereas lipid extracts from the spinal cord of mice treated with glucosylceramide synthase inhibitor-AMP-DNM failed to promote TH17 cells. Taken together, this study indicates that sensing of danger signal by Mincle on TH17 cells plays a critical role in promoting CNS inflammation. Importantly, elevated circulating concentrations of β-glucosylceramide were indeed detected in patients with MS46, implicating the therapeutic potential in targeting the β-glucosylceramide-Mincle axis for patients with MS. Notably, in addition to β-glucosylceramide, SAP130, another endogenous Mincle ligand, has also been implicated in EAE pathogenesis47. Moreover, a recent study indicates that microbiota sensing by the Mincle-Syk axis in dendritic cells regulates interleukin-17 and −22 production48. While our studies demonstrated the critical role of the T-cell-intrinsic role of Mincle in sensing these danger signals released by dying cells, Mincle on macrophage or/and dendritic cells might also contribute to EAE pathogenesis.
Methods
Mice
B6 (Cg)-Tg(Lck-Cd1d1)1Aben/J, B6 (Cg)-Tg(CD4-cre)1Cwi1/BfluJ, B6.129P2-Lyz2tm1(cre)Ifo/J and B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J mice (C57BL/6 background) and Rag1−/− were purchased from Jackson Laboratory (stock number 019418, 022071, 004781, 025524 and 002216), Nlrp3−/−, Il1b−/−, Caspase1/11−/− and Asc flox/flox mice were described previously28. All of the mice used in this study were female at 10–12 weeks of age, and age-matched littermates were used as experimental groups, at least five mice for each group. These mice were euthanized with carbon dioxide (CO2). Experimental procedures were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic and mice were housed under specific pathogen-free conditions.
Generation of Mincle flox/flox mice
The Mincle flox/flox mice was generated by Cyagen Biosciences Inc. with the Flp-Frt system. As illustrated in Supplemental Fig. 2a, the “floxed” targeting vector was generated by inserting floxed-neo after exon 5 and a loxP site after exon 2, in order to delete the floxed exon 3–5 by Cre recombinase. The mice carrying Mincle-floxed-neo allele were bred with Flp-Cre mice to delete the neo cassette; the progeny then carried only the loxP sites after exon 2 and exon 5. After backcrossing these mice with C57BL/6 J mice for 8–10 generations, progeny were crossed with mice expressing different cell-specific Cre recombinases to generate cell-specific Mincle-deficient mice. PCR genotyping and ARMS-PCR (amplification refractory mutation system-PCR) were carried out with the following primers: Genotyping (for upstream loxP site, F1: CTGGTCAGGATGAGGACACAACAATT, R1: GGGAAGTGGTTAATGCTTTGTGTCC, for downstream loxP site, F2: TGACTGAACGATA-TCGAATTCCG, R2: GAATTAGGGAAAAGCTGGCAGAA, internal control F2’: ACTCCAAFFCCAC-TTATCACC, internal control R2’: ATTGTTACCAACTGGGACGACA), ARMS-PCR (F3: CGAATT-CCGAAGTTCCTATTCTCTAG, R3: AGAGTTCCTTGGTCCTATGAGGTTCG).
Reagents
Anti-Mincle (1:1000, 1B6) was purchased from MBL. Anti-MCL(1:1000, PA5-102645) was purchased from Thermo Fisher. Anti-ASC (1:1000, N-15-R) was purchased from Santa Cruz Biotechnology. Anti-ASC (1:1000, 2EI-7) and anti-FcRγ(1:1000, 06-727) were purchased from Millipore. Anti-IL-1β (1:1000, AF-401-NA) was purchased from R&D. Anti-Caspase 8 (1:1000, 9429) was purchased from Cell Signaling Technology. Anti-NLRP3 (1:500, H-66) was purchased from SANTA CRUZ Biotechnology. Anti-Caspase 8 (1:1000, 1G12, ALX-804-447-C100) was purchased from Enzo. Anti-NLRP3 (1:500, H-66) was purchased from SANT CRUZ Biotechnology. Anti-Actin (1:5000, A-2228) was purchased from Sigma. Anti-Ki67 (1:1000, ab15580) was purchased from Abcam. Anti-CD45-APC (1:500, 103112), Anti-F4/80-FITC (1:200, 123108) Anti-mouse-CD25-PE (1:300, PC61, 102008). anti-mouse/human CD44-PE/Cy7 (1:300, IM7, 103030), anti-mouse MCH-II-PerCP/Cy5.5 (1:300, M5/114.15.2, 107626), anti-mouse CD134/OX40-PE/Cy7, OX-86, 119415), anti-mouse GM-CSF-Percp5.5 (1:300, MP1-22E9, 505409), anti-mouse IL-10-PE (1:300, JES5-16E3, 505007) anti-mouse Ki67PE (1:300, 652404), anti-mouse CD3 PE/Cy7(1:300, 100220) and anti-mouse CD8 APC(1:300, 100712) were purchased from Biolegend. Anti-CD4-FITC (1:200, L3T4), Anti-Ly6C-PE (1:300, HK1.4), Anti-IFN-γ-FITC (1:200, XMG1.2), Anti-CD3 (1:1000, 145-2C11), Anti-CD28 (1:1000, 37.51), Anti-IL-4-FITC (1:200, BVD6-24G2) and Anti-FOXP3-PE (1:300, FJK-16S) were purchased from eBioscience. Anti-IL-17A-PE (1:300, 559502), Anti-CD8-PE (1:300, 553041) and Anti-Ly6G-PE (1:300, 1A8) were purchased from BD. Luxol Fast Blue MBS Solution (26681) was purchased from Electron Microscopy Sciences. Caspase-Glo 8 assay system (Kit#G8200) was purchased from Promega. YVAD-fmk (ALX-260-154-R100) and IETD-fmk (550380) were purchased from Enzo and BD. Synthetic β-GluCer [d18:1/C24:1(15Z), C18:0, C16:0, C12:0]) were purchased from Avanti Polar Lipids. TDB was purchased from InvivoGen.
Induction of EAE and drug treatment
Active and adoptive transfer EAE model were induced and assessed as previously described49,50. Briefly, for the active EAE, mice were immunized with 200 ng MOG35-55 emulsified in CFA (1:1) subcutaneously followed by intraperitoneal injection of pertussis toxin 200 ng on Day0 and Day2. For the adoptive transfer, donor mice were immunized same as active EAE without pertussis toxin and spleen/draining lymph nodes were harvested 10 days after immunization. The cells were in vitro cultured for 5 days with MOG35-55 (15 μg/ml) under either TH1 cell–polarizing conditions (20 ng/ml IL-12, R&D Systems; 2 μg/ml anti-IL-23p19, eBiosciences) or TH17 cell–polarizing conditions (20 ng/ml IL-23, R&D Systems). For drug treatment, mice were treated with β-glucosylceramide (300 μg per mice) by intravenous on day 10 post the immunization. As for AMP-DNM treatment, mice were intraperitoneally injected with AMP-DNM (1 mg/kg) of ethanol dissolved in PBS daily since the onset of the EAE symptom.
Isolation and differentiation of T cells
Naive CD4+ T cells were purified with Mojo naive 4 T-cell isolation kit (Biolegend) and percentage of CD4+CD44lo were higher than 98% tested by flow cytometry. Sorted naive CD4 T cells were activated by plate-bound 1 mg/ml anti-CD3 and 1 mg/ml anti-CD28 under differentiation conditions for TH1 (20 ng/ml IL-12, 10 μg/ml anti-IL-4), TH2 (10 ng/ml IL-4, 10 μg/ml anti-IFNγ), TH17 (50 ng/ml IL-6, 2 ng/ml hTGFβ1, 10 μg/ml anti-IFNγ, 10 μg/ml anti-IL-4) and Treg (10 ng/ml hTGFβ1) for 3 days.
β-Glucosylceramide treatment for inflammasome activation
Polarized TH17 cells were washed and re-suspended in starvation media (0.1% of serum in RPMI-1640). Then 2 × 106 TH17 cells were plated on the β-glucosylceramide coated 48-well plates for 12 h. After stimulation, cells were washed and lysed in RIPA lysis buffer. Proteins in cell culture media were concentrated by methanol and chloroform (2:1) method as described51. In all, 20 μg of the sample was run on 12% sodium dodecyl–sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) gel and analyzed by western blot.
Quantitative real-time PCR
Total RNA was extracted from the spinal cord with TRIzol (Invitrogen) according to the manufacturer’s instructions. All gene expression results were expressed as arbitrary units relative to the expression of Actb. Fold induction of gene expression in the spinal cord after EAE induction was determined by dividing the relative abundance of experimental samples by the mean relative abundance of control samples from naive mice. Primers used for real-time PCR are listed in Table 1 in Supplementary files.
CFSE proliferation assay
Naive CD4+ T cells were labeled with 5 μM CFSE (Invitrogen) at 37°C for 15 min. Excess dye was washed away by PBS twice. Then cells were cultured in TH17 differentiation media. After 4 days of differentiation, cells were collected and CFSE dilution was assessed by flow cytometry.
Enzyme-linked immunosorbent assay (ELISA)
Supernatants were collected for enzyme-linked immunosorbent assay of cytokines with a kit from BioLegend (for IL-17A) or kits from R&D Systems (for all other cytokines). Il-1β levels were assayed by Il-1β (MLB00C) ELISA kit (R&D systems) according to the manufacturer’s instructions.
Western analysis and co-immunoprecipitation assay
Cells were lysed by lysis buffer (0.5% Triton X-100, 20 mM Hepes pH 7.4,150 mM NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 10 mM NaF, 2 mM dithiothreitol, 1 mM sodium orthovanadate, 2 mM EGTA, 20 mM aprotinin, 1 mM phenylmethylsulfonyl fluoride). 20 μg of protein lysate per lane was run on a 12% SDS-PAGE gel, followed by immune-blotting with different antibodies. Co-immunoprecipitation experiments were performed as described previously17. In brief, cell extracts were incubated overnight with antibodies and protein A beads at 4 °C After incubation, beads were washed four times with lysis buffer, resolved by SDS-PAGE, and analyzed by immunoblotting according to standard procedures.
Lipid extraction and quantification
Primary oligodendrocytes were cultured with TNF to induce cell death or pretreated with AMP-DNM to prevent glucosylceramide release. Lipids in the culture medium were extracted with a lipid extraction kit (Biovision) and dried. Lipids from plasma and spinal cord homogenate of EAE mice were also extracted with a lipid extraction kit. The extracted lipids were analyzed with Q-Orbitrap-MS.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data generated in this study are provided in the Supplementary Information and the Source Data file, or are available from the corresponding authors upon request.
References
Frohman, E. M. Multiple sclerosis. Med. Clin. North Am. 87, 867–897 (2003). viii-ix.
Noseworthy, J. H., Lucchinetti, C., Rodriguez, M. & Weinshenker, B. G. Multiple sclerosis. N. Engl. J. Med. 343, 938–952 (2000).
Pouly, S. & Antel, J. P. Multiple sclerosis and central nervous system demyelination. J. Autoimmun. 13, 297–306 (1999).
Fletcher, J. M., Lalor, S. J., Sweeney, C. M., Tubridy, N. & Mills, K. H. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 162, 1–11 (2010).
Segal, B. M. The diversity of encephalitogenic CD4+ T cells in multiple sclerosis and its animal models. J. Clin. Med. 8, 120 (2019).
Veldhoen, M., Hocking, R. J., Atkins, C. J., Locksley, R. M. & Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189 (2006).
Mangan, P. R. et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441, 231–234 (2006).
Kawasaki, T., Kawai, T. & Akira, S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol. Rev. 243, 61–73 (2011).
Vijay, K. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int. Immunopharmacol. 59, 391–412 (2018).
Mills, K. H. TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 11, 807–822 (2011).
Rogier, R., Koenders, M. I. & Abdollahi-Roodsaz, S. Toll-like receptor mediated modulation of T cell response by commensal intestinal microbiota as a trigger for autoimmune arthritis. J. Immunol. Res. 2015, 527696 (2015).
Brown, G. D., Willment, J. A. & Whitehead, L. C-type lectins in immunity and homeostasis. Nat. Rev. Immunol. 18, 374–389 (2018).
Miyake, Y. et al. C-type lectin MCL is an FcRγ-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity 38, 1050–1062 (2013).
Sancho, D. & Reis e Sousa, C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu. Rev. Immunol. 30, 491–529 (2012).
García-Vallejo, J. J. et al. CNS myelin induces regulatory functions of DC-SIGN-expressing, antigen-presenting cells via cognate interaction with MOG. J. Exp. Med. 211, 1465–1483 (2014).
Shenderov, K. et al. Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through Mincle/CARD9 signaling and the inflammasome. J. Immunol. 190, 5722–5730 (2013).
Lee, E. J. et al. Mincle activation and the Syk/Card9 signaling axis are central to the development of autoimmune disease of the eye. J. Immunol. 196, 3148–3158 (2016).
Stoppelkamp, S. et al. Murine pattern recognition receptor dectin-1 is essential in the development of experimental autoimmune uveoretinitis. Mol. Immunol. 67, 398–406 (2015).
Reynolds, J. M. et al. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 32, 692–702 (2010).
Reynolds, J. M., Martinez, G. J., Chung, Y. & Dong, C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc. Natl Acad. Sci. USA 109, 13064–13069 (2012).
Patin, E. C., Orr, S. J. & Schaible, U. E. Macrophage inducible C-type lectin as a multifunctional player in immunity. Front. Immunol. 8, 861 (2017).
Chyuan, I. T., Tsai, H. F., Wu, C. S., Sung, C. C. & Hsu, P. N. TRAIL-mediated suppression of T cell receptor signaling inhibits T cell activation and inflammation in experimental autoimmune encephalomyelitis. Front. Immunol. 9, 15 (2018).
Gaublomme, J. T. et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 163, 1400–1412 (2015).
Inoue, M. et al. An interferon-β-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat. Neurosci. 19, 1599–1609 (2016).
Nagata, M. et al. Intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc. Natl Acad. Sci. USA 114, E3285–E3294 (2017).
Kodar, K., Harper, J. L., McConnell, M. J., Timmer, M. S. M. & Stocker, B. L. The Mincle ligand trehalose dibehenate differentially modulates M1-like and M2-like macrophage phenotype and function via Syk signaling. Immun. Inflamm. Dis. 5, 503–514 (2017).
Zhou, H. et al. IRAKM-Mincle axis links cell death to inflammation: pathophysiological implications for chronic alcoholic liver disease. Hepatology 64, 1978–1993 (2016).
Martin, B. N. et al. T cell-intrinsic ASC critically promotes T(H)17-mediated experimental autoimmune encephalomyelitis. Nat. Immunol. 17, 583–592 (2016).
Levesque, S. A. et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1beta-driven neuroinflammation during autoimmune encephalomyelitis in mice. J. Exp. Med. 213, 929–949 (2016).
Mielke, M. M. et al. Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS One 8, e73094 (2013).
Mc Guire, C. et al. Oligodendrocyte-specific FADD deletion protects mice from autoimmune-mediated demyelination. J. Immunol. 185, 7646–7653 (2010).
Hisahara, S., Okano, H. & Miura, M. Caspase-mediated oligodendrocyte cell death in the pathogenesis of autoimmune demyelination. Neurosci. Res. 46, 387–397 (2003).
Hirota, K. et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12, 255–263 (2011).
Gulen, M. F. et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity 32, 54–66 (2010).
Lin, C. C. et al. IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. J. Exp. Med. 213, 251–271 (2016).
Croxford, A. L. et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 43, 502–514 (2015).
Chang, J. et al. MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling. Proc. Natl Acad. Sci. USA 110, 2270–2275 (2013).
Gulen, M. F. et al. Inactivation of the enzyme GSK3α by the kinase IKKi promotes AKT-mTOR signaling pathway that mediates interleukin-1-induced Th17 cell maintenance. Immunity 37, 800–812 (2012).
Ishikawa, E. et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 206, 2879–2888 (2009).
Drage, M. G. et al. TLR2 and its co-receptors determine responses of macrophages and dendritic cells to lipoproteins of Mycobacterium tuberculosis. Cell Immunol. 258, 29–37 (2009).
Pecora, N. D., Gehring, A. J., Canaday, D. H., Boom, W. H. & Harding, C. V. Mycobacterium tuberculosis LprA is a lipoprotein agonist of TLR2 that regulates innate immunity and APC function. J. Immunol. 177, 422–429 (2006).
Bafica, A. et al. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J. Exp. Med. 202, 1715–1724 (2005).
Underhill, D. M., Ozinsky, A., Smith, K. D. & Aderem, A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. USA 96, 14459–14463 (1999).
Schoenen, H. et al. Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J. Immunol. 184, 2756–2760 (2010).
Kerscher, B., Willment, J. A. & Brown, G. D. The Dectin-2 family of C-type lectin-like receptors: an update. Int. Immunol. 25, 271–277 (2013).
Kurz, J. et al. The relevance of ceramides and their synthesizing enzymes for multiple sclerosis. Clin. Sci. (Lond.) 132, 1963–1976 (2018).
N’Diaye, M. et al. C-type lectin receptors Mcl and Mincle control development of multiple sclerosis-like neuroinflammation. J. Clin. Invest. 130, 838–852 (2020).
Martinez-Lopez, M. et al. Microbiota sensing by Mincle-Syk axis in dendritic cells regulates interleukin-17 and −22 production and promotes intestinal barrier integrity. Immunity 50, 446–461 e449 (2019).
Kang, Z. et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 32, 414–425 (2010).
Qian, Y. et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 8, 247–256 (2007).
Lee, W. B. et al. Mincle-mediated translational regulation is required for strong nitric oxide production and inflammation resolution. Nat. Commun. 7, 11322 (2016).
Acknowledgements
This study was supported in part by grants from the National Institutes of Health: R01AA023722, to X.L. and L.E.N., P01HL103453, P01AI141350 to X.L, P50AA024333 to L.E.N, P01HL144497 to C.L. R01NS104164 to Z.K.).
Author information
Authors and Affiliations
Contributions
X.L. and Q.R.Z. conceived and coordinated the study, and wrote the manuscript. Q.R.Z. conducted the majority of experiments and analyzed the data. C.J.Z., H.W., W.L., H.Z., K.B., J.J.Z., and X.C. helped with experiments. R.L.Z. performed the mass spectrometry and analyzed the data. Z.K., C.L., R.B., G.D., D.A., T.X., and L.E.N. provided critical discussion, and X.L. and L.E.N. obtained funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Salvador Iborra and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Q., Liu, W., Wang, H. et al. TH17 cells promote CNS inflammation by sensing danger signals via Mincle. Nat Commun 13, 2406 (2022). https://doi.org/10.1038/s41467-022-30174-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-30174-1
This article is cited by
-
Inflammasomes in neurological disorders — mechanisms and therapeutic potential
Nature Reviews Neurology (2024)
-
Infiltrating myeloid cell-derived properdin markedly promotes microglia-mediated neuroinflammation after ischemic stroke
Journal of Neuroinflammation (2023)
-
USP1-regulated reciprocal differentiation of Th17 cells and Treg cells by deubiquitinating and stabilizing TAZ
Cellular & Molecular Immunology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
