
Abstract
Due to high bond dissociation energies of Csp2–F bonds, using fluorinated compounds in Csp2–Csp3 cross-coupling is difficult. Here the authors report a protocol for enantioselective Csp2–Csp3 coupling of dienyl fluorides with aldimine esters, enabled by synergistic copper and palladium catalysis. This reaction represents the first example of asymmetric Csp2–Csp3 cross-coupling involving an inert Csp2–F bond and provides expeditious access to chiral α-alkenyl α-amino acids with high enantioselectivity. Control experiments suggest that the Csp2–F bond activation occurs through a pathway involving PdH migratory insertion and subsequent allylic defluorination, rather than by direct oxidative addition of the Csp2–F bond to Pd(0). The detailed mechanism is further investigated by DFT calculation and the enantioselectivity is rationalized.
Similar content being viewed by others
Introduction
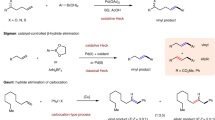
Transition-metal-catalyzed enantioselective cross-coupling reactions between Csp2–X compounds (X = halogen) and enolizable carbonyl compounds are commonly used transformations for asymmetric construction Csp2–Csp3 bonds1,2,3,4,5,6,7. Many successful examples of this method have been reported, including pioneering work by the research groups of Ma1, Hartwig2, and Buchwald3,6, who used chiral-ligand-bearing transition metals such as Cu and Pd to achieve enantiocontrol (Fig. 1a). These reactions initiated with oxidative addition of Csp2–X bond to the Pd(0) or Cu(I), followed by ligand exchange and reductive elimination to form the Csp2–Csp3 bond. Halogenated compounds with Csp2–I, Csp2–Br, and even Csp2–Cl bonds are suitable reaction substrates. However, because Csp2–F bonds have high energies (120–129 kcal/mol for olefinic C–F bonds), fluorinated compounds have rarely been used as coupling partners8,9,10,11,12,13,14,15.

a Enantioselective Csp2–Csp3 cross-coupling of Csp2–I (Br, Cl) Bonds. b C–F bond Activation by Nu-M Insertion/β-fluorine elimination. c This work: C–F bond activation by Pd-H insertion/allylic defluorination for Csp2–Csp3 cross-coupling.
Recently, C–F activation has been an important research topic in synthetic organic chemistry16,17,18,19,20. One of the most successful strategies in this area is using transition-metal-mediated or -catalyzed metal-Nu insertion/β–F elimination process21. Trifluoro-, difluoroalkenes have been intensively investigated for this purpose during the past few years22,23,24,25,26,27,28,29,30; however, monofluoroalkenes are rarely explored class of compounds for similar C–F activation reactions (Fig. 1b). Moreover, despite those achievements in defluorinative carbon–carbon and carbon–heteroatom bond formation, enantioselective variants have seldom been realized. We envisioned that if an ingenious insertion/β–F elimination mechanism was designed together with a suitable chiral induction strategy, the aforementioned challenged defluorinative Csp2–Csp3 coupling might be achieved.
In this work, we report a Cu/Pd cooperative system31 for enantioselective Csp2–Csp3 cross-coupling between dienyl fluorides and aldimine esters (Fig. 1c). Experimental and computational studies revealed that this reaction involved a unique Pd-H insertion/allylic defluorination process. This work not only represents the first example of enantioselective defluorinative Csp2–Csp3 coupling but also provides a highly efficient catalytic method to prepare chiral α-alkenyl α-amino acids (α-AAs), which are important synthetic targets32,33,34,35,36 because of their potential biochemical and pharmacological activities37.
Results and discussion
Reaction development
Our studies begin with investigating the reaction of dienyl fluoride E-1a with aldimine ester38,39,40 2a to generate α-alkenyl, α-methyl α-AA 3aa. Using the stereocontrol exhibited by Cu-azomethine ylides in two-metal catalytic systems41,42,43, we designed a synergistic Pd/Cu catalyst system44,45,46,47,48,49,50,51,52,53,54,55 for controlling the stereochemistry of the newly formed chiral center by means of an appropriate combination of ligands on the two metals during the coupling step (Table 1). First, we tested Phosferrox Cu complex L1-Cu with Pd catalysts bearing a bisphosphine ligand (dppe, dppp, Xantphos, or DPEphos; L4-Pd–L7-Pd, respectively) and found that none of these combinations catalyzed the desired reaction (entries 1–4). In contrast, BINAP-ligated catalyst L8-Pd afforded 3aa in 53% yield with 96% ee (entry 5). SEGPHOS- and Biphep-derived catalysts (L9-Pd–L11-Pd, entries 6–8) were also examined, and L11-Pd gave the best yield of the product. Subsequent tests of L11-Pd in combination with other Cu catalysts (L2-Cu and L3-Cu, entries 9 and 10) revealed that L11-Pd/L3-Cu gave the best results (86% yield, 99% ee). A slight decrease in enantioselectivity was observed when the opposite enantiomer of the Pd catalyst (ent-L11-Pd) was used together with L3-Cu (entry 11). This result implies the enantioselectivity was mainly controlled by the chiral Cu catalyst but the mismatched chirality between the two catalysts was slightly deleterious to the enantiocontrol.
Substrate scope
Having developed an effective dual-metal catalyst system, we investigated the substrate scope of the reaction, starting with dienyl fluorides 1 bearing various R1 and R2 substituents (Fig. 2). Phenyl rings with a methyl group (3ba, 3ca), a fluorine atom (3da–3fa), a chlorine atom (3ga), a trifluoromethyl group (3 ha), or a methoxy group (3ia) were well tolerated, regardless of the location of the substituent, affording the corresponding coupling products in 71–87% yields with enantioselectivities exceeding 98% ee. Replacing the phenyl ring with a different aromatic ring—naphthyl (3ja), furyl (3ka), thiophenyl (3la), or indolyl (3ma)—had little influence on the reaction outcome, the corresponding α-alkenyl, α-alkyl α-AAs were obtained with excellent enantioselectivities. An alkyl-substituted substrate (R1 = cyclohexyl) furnished 3na in 63% yield, albeit with a reduced ee (94%). Even though allylic ethers are commonly sensitive to Pd owing to the possibility of C–O bond cleavage, a substrate with an allylic BnO ether moiety was well tolerated in this reaction system, giving 3oa in 60% yield with 99% ee. A 1,4-disubstituted dienyl fluoride (R1 = Ph, R2 = Me) was also investigated, which afforded the desired product 3pa in 50% yield with >99% ee. To determine the stereochemistry of the product, we converted 3aa to its p-toluenesulfonamide derivative and confirmed its structure by means of X-ray crystallographic analysis, which allowed us to assign the absolute configuration as 2 S.

Reaction conditions: (i) 1a (0.4 mmol), 2a (0.2 mmol), L11-Pd (4 mol%), L3-Cu (5 mol%), Et3N (200 mol%), THF (0.5 mL), 30 °C, 24 h; (ii) citric acid (10 wt%, 4 mL). Isolated yields are provided. The ee values were determined by HPLC on a column with a chiral stationary phase. aL11-Pd (8 mol%), L3-Cu (10 mol%), THF (0.2 mL), 40 °C, 120 h.
Next, we probed the scope of aldimine esters substrate (Fig. 3). When R4 was methyl, R3 could be Et (3ab), nBu (3ac), or phenylethyl (3ad). Heteroatom-tethered alkyl chains were also well-tolerated, as indicated by the formation of coupling products 3ae–3ag in moderate to good yields with good enantioselectivities. In addition, we investigated compounds with various alkyl R4 groups (3ah–3aj), revealing that the reaction was not sensitive to the steric bulk of the ester. An aldimine ester derived from glutamic acid also reacted smoothly but gave lactam 3ak in 58% yield with 92% ee, as a result of cyclization during the acidic workup. α-Amino-γ-butyrolactone derived imine underwent reaction with 1a to give 3al in moderate yield. In addition to aldimine ester, cyclic ketimine ester and oxazoline ester were also compatible reaction partners. As shown in the formation of 3am and 3an, both the yields and enantioselectivities were well maintained for these types of nucleophiles. Phenylalanine derived aldimine ester 2o failed to undergo this transformation due to the steric bulk, and substrate 2p bearing allyl group only gave complexed products, probably because the isomerization of the terminal olefin moiety.

Reaction conditions: (i) 1a (0.4 mmol), 2a (0.2 mmol), L11-Pd (4 mol%), L3-Cu (5 mol%), Et3N (200 mol%), THF (0.5 mL), 30 °C, 48 h; (ii) citric acid (10 wt%, 4 mL). Isolated yields are provided. The ee values were determined by HPLC on a column with a chiral stationary phase.
Synthetic application
The reaction was scaled up to more than one mmol scale with a reduced catalyst loading [L11-Pd (2 mol%) and L3-Cu (2.5 mol%)] and the yield and enantioselectivity were well maintained (Fig. 4a). To demonstrate the utility of the reaction, the coupling products were transformed to other chiral scaffolds. Protection (S)-3aa with p-tolylsulfonyl group gave (S)-4aa, and the latter was treatment with NBS/Na2CO3 in acetonitrile to elaborate the bromoamination product 5aa in 75% yield (Fig. 4b). The internal alkene moiety of (S)-4aa could be selectively cleaved with K2Os(OH)4/NMO, followed by NaIO4, affording aldehyde (S)-6aa in 86% yield (Fig. 4c). Moreover, protection the amine of (S)-3oa with benzoyl group gave (S)-4oa, which was further subjected to sequential dihydroxylation/lactonization conditions to furnish densely functionalized lactone 5oa with good diastereoselectivity (Fig. 4d).

a Scale-up reaction at lower catalyst loading. b Intramolecular bromoamination reaction. c Selective cleavage of the internal alkene. d Dihydroxylation/lactonization reaction.
Mechanistic studies
To gain insight into the mechanism, we carried out some control experiments. We found that reactions of both >20:1 and 1:1 E/Z-1a gave >20:1 E/Z-3aa with essentially identical yields and ee values (Fig. 5a). On the other hand, when 1a with the E/Z ratio of 1:1.1 was subjected to the reaction, the absolute amount of the Z-1a and E-1a was monitored (Fig. 5b). Interestingly, the concentration of Z-1a and E-1a are both decreased during the reaction; however, the E-1a has a faster consumption rate than Z-1a did. Most importantly, an inflection point appeared around 5 h in the E-1a consumption curve. These results indicate a possibility that E-1a preferentially reacted under the optimized conditions at the early stage, and Z-1a gradually isomerized to E-1a. In addition, when dienyl bromide 4a or dienyl chloride 4b was subjected to the reaction conditions, almost none of the coupling product (3aa) was observed (Fig. 5c). Consequently, we reasoned that direct oxidative addition of the Csp2–F bond to Pd(0) was probably not involved in the reaction pathway56,57. Finally, the reaction between 1a and deuterium-labeled 2a resulted in the incorporation of a total 30% of the deuterium at the terminal carbon of the double bond in the coupling product (Fig. 5d). As a result, we speculated that a Pd–D insertion process might be involved in the reaction, which would lead to deuterium enriching at the terminus of the alkenes58.

a Relationship between the ratio of Z/E-1a and ratio of Z/E-3aa. b The absolute amount the Z-1a and E-1a during the reaction. c Reaction of dienyl bromide 4a or dienyl chloride 4b under the standard conditions. d Deuterium atom scramble experiment.
Based on the above-described results, as well as our previous studies59,60 on synergistic Pd/Cu-catalyzed coupling reactions of aldimine esters with unsaturated compounds, we proposed that this coupling reaction proceeds based on a mechanism involving the Cu and PdH cycles shown in Fig. 6. In the Cu catalytic cycle, Cu acts as a Lewis acid to activate aldimine ester 2a to form metallated azomethine ylide II, which serves as a nucleophile in the coupling reaction. Allylation of II with pre-palladium catalyst L11-Pd forms the L11-Pd(0), which then undergoes oxidative addition with Et3NH+ to generate the PdH catalyst (Fig. 6a). In the PdH catalytic cycle (Fig. 6b), PdH migratory insertion into the C = C bond of the vinyl fluoride moiety of E-1a generates fluorinated Pd-allyl III61,62,63,64. An allylic substitution reaction between III and metallated azomethine ylide II affords Pd intermediate IV and regenerates the Cu catalyst. Intermediate IV undergoes rapid allylic defluorination, giving allyl-Pd V, which is converted to VI via β-H elimination. Product 3aa dissociates from VI, and the PdH regenerated65,66,67. For Z-1a, the anti-anti η3-Pd-allyl VII is generated after PdH migratory insertion, which equilibrates to thermodynamically more stable syn-anti η3-Pd-allyl III through η3-η1-η3 isomerization (Fig. 6c). Therefore Z-1a would then undertake the same catalytic cycle to form the same E-type product 3aa. This proposed mechanism was highly consistent with the controlled experiments in Fig. 3. Moreover, we observed a [M + H]+ signal at 252.1393 in the HRMS spectrum of the reaction mixtures. This result indicates the formation of IX, which comes from the dissociation of Pd(0) from intermediate IV (Fig. 6d).

a Initial generation of PdH catalyst from the precatalyst L11-Pd. b Proposed catalytic cycle. c Isomerization of anti-anti π-allyl-Pd to syn-anti π-allyl-Pd. d HRMS detection of [M + H]+ for Intermediate IX. The ligands were omitted for clarity.
Computational studies
To further understand the mechanism, we performed DFT calculation to investigate the energy profile of this reaction. Stoichiometric reaction of L3-Cu and substrate 2a with DBU, only one isomeric Cu-azomethine ylide was formed, as indicated by a single 31P NMR signal at −16.09 (Fig. 7). The structures of Cu-azomethine ylide featuring a (R)- or (S)- metal chirality were calculated and we found the former is 2.9 kcal/mol stable than the latter. Therefore, we rationalized that Cu(R)-Nu rather than Cu(S)-Nu was the form for nucleophile and therefore its structure was adopted for the remaining DFT calculation (Fig. 8).

a Observation of the Cu-zaomethine ylide by 31P NMR. b Energy comparison of Cu(R)-Nu and Cu(S)-Nu.

Calculations were carried out at the M06-2x(SMD)/def2-TZVP//B3LYP-D3BJ/6-31 g(d)/Lanl2dz level of theory.
As proposed in Fig. 8, the PdH was initially formed via oxidative protonation of Pd(0) with Et3NH+. The transition state for this step was located as TS-1, which has an energy barrier of 15.0 kcal/mol. After being coordinated by substrate 2a, the resulting intermediate Int-2 occurs migratory insertion of the Pd–H bond into the terminal olefin moiety to afford π-allyl-Pd species Int-4. The energy barrier for this step is only 1.1 kcal/mol; however, the reserved β–H elimination step requires activation energy of 29.7 kcal/mol (Int-4 → TS-3 → Int-2). Therefore, the Pd–H migratory insertion is not a reversible process. The resulting Int-4 accepts nucleophilic attach from the si-face of the metallated azomethine ylide Cu-Nu, to give C–C bond formation intermediate Int-6. Dissociation of LCu from Int-6 affords species Int-7, which then undergoes allylic defluorination. The direct nucleophilic displacement/ionization mechanism for the defluorination transition state (TS-8A) has a high energy barrier of 28.0 kcal/mol. But a Et3NH+ mediated process, of which the transition state was located as TS-8B, only requires an activation barrier of 12.2 kcal/mol. The resulting Int-9 then occurs β–H elimination step via TS-10 with an energy barrier of 27.5 kcal/mol to elaborate product (2 S)-3aa and regenerates the PdH catalyst.
To elucidate the enantioselectivity, the approach from the re-face of Cu-Nu during the C–C bond formation was also calculated and the transition state is identified as TS-5-II (Fig. 9a). Comparing the energies of TS-5 and TS-5-II, the latter is disfavored over the former by 5.0 kcal/mol, which is in agreement with the experimental result that (2 S)-3aa is formed with 99% ee. In the transition state structure TS-5-II, the oxazoline ring group on the phosferrox ligand has a very short distance away from t-Bu groups of DTB-Biphep and which leads to significant steric interaction between the two ligands (Fig. 9b). This repulsive interaction likely contributes to the higher energy observed for this competing transition state. In contrast, in the transition state structure TS-5, the electrophile approaches from the si-face of Cu-Nu which avoids the steric repulsion between the oxazoline ring and t-Bu groups. Moreover, an attractive C–H ∙ ∙ ∙ Ar interaction is observed in TS-5, which also contributes to a lower energy (Fig. 9c).

a Comparison of Transition states TS-5 and TS-5-II. b Steric interaction between the oxazoline ring and t-Bu group in TS-5-II. c Attractive C–H⋯Ar interaction in TS-5.
In summary, the first enantioselective defluorinative Csp2–Csp3 cross-coupling was achieved by means of synergistic Cu/Pd-catalyzed asymmetric coupling between aldimine esters and dienyl fluorides. This reaction has a wide substrate scope and shows good to excellent enantioselectivities and it provide an efficient catalytic method for preparing chiral α-vinyl, α-alkyl α-amino acid derivatives. Both experimental and computational studies revealed that the reaction is initiated by a PdH migratory insertion, which is followed by nucleophilic allylic substitution by a Cu-azomethine ylide to form the C–C bond. Then a Pd/Et3NH+-mediated allylic defluorination undergoes, subsequently followed by a β–H elimination to elaborate the coupling product and regenerate the PdH catalyst.
Methods
General procedure for coupling of dienyl flurides and aldimine esters
In glove box, Cu(MeCN)4PF6 (3.7 mg, 0.01 mmol, 5 mol%) and chiral ligand (S,Sp)-L3 (5.3 mg, 0.011 mmol, 5.5 mol%) were dissolved in dry THF (0.4 M, 0.5 mL) and stirred at room temperature for 0.5 h. To the solution, substrate aldimine esters 2 (0.2 mmol), Et3N (0.4 mmol), dienes 1 (0.4 mmol) and palladium catalyst L11-Pd (10.1 mg, 0.008 mmol, 4 mol%) were added sequentially. The reaction mixture was stirred at 30 °C for 24 h. To the reaction mixture was added citric acid solution (4 mL, 10 wt%) and the mixture was stirred for 2 h. The mixture was neutralized with solid K2CO3 and extracted with EtOAc (10 mL × 3). The combined extracts were dried over MgSO4 and concentrated in vacuo to afford a residue. The residue was then purified by SiO2 column chromatography to give the desired product.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information Files as well as from the corresponding author on request. The Cartesian coordinates for the calculated structures are available within the Supplementary Data 1. The X-ray crystallographic coordinates for structure reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2036502 (p-toluenesulfonamide derivative of 3aa). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Xie, X., Chen, Y. & Ma, D. Enantioselective arylation of 2-methylacetoacetates catalyzed by CuI/trans-4-Hydroxy-l-proline at low reaction temperatures. J. Am. Chem. Soc. 128, 16050–16051 (2006).
Ge, S. & Hartwig, J. F. Nickel-catalyzed asymmetric α-arylation and heteroarylation of ketones with chloroarenes: effect of halide on selectivity, oxidation state, and room-temperature reactions. J. Am. Chem. Soc. 133, 16330–16333 (2011).
Zhu, Y. & Buchwald, S. L. Ligand-controlled asymmetric arylation of aliphatic α-amino anion equivalents. J. Am. Chem. Soc. 136, 4500–4503 (2014).
Jiao, Z., Chee, K. W. & Zhou, J. S. Palladium-catalyzed asymmetric α-arylation of alkylnitriles. J. Am. Chem. Soc. 138, 16240–16243 (2016).
Zhu, C., Wang, D., Zhao, Y., Sun, W.-Y. & Shi, Z. Enantioselective palladium-catalyzed intramolecular α-arylative desymmetrization of 1, 3-diketones. J. Am. Chem. Soc. 139, 16486–16489 (2017).
Taylor, A. M., Altman, R. A. & Buchwald, S. L. Palladium-catalyzed enantioselective α-arylation and α-vinylation of oxindoles facilitated by an axially chiral P-stereogenic ligand. J. Am. Chem. Soc. 131, 9900–9901 (2009).
Chieffi, A., Kamikawa, K., Åhman, J., Fox, J. M. & Buchwald, S. L. Catalytic asymmetric vinylation of ketone enolates. Org. Lett. 3, 1897–1900 (2001).
Ackermann, L., Born, R., Spatz, J. & Meyer, H. D. Efficient Aryl–(Hetero)Aryl coupling by activation of C–Cl and C–F Bonds Using Nickel Complexes of Air-Stable Phosphine Oxides. Angew. Chem. Int. Ed. 44, 7216–7219 (2005).
Yoshikai, N., Mashima, H. & Nakamura, E. Nickel-catalyzed cross-coupling reaction of aryl fluorides and chlorides with grignard reagents under Nickel/Magnesium Bimetallic Cooperation. J. Am. Chem. Soc. 127, 17978–17979 (2005).
Yoshikai, N., Matsuda, H. & Nakamura, E. Hydroxyphosphine Ligand for Nickel-Catalyzed Cross-Coupling through Nickel/Magnesium Bimetallic Cooperation. J. Am. Chem. Soc. 131, 9590–9599 (2009).
Liu, X.-W., Echavarren, J., Zarate, C. & Martin, R. Ni-catalyzed borylation of aryl fluorides via C–F cleavage. J. Am. Chem. Soc. 137, 12470–12473 (2015).
Niwa, T., Ochiai, H., Watanabe, Y. & Hosoya, T. Ni/Cu-catalyzed defluoroborylation of fluoroarenes for diverse C–F bond functionalizations. J. Am. Chem. Soc. 137, 14313–14318 (2015).
Tobisu, M., Xu, T., Shimasaki, T. & Chatani, N. Nickel-catalyzed Suzuki–Miyaura reaction of aryl fluorides. J. Am. Chem. Soc. 133, 19505–19511 (2011).
Ho, Y. et al. Nickel-Catalyzed Csp2–Csp3 bond formation via C–F bond activation. Org. Lett. 20, 5644–5647 (2018).
Butcher, T. W. et al. Desymmetrization of difluoromethylene groups by C–F bond activation. Nature 583, 548–553 (2020).
Amii, H. & Uneyama, K. C−F Bond Activation in Organic Synthesis. Chem. Rev. 109, 2119–2183 (2009).
Ahrens, T., Kohlmann, J., Ahrens, M. & Braun, T. Functionalization of fluorinated molecules by transition-metal-mediated C–F bond activation to access fluorinated building blocks. Chem. Rev. 115, 931–972 (2015).
Unzner, T. A. & Magauer, T. Carbon–fluorine bond activation for the synthesis of functionalized molecules. Tetrahedron Lett. 56, 877–883 (2015).
Shen, Q. et al. Review of recent advances in C–F bond activation of aliphatic fluorides. J. Fluor. Chem. 179, 14–22 (2015).
Fu, L., Chen, Q. & Nishihara, Y. Recent advance in transition-metal-catalyzed C–C bond formation via C(sp2)–F bond cleavage. Chem. Rec. 21, 1–18 (2021).
Fujita, T., Fuchibe, K. & Ichikawa, J. Transition-metal-mediated and -catalyzed C−F bond activation by fluorine elimination. Angew. Chem. Int. Ed. 58, 390–402 (2019).
Ichitsuka, T., Fujita, T., Arita, T. & Ichikawa, J. Double C−F bond activation through β-fluorine elimination: Nickel-Mediated [3+2] cycloaddition of 2-Trifluoromethyl-1-alkenes with alkynes. Angew. Chem. Int. Ed. 53, 7564–7568 (2014).
Thornbury, R. T. & Toste, F. D. Palladium-catalyzed defluorinative coupling of 1-Aryl-2,2-difluoroalkenes and boronic acids: stereoselective synthesis of monofluorostilbenes. Angew. Chem. Int. Ed. 55, 11629–11632 (2016).
Tian, P., Feng, C. & Loh, T.-P. Rhodium-catalysed C(sp2)–C(sp2) bond formation via C–H/C–F activation. Nat. Commun. 6, 7472 (2015).
Wu, J.-Q. et al. Experimental and theoretical studies on rhodium-catalyzed coupling of benzamides with 2,2-difluorovinyl tosylate: diverse synthesis of fluorinated heterocycles. J. Am. Chem. Soc. 139, 3537–3545 (2017).
Lu, X. et al. Nickel-catalyzed defluorinative reductive cross-coupling of gem-difluoroalkenes with unactivated secondary and tertiary alkyl halides. J. Am. Chem. Soc. 139, 12632–12637 (2017).
Sakaguchi, H. et al. Copper-catalyzed regioselective monodefluoroborylation of polyfluoroalkenes en route to diverse fluoroalkenes. J. Am. Chem. Soc. 139, 12855–12862 (2017).
Zell, D. et al. Mild Cobalt(III)-Catalyzed Allylative C−F/C−H functionalizations at room temperature. Chem. Eur. J. 23, 12145–12148 (2017).
Ma, Q., Wang, Y. & Tsui, G. C. Stereoselective palladium-catalyzed C−F bond alkynylation of tetrasubstituted gem-difluoroalkenes. Angew. Chem. Int. Ed. 59, 11293–11297 (2020).
Dai, W., Shi, H., Zhao, X. & Cao, S. Sterically controlled Cu-catalyzed or transition-metal-free cross-coupling of gem-Difluoroalkenes with tertiary, secondary, and primary alkyl grignard reagents. Org. Lett. 18, 4284–4287 (2016).
Wu, Y., Huo, X. & Zhang, W. Synergistic Pd/Cu catalysis in organic synthesis. Chem. Eur. J. 26, 4895–4916 (2020).
Armstrong, A. & Emmerson, D. P. G. Enantioselective synthesis of α-Alkyl,α-Vinyl Amino Acids via [2,3]-sigmatropic rearrangement of selenimides. Org. Lett. 13, 1040–1043 (2011).
Berkowitz, D. B., Wu, B. & Li, H. A Formal [3,3]-sigmatropic rearrangement route to quaternary α-Vinyl Amino Acids: Use of Allylic N-PMP Trifluoroacetimidates. Org. Lett. 8, 971–974 (2006).
Shea, R. G. et al. Allylic selenides in organic synthesis: new methods for the synthesis of allylic amines. J. Org. Chem. 51, 5243–5252 (1986).
Genna, D. T., Hencken, C. P., Siegler, M. A. & Posner, G. H. α-Chloro-β,γ-ethylenic Esters: enantiocontrolled synthesis and substitutions. Org. Lett. 12, 4694–4697 (2010).
Panahi, F., Khosravi, H., Bauer, F. & Breit, B. Asymmetric hydroalkylation of alkynes and allenes with imidazolidinone derivatives: α-alkenylation of α-amino acids. Chem. Sci. 12, 7388–7392 (2021).
Berkowitz, D. B., Charette, B. D., Karukurichi, K. R. & McFadden, J. M. α-Vinylic amino acids: occurrence, asymmetric synthesis, and biochemical mechanisms. Tetrahedron.: Asymmetry 17, 869–882 (2006).
Coldham, I. & Hufton, R. Intramolecular dipolar cycloaddition reactions of azomethine ylides. Chem. Rev. 105, 2765–2810 (2005).
Narayan, R., Potowski, M., Jia, Z.-J., Antonchick, A. P. & Waldmann, H. Catalytic Enantioselective 1,3-Dipolar cycloadditions of azomethine ylides for biology-oriented synthesis. Acc. Chem. Res. 47, 1296–1310 (2014).
Wei, L., Chang, X. & Wang, C.-J. Catalytic asymmetric reactions with N-metallated azomethine ylides. Acc. Chem. Res. 53, 1084–1100 (2020).
Huo, X., Zhang, J., Fu, J. & Zhang, W. Ir/Cu dual catalysis: enantio- and diastereodivergent access to α,α-disubstituted α-amino acids bearing vicinal stereocenters. J. Am. Chem. Soc. 140, 2080–2084 (2018).
Wei, L., Zhu, Q., Xu, S.-M., Chang, X. & Wang, C.-J. Stereodivergent synthesis of α,α-disubstituted α-amino acids via synergistic Cu/Ir catalysis. J. Am. Chem. Soc. 140, 1508–1513 (2018).
Peng, L., He, Z., Xu, X. & Guo, C. Cooperative Ni/Cu-catalyzed asymmetric propargylic alkylation of aldimine esters. Angew. Chem. Int. Ed. 59, 14270–14274 (2020).
Nahra, F., Macé, Y., Lambin, D. & Riant, O. Copper/Palladium-Catalyzed 1,4 reduction and asymmetric allylic alkylation of α,β-unsaturated ketones: enantioselective dual catalysis. Angew. Chem. Int. Ed. 52, 3208–3212 (2013).
Semba, K. & Nakao, Y. Arylboration of alkenes by cooperative palladium/copper catalysis. J. Am. Chem. Soc. 136, 7567–7570 (2014).
Jia, T. et al. A Cu/Pd cooperative catalysis for enantioselective allylboration of alkenes. J. Am. Chem. Soc. 137, 13760–13763 (2015).
Friis, S. D., Pirnot, M. T. & Buchwald, S. L. Asymmetric hydroarylation of vinylarenes using a synergistic combination of CuH and Pd catalysis. J. Am. Chem. Soc. 138, 8372–8375 (2016).
Logan, K. M. & Brown, M. K. Catalytic enantioselective arylboration of alkenylarenes. Angew. Chem. Int. Ed. 56, 851–855 (2017).
Saito, A., Kumagai, N. & Shibasaki, M. Cu/Pd synergistic dual catalysis: asymmetric α-allylation of an α-CF3 amide. Angew. Chem. Int. Ed. 56, 5551–5555 (2017).
Huo, X. et al. Stereoselective and site-specific allylic alkylation of amino acids and small peptides via a Pd/Cu dual catalysis. J. Am. Chem. Soc. 139, 9819–9822 (2017).
Wei, L., Xu, S.-M., Zhu, Q., Che, C. & Wang, C.-J. Synergistic Cu/Pd catalysis for enantioselective allylic alkylation of aldimine esters: access to α,α-Disubstituted α-Amino Acids. Angew. Chem. Int. Ed. 56, 12312–12316 (2017).
Yabushita, K., Yuasa, A., Nagao, K. & Ohmiya, H. Asymmetric catalysis using aromatic aldehydes as chiral α-alkoxyalkyl anions. J. Am. Chem. Soc. 141, 113–117 (2019).
Liao, Y. et al. Enantioselective synthesis of multisubstituted allenes by cooperative Cu/Pd-Catalyzed 1,4-Arylboration of 1,3‐Enynes. Angew. Chem. Int. Ed. 59, 1176–1180 (2020).
Bai, X., Wu, C., Ge, S. & Lu, Y. Pd/Cu-catalyzed enantioselective sequential Heck/Sonogashira coupling: asymmetric synthesis of oxindoles containing Trifluoromethylated Quaternary Stereogenic Centers. Angew. Chem. Int. Ed. 59, 2764–2768 (2020).
He, R. et al. Stereodivergent Pd/Cu catalysis for the dynamic kinetic asymmetric transformation of racemic unsymmetrical 1,3-disubstituted allyl acetates. J. Am. Chem. Soc. 142, 8097–8103 (2020).
de Jong, G. T. & Bickelhaupt, F. M. Oxidative addition of the fluoromethane C−F Bond to Pd. An ab Initio Benchmark and DFT validation study. J. Phys. Chem. A 109, 9685–9699 (2005).
Rekhroukh, F., Chen, W., Brown, R. K., White, A. J. P. & Crimmin, M. R. Palladium-catalysed C–F alumination of fluorobenzenes: mechanistic diversity and origin of selectivity. Chem. Sci. 11, 7842–7849 (2020).
Wang, H., Zhang, R., Zhang, Q. & Zi, W. Synergistic Pd/Amine-catalyzed stereodivergent hydroalkylation of 1,3-dienes with aldehydes: reaction development, mechanism, and stereochemical origins. J. Am. Chem. Soc. 143, 10948–10962 (2021).
Zhang, Q., Yu, H. & Zi, W. Stereodivergent coupling of 1,3-dienes with aldimine esters enabled by synergistic Pd and Cu catalysis. J. Am. Chem. Soc. 141, 14554–14559 (2019).
Zhu, M., Zhang, Q. & Zi, W. Diastereodivergent synthesis of β-Amino alcohols by dual-metal-catalyzed coupling of alkoxyallenes with aldimine esters. Angew. Chem. Int. Ed. 60, 6545–6552 (2021).
Liao, L. & Sigman, M. S. Palladium-catalyzed hydroarylation of 1,3-dienes with boronic esters via reductive formation of π-Allyl palladium intermediates under oxidative conditions. J. Am. Chem. Soc. 132, 10209–10211 (2010).
Zhou, H., Wang, Y., Zhang, L., Cai, M. & Luo, S. Enantioselective terminal addition to allenes by dual chiral primary amine/palladium catalysis. J. Am. Chem. Soc. 139, 3631–3634 (2017).
Adamson, N. J., Wilbur, K. C. E. & Malcolmson, S. J. Enantioselective intermolecular Pd-catalyzed hydroalkylation of acyclic 1,3-dienes with activated pronucleophiles. J. Am. Chem. Soc. 140, 2761–2764 (2018).
Park, S., Adamson, N. J. & Malcolmson, S. J. Brønsted acid and Pd–PHOX dual-catalysed enantioselective addition of activated C-pronucleophiles to internal dienes. Chem. Sci. 10, 5176–5182 (2019).
Adamson, N., Hull, E. & Malcolmson, S. J. Enantioselective intermolecular addition of aliphatic amines to acyclic dienes with a Pd–PHOX catalyst. J. Am. Chem. Soc. 139, 7180–7183 (2017).
Nie, S.-Z., Davison, R. T. & Dong, V. M. Enantioselective coupling of dienes and phosphine oxides. J. Am. Chem. Soc. 140, 16450–16454 (2018).
Zhang, Q., Dong, D. & Zi, W. Palladium-catalyzed regio- and enantioselective hydrosulfonylation of 1,3-dienes with sulfinic acids: scope, mechanism, and origin of selectivity. J. Am. Chem. Soc. 142, 15860–15869 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (nos. 21871150, 22071118, for W.Z. and 22001130 for Q.Z.) and the Fundamental Research Funds for Central University. H.Y. was supported by “Tianjin Research Innovation Project for Postgraduate Students” (no. 2019YJSB072). Q.Z. was supported by the China Postdoctoral Science Foundation (no. 2019M660973). We thank the Haihe Laboratory of Sustainable Chemical Transformations for financial support (no. ZYTS202101).
Author information
Authors and Affiliations
Contributions
W.Z. conceived and tutored this work. Q.Z. conducted preliminary studies and co-tutored this work. H.Y. performed all the experiments and prepared the Supplementary Information. All the authors co-wrote this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, H., Zhang, Q. & Zi, W. Synergistic Pd/Cu-catalyzed enantioselective Csp2–F bond alkylation of fluoro-1,3-dienes with aldimine esters. Nat Commun 13, 2470 (2022). https://doi.org/10.1038/s41467-022-30152-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-30152-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
