
Abstract
Organic semiconductors with high-spin ground states are fascinating because they could enable fundamental understanding on the spin-related phenomenon in light element and provide opportunities for organic magnetic and quantum materials. Although high-spin ground states have been observed in some quinoidal type small molecules or doped organic semiconductors, semiconducting polymers with high-spin at their neutral ground state are rarely reported. Here we report three high-mobility semiconducting polymers with different spin ground states. We show that polymer building blocks with small singlet-triplet energy gap (ΔES-T) could enable small ΔES-T gap and increase the diradical character in copolymers. We demonstrate that the electronic structure, spin density, and solid-state interchain interactions in the high-spin polymers are crucial for their ground states. Polymers with a triplet ground state (S = 1) could exhibit doublet (S = 1/2) behavior due to different spin distributions and solid-state interchain spin-spin interactions. Besides, these polymers showed outstanding charge transport properties with high hole/electron mobilities and can be both n- and p-doped with superior conductivities. Our results demonstrate a rational approach to obtain high-mobility semiconducting polymers with different spin ground states.
Similar content being viewed by others

Introduction
π-Conjugated organic molecules with open-shell or high-spin ground state have attracted increasing interests due to their unique optoelectronic and magnetic properties1,2,3. However, these molecules contain unpaired electrons and are often unstable compared with the closed-shell molecules because they can easily form dimers, or being quenched due to their high reactivity. Recently, many efforts have been devoted to the development of stable open-shell conjugated molecules. Related strategies include the use of large steric hindrance groups to protect unpaired electrons, and delocalization of unpaired electrons to large conjugated systems4. Based on these strategies, some open-shell π-conjugated organic molecules, including p-quinodimethanes5, polycyclic aromatic hydrocarbons, quinoidal oligothiophene derivatives6 were investigated (Fig. 1a). Recent works on the design and precise synthesis of open-shell small molecules and oligomers have greatly expanded the open-shell small π-conjugated organic molecule library and provided a more in-depth understanding on their optoelectronic and magnetic properties7,8,9,10,11.

a Resonance structures of several open-shell π-conjugated organic molecules. b Some representative open-shell or high-spin organic semiconductors and their charge carrier mobilities.
Charge carrier mobility is a critical parameter for an optoelectronic device, which reflects the speed at which the charge carriers move in a material. Open-shell organic molecules with high mobility will enable long spin transport distance which is crucial for spin-related applications12,13,14. However, there are few reports of small open-shell molecules with high charge carrier mobilities10,15,16,17. This is because the use of steric hindrance groups prevents close packing of molecules, and increasing the conjugation length requires more complex synthetic steps. Figure 1b shows some representative π-conjugated open-shell molecules, which usually show low charge carrier mobilities and only a few show mobilities over 0.1 cm2 V−1 s−1 17. The low charge carrier mobilities of these molecules greatly restrict their applications in optoelectronic and spin-related devices.
Conjugated polymers have been intensively studied for various optoelectronic applications because of their high mechanical flexibility/stretchability and large-scale solution processability18,19,20. In the past decade, the development of high-performance polymer building blocks, including indacenodithiophene19, naphthalene diimide (NDI)21, diketopyrrolopyrrole (DPP)22, isoindigo (IID)23, and benzodifurandione-based oligo(p-phenylene vinylene) (BDOPV)24, have greatly promoted the charge carrier mobility enhancement in conjugated polymers. Conjugated polymers with open-shell character can be obtained by chemical doping25. However, the doped polymers are unstable and the ionized dopants always result in large structural and energetic disorders26. In addition, doped semiconducting polymers are always conductors while losing their semiconducting properties. Recently, conjugated polymers at their neutral ground state were reported to have open-shell high-spin character27,28, but these polymers showed conducting properties with limited applications in semiconducting devices. To date, the guidelines for the design of these polymers are rarely explored, and the influential factors for their spin properties are still obscure.
Here we report a strategy to design high-spin ground state and high-mobility semiconducting polymers. This approach is based on three assumptions: (i) Most high-mobility conjugated polymers are based on several high-performance building blocks (e.g., NDI, IID, and DPP etc. in Fig. 2a), and thus these building blocks are needed; (ii) The single-triplet energy gap (ΔES-T = ES−ET) is the energy difference between open-shell singlet state and open-shell triplet state1; as the singlet-triplet energy gap (ΔES-T) value increase, the stability of the high-spin triplet state will increase, and the triplet state may become the ground state5; (iii) the polymers need to have a highly planar backbone, which can lead to better conjugation, smaller ΔES-T, and also benefits charge transport. Based on these assumptions, we screened currently available high-performance polymer building blocks by calculating their ΔES-T values and planarity indexes. Three polymers were synthesized for comparison. These polymers exhibit distinct magnetic properties and solid-state spin-spin interactions. Among them, p(TDPP-TQ) and p(TDPP-BBT) exhibit an air-stable triplet ground state. Moreover, p(TDPP-TQ) displays high electron mobilities of up to 7.76 cm2 V−1 s−1 and high hole mobilities of up to 6.16 cm2 V−1 s−1. Interestingly, the polymer can also be effectively n-doped and p-doped, showing outstanding n-type and p-type electrical conductivities (σe = 16.1 S/cm and σh = 348.3 S/cm). We believe that our design strategy could help to discover more high-mobility semiconducting polymers with different spin ground states.

DFT Calculated ΔES-T values of a “large fused aromatic” and b “small-size aromatic” building blocks used in high-mobility semiconducting polymers; c The closed-shell and open-shell resonance structures of TDPP. d The closed-shell and open-shell resonance structures of BT, TQ, and BBT.
Results
Polymer design and synthesis
To date, many high-mobility semiconducting polymers are based on the copolymerization of “large fused aromatics” and “small-size aromatics”. Thus, we collected potential polymer building blocks from recently published reviews22,29 and separate them into two groups: “large fused aromatics” and “small-size aromatics” (Fig. 2a, b). Based on the above assumptions, we first performed DFT calculations to obtain their ΔES-T values. Among all the building blocks, TDPP, BDOPV, TQ, and BBT exhibit the smallest |ΔES-T|. Then, we performed DFT calculations to estimate the planarity of the polymer building block combinations by using a recently developed planarity index 〈cos2φ〉30. We found that TDPP can form planar polymer backbone with TQ and BBT with high torsional barriers and large 〈cos2φ〉 (Fig. 1c). Whereas when BDOPV was copolymerized with TQ and BBT, the resulting polymers show small 〈cos2φ〉 values, suggesting the polymers significantly deviate from planarity (Supplementary Fig. 1). Note that the backbone planarity also strongly affects the ΔES-T values (Supplementary Table 1). Better backbone planarity could lead to smaller |ΔES-T| values and more stable triplet states. Therefore, backbone planarity is crucial for both efficient intrachain and interchain charge transport, as well as for realizing high-spin ground states. To have a systematic understanding of the relationship between molecular structure and open-shell property, we choose TDPP as the “large fused aromatic”, and BT, TQ and BBT with decreased ΔES-T values, as the “small-size aromatic” to construct polymers. Note that TDPP, BT, TQ and BBT are usually considered as “acceptors” in conjugated polymers. Unlike traditional donor-acceptor design27, this “acceptor-acceptor” design allows the polymers to have enough bandgap to show semiconducting rather than conducting property.
The three polymers, p(TDPP-BT), p(TDPP-TQ), and p(TDPP-BBT), were synthesized via Pd-catalyzed Stille polymerization reaction between the trimethyltin TDPP and the dibromo compounds of BT, TQ, and BBT (Fig. 3a). The synthesis and purification procedures are detailed in Supplementary Materials. The stability of those polymers was proved by thermogravimetric analysis, and all the polymers showed high decomposition temperatures over 350 °C (Supplementary Fig. 2). Three polymers show gradually red-shifted absorption spectra as decreasing the ΔES-T of the “small-size aromatic” (Fig. 3b). The HOMO/LUMO energy levels of p(TDPP-BT), p(TDPP-TQ) and p(TDPP-BBT) obtained by cyclic voltammogram measurements are −5.34/−3.55, −5.23/−3.94, and −5.20/−4.17 eV, respectively (Supplementary Fig. 3). The increase of the HOMO and the decrease of the LUMO level is probably due to the enhanced planarity, better conjugation, and more readily aromatic-to-quinoidal transformation (Fig. 3c and Supplementary Fig. 3).

a Molecular structures of three semiconducting polymers. “Ar” is the aromatic building block. b UV–vis–NIR absorption spectra of the polymers in ODCB solution (1 × 10−5 M). c Calculated potential energy scans (PES) of the dihedral angles φ between the TDPP and three BT derivatives.
Characterization of magnetic properties
The high-spin characteristics of the polymers were investigated by electron paramagnetic resonance (EPR) and superconducting quantum interference device (SQUID) both in solution and in the solid state. At room temperature, the solid-state EPR intensity increases dramatically in the order of p(TDPP-BT), p(TDPP-TQ), and p(TDPP-BBT) (Fig. 4a), which reflects an increase of spin density. More importantly, the EPR intensities showed negligible change after storing the polymers in air for 90 days, suggesting the high air stability of these open-shell polymers (Supplementary Fig. 4). The EPR intensity of p(TDPP-BT) is very weak and shows a close-shell feature, and thus we will not study this polymer in detail. To compare the spin dynamics of p(TDPP-TQ) and p(TDPP-BBT), temperature-dependent EPR measurements in the solid state were performed (Fig. 4b, Supplementary Fig. 5a). Their EPR intensities decrease with the increase of temperature, suggesting that they probably have a triplet ground state1. It is known that if there exists a triplet state ground state, the EPR signal of |Δms| = 2 forbidden transition can be observed, even with very weak intensity1. To determine the existence of triplet ground state in the polymers, the |Δms| = 2 forbidden transition was measured. For p(TDPP-BBT), a low intensity but clear |Δms| = 2 signal (g factor = 3.9981) is observed (Fig. 4c), suggesting the existence of triplet ground state. For p(TDPP-TQ), the forbidden transition could not be observed, which is largely due to its relatively weak EPR intensity compared to p(TDPP-BBT). Furthermore, the variable temperature EPR of both polymers in solution was also measured and showed a similar behavior compared to solid state EPR. The EPR intensities of p(TDPP-TQ) and p(TDPP-BBT) decrease with the increase of temperature (Fig. 4d, Supplementary Fig. 5b). The EPR data of p(TDPP-BBT) from 4.9 K to 50 K were fitted by the Bleaney-Bowers equation, which provides an energy gap between singlet and triplet (ΔES-T) of 4.92 × 10−3 kcal mol−1 (J = 2.48 K) (Fig. 4e).

a Room temperature EPR signals of the polymers in the solid state; b variable temperature EPR of p(TDPP-BBT) in the solid state; c the half field line of p(TDPP-BBT) in the solid state: |Δms| = 2 signal (g factor = 3.9981, the theoretical value: g = 4.0046); d temperature-dependent EPR of p(TDPP-BBT) in 1 × 10−3 M o-xylene and e the corresponding Bleaney-Bowers equation fitting result; f temperature-dependent magnetic susceptibility from 2 K to 300 K. Solid squares are data, solid lines are linear fitting lines. g spin density distribution of the triplet states of the oligomers (n = 6) of p(TDPP-BT) (top), p(TDPP-TQ) (middle), p(TDPP-BBT) (bottom). DFT calculations were performed at the UB3LYP/6-31 G** level. Atoms’ colors: gray for C, red for O, blue for N, yellow for S, and white for H.
To further investigate the static magnetic properties of the polymers, temperature-dependent magnetic susceptibility was measured by SQUID from 2 K to 300 K under DC field (Fig. 4f). The values of the product of magnetic susceptibility and temperature (χMT) increase linearly with temperature. This feature usually suggests strong antiferromagnet coupling or temperature-independent paramagnetism (TIP). Due to the relatively low calculated spin density and weak spin-orbit coupling in organic polymers, this phenomenon is usually attributed to TIP31,32. TIP comes from Curie and Pauli paramagnetism (χtotalT = C + χPauliT), where C is Curie paramagnetism and T is temperature. After fitted the χT–T plot, the χPauli of p(TDPP-TQ) and p(TDPP-BBT) were determined to be 8.2 × 10−4 and 3.5 × 10−3 cm3 mol−1, and Curie constants are 3.2 × 10−3 and 4.0 × 10−2 cm3 mol−1 K, respectively. From the relation χPauli = μB2N(EF), where μB is the Bohr magneton, the densities of states at the Fermi energy N(EF) for those p(TDPP-TQ) and p(TDPP-BBT) were calculated to be 1.5 × 1022 and 6.5 × 1022 eV−1 cm−3. The Pauli paramagnetism and large value of N(EF) reveal that the spins in these polymers are highly delocalized32,33 (see detailed discussions in the Supplementary Information Supplementary Note 4). In particular, for p(TDPP-BBT), the χMT and T are slightly deviated from the linear relationship at low temperature (red circle in Fig. 4f). This behavior could be due to the weak antiferromagnetic coupling between polymer chains, which we will prove later by studying the aggregation behavior of the polymers and the DFT calculations of oligomers and dimers.
Understanding of the different spin ground states
To determinate the spin ground state of those polymers, field-dependent magnetization measurements (M–H) were performed. For p(TDPP- TQ), it shows a close to S = 1 triplet state (Fig. 5a), consistent with EPR results; while for p(TDPP-BBT), it shows a close to S = 1/2 doublet state in the measurement (Fig. 5b). It seems that this result conflicts with the EPR measurement. However, we will show that this phenomenon can be well explained as following.

Field dependent magnetization measurement of a p(TDPP-TQ) and b p(TDPP-BBT) from 0 Oe to 50,000 Oe. The solid squares are the experimental data, the solid lines stand for the simulated curve by Brillouin equation. The simulation results exhibit the S = 1 and S = 1/2 ground states of p(TDPP-TQ) and p(TDPP-BBT). c Schematic illustration of the mechanism of the different ground states of p(TDPP-TQ) and p(TDPP-BBT). Left image shows the microstructure of a typical polymer film, which contains ordered crystalline regions (highlighted in darker yellow) and amorphous regions. Right image illustrates the spin-spin interactions in solid state. The orange shadings on the polymers indicate where the spins mostly distribute. In p(TDPP-TQ), the low spin density makes the spins mostly interact within a chain. While in p(TDPP-BBT), we observed interchain spin-spin interactions because of its high spin density.
First, p(TDPP-BBT) has a long polymer chain (~20 repeat units for one chain) with spins distributed at the chain ends. We performed density functional theory (DFT) calculations for their oligomers (n = 6). The calculated ΔES-T of the oligomers were −19.50 kcal mol−1 for p(TDPP-BT), −3.69 kcal mol−1 for p(TDPP-TQ), and −0.14 kcal mol−1 for p(TDPP-BBT), which is consistent with the above observed trend that p(TDPP-BBT) could have very small ΔES-T value and a triplet ground state. The significant decrease of |ΔES-T| indicates the increased stability of the open-shell triplet ground state, which agrees well with the observed EPR results and magnetic property. The large |ΔES-T| value of p(TDPP-BT) leads to the inaccessibility of the triplet state even at room temperature. While for p(TDPP-TQ) and p(TDPP-BBT), the small |ΔES-T| value of their oligomers could make the polymers have triplet ground state if the polymers have high degree of polymerization and strong interchain interactions. The singlet diradical character index (y0), which is often used to estimate the diradical property, was also calculated34,35. The value of y0 ranges from 0 to 1; 0 means closed-shell, while y0 = 1 stands for pure diradical. The calculated y0 values of (TDPP-BT)6, (TDPP-TQ)6, and (TDPP-BBT)6 are 0.012, 0.350, and 0.996 (Supplementary Tables 4–6). The difference of the spin triplet states for each polymer can be visualized by the spin density distribution in Fig. 4g. From (TDPP-BT)6 to (TDPP-BBT)6, the triplet spin density distribution changes from localized in the center, to uniformly distributed along the chain, and to mostly distributed at the end of the chain. This result agrees well with the increase of the y0 value (Supplementary Figs. 14–16). The bond length alternation analysis (Supplementary Figs. 17, 19) also shows that the bond length difference between open-shell singlet and triplet of (TDPP-BT)6 become obvious only in the middle of the molecule. For (TDPP-TQ)6, it emerges throughout the entire oligomer. For (TDPP-BBT)6, the bond length difference is negligible, which is consistent with its small |ΔES-T| value. It has been proposed that when the two spins are separated at a long distance, the two spins have weak interactions and can be regarded as two radicals (or diradical)1. Similar results have been observed in a quinoidal oligomer system36. They found that when oligomer number ≥4, the triplet state of the oligomer is better to be described by two individual radicals (also known as biradicals).
Second, the strong interchain interactions between p(TDPP-BBT) chains might also lead to the doublet ground state. It has been reported that conjugated polymers with rigid backbones are strongly aggregated even in dilute solutions37,38. The strong intermolecular interaction will affect spin delocalization31. To understand the polymer chain interactions, we performed the temperature-dependent UV–vis absorption spectra of p(TDPP-TQ) and p(TDPP-BBT) (Supplementary Figs. 10 and 11). We and others have shown that in good solvent (dissolving polymers better), conjugated polymers can be more readily disaggregated at elevated temperature37,39. Therefore, three solvents, toluene, o-dichlorobenzene (o-DCB), and 1-chloronaphthalene (1-CN), with increased solubility for conjugated polymers were used. For both polymers, the maximum absorption peak decreases with increasing temperature and decreases more significantly in a good solvent, such as 1-CN, suggesting that both polymers are strongly aggregated in solution. The strong aggregation of the polymer supports the observed large N(EF) values, which could explain the TIP phenomenon. Previous study has shown that strong spin-spin interactions in triplet small molecules could result in doublet state in magnetization measurements15. p(TDPP-TQ) has a low spin density and direct spin-spin interaction can hardly happen (Fig. 5c), thus exhibiting a triplet ground state in solid state. According to the quantitative ESR measurement (Supplementary Fig. 6), we can obtain the number of spins per polymer chain is 0.98, close to 1, at room temperature for p(TDPP-BBT). The high spin density makes the spin at the chain end can directly interact with each other, leading to an apparent doublet ground state, consistent with the field-dependent magnetization measurement.
To support this hypothesis, we also performed DFT calculations for the polymer dimers of the n = 4 oligomer to compare the energy, molecular orbitals, and spin density between a single polymer chain and dimers with different interchain packings (Supplementary Figs. 20, 23). As expected, all the stacked dimers of three polymers are more stable than two single chains, indicating the strong π-π stacking interactions between the polymer chains (Supplementary Fig. 21). p(TDPP-BT) dimers showed a large negative ΔES-T value, making it hard to access the triplet state. This is consistent with our result that p(TDPP-BT) is a close-shell polymer with a singlet ground state. We then focus on the difference between p(TDPP-BBT) and p(TDPP-TQ). As shown in Supplementary Fig. 22, the frontier orbitals of p(TDPP-BBT) and p(TDPP-TQ) dimers have shown obvious overlaps between two chains, suggesting the two polymer chains have strong electronic couplings40. Spin density redistribution is observed for both p(TDPP-TQ) and p(TDPP-BBT), indicating the existence of strong intermolecular spin-spin interaction (Supplementary Fig. 23). Interestingly, the two polymers exhibit significantly different spin distributions. The intermolecular electron coupling through π-π stacking decreases the spin density in the middle of the p(TDPP-BBT) dimer. This feature increases the distance of the two unpaired spins located at the end of the dimer and diminishes the electron exchange interaction, leading to the formation of two independent radicals (doublet state). However, for p(TDPP-TQ), the spin is distributed all along the polymer chain for both the single chain and the dimer. Thus, for p(TDPP-TQ), the two radicals are closer with stronger interactions, forming diradicals and exhibiting triplet ground state in solid-state.
Based on the magnetic properties and DFT calculations, the following conclusion can be obtained: (1) p(TDPP-BT) has a singlet ground state because of its large ΔES-T value; (2) p(TDPP-TQ) and p(TDPP-BBT) have triplet ground state because of their intrinsically small ΔES-T values; (3) because of high spin density, separately distributed spin-density, and interchain spin-spin interactions in p(TDPP-BBT), it shows doublet state in solid state; whereas the low spin density and relatively uniformly distributed spin density in p(TDPP-TQ) make it exhibit triplet ground state.
Charge-transport properties and thin-film characterization
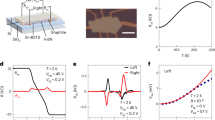
The charge transport properties of the polymers were evaluated by field-effect transistors with a top-gate/bottom-contact configuration (Fig. 6d, Supplementary Figs. 24, 27). Unlike previously reported high-spin polymers with conducting properties27, our polymers show typical and close to ideal semiconducting properties with good ambipolar charge transport properties. The polymers also showed good on/off ratios if the VDS is small. p(TDPP-BT) exhibits good electron mobilities of up to 3.83 ± 0.65 cm2 V−1 s−1 and hole mobilities of up to 2.77 ± 0.15 cm2 V−1 s−1 in the saturation regime. p(TDPP-TQ) exhibits high electron mobilities of up to 7.76 ± \(0\).86 cm2 V−1 s−1 and high hole mobilities of up to 6.16 ± \(0\).68 cm2 V−1 s−1 in the saturation regime. These values are the highest reported to date in high-spin ground state polymers, and also among the highest in all reported organic semiconductors22. p(TDPP-BBT) shows relatively lower electron mobilities of 0.35 ± 0.08 cm2 V−1 s−1 and hole mobilities of 0.25 ± 0.06 cm2 V−1 s−1. Their charge carrier mobilities in the linear regime are also calculated (Supplementary Table 7). To date, only a few reported open-shell small molecules have shown moderate charge carrier mobilities, usually on the order of 10−3 cm2 V−1 s−1 (Fig. 1b and Supplementary Table 8)10,15,16,41,42. Therefore, our approach successfully addressed the challenge to design high-mobility and high-spin ground-state organic semiconductors.

2D-grazing incidence wide-angle X-ray scattering (GIWAXS) pattern of a p(TDPP-BT), b p(TDPP-TQ), c p(TDPP-BBT). d Typical transfer characteristics of a p(TDPP-TQ) FET device. e N-type electrical conductivities of p(TDPP-TQ) doped by N-DMBI with different ratios; f P-type electrical conductivities of p(TDPP-TQ) doped by immersing in 10 mM FeCl3 solution. Error bars indicate the standard deviation of ten experimental replicates.
Grazing incidence wide-angle X-ray scattering (GIWAXS) and atomic force microscopy (AFM) were employed to study the microstructures and morphology of the polymer films. (Fig. 6a–c, Supplementary Fig. 30). All the polymers show typical face-on dominated molecular packing in solid state. p(TDPP-BT) shows the narrower full width at half maximum (FWHM) (Supplementary Fig. 32 and Supplementary Table 9) with a lamellar distance of 21.67 Å and a π–π stacking distance of 3.57 Å, suggesting its higher ordered crystallite domain. p(TDPP-TQ) shows wider FHWM with a lamellar distance of 20.94 Å and a π–π stacking distance of 3.70 Å. For p(TDPP-BBT) (Fig. 6c), it shows the widest FWHM and indistinguishable π–π stacking peak. After re-measuring its film at Shanghai Synchrotron Radiation Facility (Supplementary Fig. 33), we finally identified that p(TDPP-BBT) has a lamellar distance of 20.27 Å and a π–π stacking distance of 3.69 Å. The poor crystallinity of p(TDPP-BBT) might explain its relatively lower charge carrier mobilities. AFM height images show that all the polymer films have a very smooth surface with root-mean-square surface roughness <1 nm (Supplementary Fig. 30). Note that the charge transport properties of the three polymers are not strongly correlated with their molecular packings. This phenomenon is common in conjugated polymers, because many studies have shown that crystallinity and π–π stacking distance of polymers are not the only parameters affecting their charge transport properties, and other parameters, such as interchain short contacts43, packing conformation44, and energetic disorders45, also strongly influences charge carrier mobilities.
Previous studies suggest that spin-spin interactions in materials could lead to enhanced thermopower in some thermoelectric materials46,47. Since organic thermoelectric materials are usually achieved under doped states, we explored the charge transport properties after doping. We found that p(TDPP-TQ) can be both effectively n-doped and p-doped. For n-doping, a commonly used n-dopant, N-DMBI, was used. After optimizing the doping concentration and annealing temperature, the n-type electrical conductivity of p(TDPP-TQ) polymer achieved 16.1 S/cm (Fig. 6e), which is among the highest in n-doped organic semiconducting polymers48,49. P-doping was performed by immersing p(TDPP-TQ) films in a 10 mM FeCl3 solution. The polymer can be easily p-doped as observed in the UV−vis−NIR absorption spectra (Supplementary Fig. 31). By varying the immersion time, the polymer showed a p-type electrical conductivity of 348.3 S/cm (Fig. 6f), which is also among the highest in p-doped conjugated polymers. One polymer that can achieve both high n-type conductivity and p-type conductivity after doping is rare in literature50,51. Compared with other polymers, both n- and p-doped p(TDPP-TQ) polymer films exhibited mediate stability in the ambient conditions (Supplementary Fig. 34). Such unique doping behavior of p(TDPP-TQ) could be probably due to the ease of accepting and donating electrons of the triplet state. These results also suggest the great potential of using high-spin semiconducting polymers in the applications requiring heavy doping, e.g., organic thermoelectrics and organic bioelectronics50,52,53.
Discussion
In summary, we have developed a rational strategy to screen the potential polymer building blocks for designing high-spin ground state and high mobility semiconducting polymers. Based on the strategy, three polymers with different spin ground states were obtained. We found that the spin distributions and interchain interactions could lead to different spin behaviors for these polymers. p(TDPP-BT) has a singlet ground state, p(TDPP-TQ) has a triplet ground state, while p(TDPP-BBT) has a doublet ground state. More importantly, all these polymers showed high charge carrier mobilities among all the reported high-spin organic semiconductors, and they could also be readily p- and n-doped to show high electrical conductivities, suggesting their broad applications for versatile organic magnetic and electronic devices. We believe that the molecular design strategy developed in this work could bring more air-stable, high-mobility semiconducting polymers with different spin ground states, which could enable broad spin-related applications beyond the current scope of organic electronics.
Methods
Polymerization methods
The distannyl TDPP (0.05 mmol), dibromo BT or TQ or BBT (0.05 mmol), Pd(PPh3)4 (8 mol%, 4 μmol), and 0.2 mL xylene were put into a 0.5 mL microwave reaction tube. The reaction was drove by microwave with the following procedure: 120 °C for 5 min, 140 °C for 5 min, 170 °C for 30 min. A deep blue solution was formed after the reaction. After the reaction was finished, N,N’-diethylphenylazothioformamide (0.01 mmol) was added and stirred at 90 °C for 30 min to remove palladium or other residue metals. The mixture was poured to 100 mL methanol to precipitate the polymers and then filtered. The solid product was put into a soxhlet extraction and extracted by methanol (4 h), acetone (4 h), hexane (10 h), tetrahydrofuran (5 h), dichloromethane (5 h), chloroform (12 h) and finally collected by chlorobenzene (24 h). 1H NMR of three polymers is shown in Supporting Information.
Electron paramagnetic resonance spectroscopy
Electron paramagnetic resonance (EPR) spectroscopy was conducted on a Bruker E580 spectrometer using ER 4122 SHQE highly sensitive EPR cavity. Oxford EPR900 cryostat was used for temperature control. The microwave frequency is at 9.37 GHz.
SQUID measurement
The static magnetic property measurement was performed on a Quantum Design MPMS XL SQUID magnetometer from 2 to 300 K. The magnetization measurements were performed at 2 K over the magnetic field range of 250–70,000 Oe. The sample was packeted by aluminum foil and placed in a nonmagnetic holder. The diamagnetic contribution of the sample holder is negligible. The diamagnetic contribution of aluminum foil was corrected with blank holders. The diamagnetic contribution of the polymer was corrected by the Pascal’s constants.
Data availability
The source data generated in this study have been deposited in the materials cloud database (https://archive.materialscloud.org/record/2022.46) and are also available from the corresponding author upon request.
Code availability
The codes that support the findings of this study are available from the corresponding authors upon request.
References
Abe, M. Diradicals. Chem. Rev. 113, 7011–7088 (2013).
Ai, X. et al. Efficient radical-based light-emitting diodes with doublet emission. Nature 563, 536–540 (2018).
Rugg, B. K. et al. Photodriven quantum teleportation of an electron spin state in a covalent donor-acceptor-radical system. Nat. Chem. 11, 981–986 (2019).
Ji, L., Shi, J., Wei, J., Yu, T. & Huang, W. Air-stable organic radicals: new-generation materials for flexible electronics? Adv. Mater. 32, 1908015–1908030 (2020).
Casado, J. Para-quinodimethanes: a unified review of the quinoidal-versus-aromatic competition and its implications. Top. Curr. Chem. 375, 73 (2017).
Zhang, C. et al. Stable cross-conjugated tetrathiophene diradical. Angew. Chem. Int. Ed. 58, 11291–11295 (2019).
Gallagher, N. M., Olankitwanit, A. & Rajca, A. High-spin organic molecules. J. Org. Chem. 80, 1291–1298 (2015).
Sun, Z., Zeng, Z. & Wu, J. Zethrenes, extended p-quinodimethanes, and periacenes with a singlet biradical ground state. Acc. Chem. Res. 47, 2582–2591 (2014).
Liu, C., Ni, Y., Lu, X., Li, G. & Wu, J. Global aromaticity in macrocyclic polyradicaloids: Hückel’s rule or Baird’s rule? Acc. Chem. Res. 52, 2309–2321 (2019).
Rudebusch, G. E. et al. Diindeno-fusion of an anthracene as a design strategy for stable organic biradicals. Nat. Chem. 8, 753–759 (2016).
Li, Y., Li, L., Wu, Y. & Li, Y. A review on the origin of synthetic metal radical: singlet open-shell radical ground state? J. Phys. Chem. C. 121, 8579–8588 (2017).
Dediu, V. A., Hueso, L. E., Bergenti, I. & Taliani, C. Spin routes in organic semiconductors. Nat. Mater. 8, 707–716 (2009).
Tsurumi, J. et al. Coexistence of ultra-long spin relaxation time and coherent charge transport in organic single-crystal semiconductors. Nat. Phys. 13, 994–998 (2017).
Fratini, S., Nikolka, M., Salleo, A., Schweicher, G. & Sirringhaus, H. Charge transport in high-mobility conjugated polymers and molecular semiconductors. Nat. Mater. 19, 491–502 (2020).
Kubo, T. et al. Synthesis, intermolecular interaction, and semiconductive behavior of a delocalized singlet biradical hydrocarbon. Angew. Chem. Int. Ed. 44, 6564–6568 (2005).
Zhang, Y. et al. Temperature tunable self-doping in stable diradicaloid thin-film devices. Adv. Mater. 27, 7412–7419 (2015).
Zong, C. et al. Isomeric dibenzoheptazethrenes for air-stable organic field-effect transistors. Angew. Chem. Int. Ed. 60, 16230–16236 (2021).
Guo, X. & Facchetti, A. The journey of conducting polymers from discovery to application. Nat. Mater. 19, 922–928 (2020).
Venkateshvaran, D. et al. Approaching disorder-free transport in high-mobility conjugated polymers. Nature 515, 384–388 (2014).
Wang, S. et al. Skin electronics from scalable fabrication of an intrinsically stretchable transistor array. Nature 555, 83–88 (2018).
Guo, X., Facchetti, A. & Marks, T. J. Imide- and amide-functionalized polymer semiconductors. Chem. Rev. 114, 8943–9021 (2014).
Yang, J., Zhao, Z., Wang, S., Guo, Y. & Liu, Y. Insight into high-performance conjugated polymers for organic field-effect transistors. Chem 4, 2748–2785 (2018).
Lei, T., Wang, J.-Y. & Pei, J. Design, synthesis, and structure-property relationships of isoindigo-based conjugated polymers. Acc. Chem. Res. 47, 1117–1126 (2014).
Lei, T., Xia, X., Wang, J.-Y., Liu, C.-J. & Pei, J. “Conformation locked” strong electron-deficient poly(p-phenylene vinylene) derivatives for ambient-stable n-type field-effect transistors: synthesis, properties, and effects of fluorine substitution position. J. Am. Chem. Soc. 136, 2135–2141 (2014).
Schott, S. et al. Polaron spin dynamics in high-mobility polymeric semiconductors. Nat. Phys. 15, 814–822 (2019).
Boyle, C. J. et al. Tuning charge transport dynamics via clustering of doping in organic semiconductor thin films. Nat. Commun. 10, 2827 (2019).
London, A. E. et al. A high-spin ground-state donor-acceptor conjugated polymer. Sci. Adv. 5, eaav2336 (2019).
Vella, J. H. et al. Broadband infrared photodetection using a narrow bandgap conjugated polymer. Sci. Adv. 7, eabg2418 (2021).
Sun, H., Guo, X. & Facchetti, A. High-performance n-type polymer semiconductors: applications, recent development, and challenges. Chem 6, 1310–1326 (2020).
Che, Y. & Perepichka, D. F. Quantifying planarity in the design of organic electronic materials. Angew. Chem. Int. Ed. 60, 1364–1373 (2021).
Ji, X. et al. Pauli paramagnetism of stable analogues of pernigraniline salt featuring ladder-type constitution. J. Am. Chem. Soc. 142, 641–648 (2020).
Kang, K. et al. 2D coherent charge transport in highly ordered conducting polymers doped by solid state diffusion. Nat. Mater. 15, 896–902 (2016).
Tanaka, H., Hirate, M., Watanabe, S. & Kuroda, S.-I. Microscopic signature of metallic state in semicrystalline conjugated polymers doped with fluoroalkylsilane molecules. Adv. Mater. 26, 2376–2383 (2014).
Yamaguchi, K., Fueno, T. & Fukutome, H. Molecular-orbital theoretical classification of reactions of singlet ground-state molecules. Chem. Phys. Lett. 22, 461–465 (1973).
Zeng, W. et al. Rylene ribbons with unusual diradical character. Chem 2, 81–92 (2017).
Zeng, Z. et al. Pushing extended p-quinodimethanes to the limit: stable tetracyano-oligo(n-annulated perylene)quinodimethanes with tunable ground states. J. Am. Chem. Soc. 135, 6363–6371 (2013).
Xiong, M. et al. Efficient n-doping of polymeric semiconductors through controlling the dynamics of solution-state polymer aggregates. Angew. Chem. Int. Ed. 60, 8189–8197 (2021).
Hu, H. et al. Design of donor polymers with strong temperature-dependent aggregation property for efficient organic photovoltaics. Acc. Chem. Res. 50, 2519–2528 (2017).
Steyrleuthner, R. et al. Aggregation in a high-mobility n-type low-bandgap copolymer with implications on semicrystalline morphology. J. Am. Chem. Soc. 134, 18303–18317 (2012).
Small, D. et al. Lntermolecular,pi-to-pi bonding between stacked aromatic dyads. Experimental and theoretical binding energies and near-IR optical transitions for phenalenyl radical/radical versus radical/cation dimerizations. J. Am. Chem. Soc. 126, 13850–13858 (2004).
Koike, H. et al. Stable delocalized singlet biradical hydrocarbon for organic field-effect transistors. Adv. Funct. Mater. 26, 277–283 (2016).
Wang, W. et al. Fine-tuning the diradical character of molecular systems via the heteroatom effect. Chem. Commun. 56, 1405–1408 (2020).
Noriega, R. et al. A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nat. Mater. 12, 1038–1044 (2013).
Lei, T., Dou, J.-H. & Pei, J. Influence of alkyl chain branching positions on the hole mobilities of polymer thin-film transistors. Adv. Mater. 24, 6457–6461 (2012).
Fornari, R. P., Blom, P. W. M. & Troisi, A. How many parameters actually affect the mobility of conjugated polymers? Phys. Rev. Lett. 118, 086601 (2017).
Wang, Y., Rogado, N. S., Cava, R. J. & Ong, N. P. Spin entropy as the likely source of enhanced thermopower in NaxCo2O4. Nature 423, 425–428 (2003).
Tam, T. L. D. et al. High spin pro-quinoid benzo[1,2-c;4,5-c']bisthiadiazole conjugated polymers for high-performance solution-processable polymer thermoelectrics. ACS Mater. Lett. 2, 147–152 (2020).
Yan, X. et al. Pyrazine-flanked diketopyrrolopyrrole (DPP): a new polymer building block for high-performance n-type organic thermoelectrics. J. Am. Chem. Soc. 141, 20215–20221 (2019).
Lu, Y. et al. The critical role of dopant cations in electrical conductivity and thermoelectric performance of n-doped polymers. J. Am. Chem. Soc. 142, 15340–15348 (2020).
Russ, B., Glaudell, A., Urban, J. J., Chabinyc, M. L. & Segalman, R. A. Organic thermoelectric materials for energy harvesting and temperature control. Nat. Rev. Mater. 1, 16050 (2016).
Zhang, F. & Di, C.-A. Exploring thermoelectric materials from high mobility organic semiconductors. Chem. Mater. 32, 2688–2702 (2020).
Rivnay, J. et al. Organic electrochemical transistors. Nat. Rev. Mater. 3, 17086 (2018).
Zeglio, E. & Inganas, O. Active materials for organic electrochemical transistors. Adv. Mater. 30, 1800941 (2018).
Acknowledgements
This work is supported by National Natural Science Foundation of China (22075001 and 92156019). The computational part is supported by High-performance Computing Platform of Peking University. X.G. and Y.W. thank NSF (OIA-1757220) for providing support to perform the scattering portion of this work. The authors thank Prof. Zhe-ming Wang, Prof. Hao-ling Sun, Ye-xin Wang, Dr. Shen Zhou for their kind help in SQUID and EPR measurement. T.L. thanks the beamline BL14B1 (Shanghai Synchrotron Radiation Facility) for providing beam time for GIWAXS measurement of the p(DPP-BBT) film.
Author information
Authors and Affiliations
Contributions
X.X.C. and G.C.L. synthesized the polymers. X.X.C. and X.Y.D. performed polymer property characterization and DFT calculation. J.T.L. and X.Q.W. performed device fabrication and characterization. Y.W. and X.G. performed the X-ray scattering measurement and data analysis. Y.H.F. and S.D.J. performed EPR measurement. X.X.C., J.T.L., and T.L. wrote the manuscript. All the authors revised and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wenlan Liu, Shun Watanabe and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, XX., Li, JT., Fang, YH. et al. High-mobility semiconducting polymers with different spin ground states. Nat Commun 13, 2258 (2022). https://doi.org/10.1038/s41467-022-29918-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-29918-w
This article is cited by
-
Open-Shell Oligomers and Polymers: Theory, Characterization Methods, Molecular Design, and Applications
Chinese Journal of Polymer Science (2024)
-
Diketopyrrolopyrrole-based Conjugated Polymers as Representative Semiconductors for High-Performance Organic Thin-Film Transistors and Circuits
Chinese Journal of Polymer Science (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
