
Abstract
Structural snapshots of protein/ligand complexes are a prerequisite for gaining atomic level insight into enzymatic reaction mechanisms. An important group of enzymes has been deprived of this analytical privilege: members of the protein tyrosine phosphatase (PTP) superfamily with catalytic WPD-loops lacking the indispensable general-acid/base within a tryptophan-proline-aspartate/glutamate context. Here, we provide the ligand/enzyme crystal complexes for one such PTP outlier: Arabidopsis thaliana Plant and Fungi Atypical Dual Specificity Phosphatase 1 (AtPFA-DSP1), herein unveiled as a regioselective and efficient phosphatase towards inositol pyrophosphate (PP-InsP) signaling molecules. Although the WPD loop is missing its canonical tripeptide motif, this structural element contributes to catalysis by assisting PP-InsP delivery into the catalytic pocket, for a choreographed exchange with phosphate reaction product. Subsequently, an intramolecular proton donation by PP-InsP substrate is posited to substitute functionally for the absent aspartate/glutamate general-acid. Overall, we expand mechanistic insight into adaptability of the conserved PTP structural elements.
Similar content being viewed by others


Introduction
Considerable efforts continue to be made to understand the molecular basis of enzyme-catalyzed hydrolysis of phosphate esters and anhydrides1. A particular challenge for this objective is presented by the protein tyrosine phosphatase (PTP) family, in no small part because phosphotyrosine phosphatase activity is not the only function for this family of enzymes2. A significant number of PTPs dephosphorylate alternate substrates such as RNA, phosphatidylglycerophosphate, inositol phospholipids, and a specialized class of signaling molecules known as diphospho-myo-inositol polyphosphates (inositol pyrophosphates, or PP-InsPs; Fig. 1a, b)2,3,4,5,6,7,8. Despite the evolution of these catalytic differences, there has been a high degree of conservation of key structural features of the PTP active site (Supplementary Fig. 1)2,3,4,5,9. One of these prominent structural elements is a flexible loop named WPD after its three most highly conserved residues, which includes an Asp (or occasionally Glu) that is typically described as an indispensable proton-donor to the leaving group (Supplementary Figs. 1, 2a). This catalytic acid is inserted into the active site by the closure of the WPD loop. Much attention is being devoted to determining how differences in conformational dynamics of this loop can contribute to catalytic versatility within the PTP family9,10.

Michaelis–Menten kinetic plots are shown for the phosphatase activities of AtPFA-DSP1 towards: (a), 5-InsP7, (b), 1,5-InsP8, (c), 6-InsP7, (d), 5-PP-InsP4 and (e), 4-InsP7. Activity data (circles, some overlapping) are from each independent experiment at which the indicated substrate concentration was tested; the total number of such experiments is given above each data set in blue font. Km values were calculated when statistically appropriate. The insets in panels (a–e) depict chair conformations of each substrate; the positions of each β-phosphate are emphasized in red. In panel (f), vertical bars represent mean values of activities against the weakest substrates when all were assayed at 10 µM concentrations. Activity data (circles, some overlapping) are from each independent experiment; the total number of such experiments is given above each data set in blue font. Phenylphosphate is abbreviated as Phenyl-P. Structures of the inositol phosphates are given as chair conformations in panels (g) (1-InsP7), (h) (2-InsP7, (i) (3-InsP7) and (j) (InsP6). Locants (using standard nomenclature for myo-inositol) are provided with the structures of 5-InsP7 and InsP6. Source data are provided as a Source Data file.
Nevertheless, there are examples of PTPs that show intriguing departures from the canonical aspects of the WPD loop. For example, the human DUSP23 gene encodes a VH1-like member Z (VHZ) protein in which the WPD-loop catalytic-acid, Asp65, is subservient to a remote Glu134 fulfilling the primary general acid function11. The ability of substrate to enter the catalytic pocket in either of two binding modes allows Asp65 to substitute as the catalytic acid when Glu134 is mutated11. Other similar dual general acid PTPs have been identified (e.g., TkPtp expressed by the hyperthermophilic archaeon Thermococcus kodakaraensis KOD112). Furthermore, there are some important PTPs in which the WPD motif is entirely absent from the host loop (Supplementary Fig. 2a). These enzymes include human CDC25 phosphatase, which supervises cell-cycle checkpoints13, the phosphoinositide/protein phosphatase PTEN14,15, MCE1, an mRNA capping enzyme that is essential for mRNA processing16, Baculovirus RNA 5ˈ-phosphatase8 and its human ortholog, DUSP11/PIR17, which participates in innate immune responses to viral infection17, and Siw14, a PTP-type PP-InsP phosphatase in Saccharomyces cerevisiae6 (Supplementary Fig. 2a,b). There has been only limited speculation in the literature as to how PTP reactions might proceed in the complete absence of a classical WPD-loop catalytic acid7,8,13,14,18,19. Practical progress towards resolving this deeply puzzling situation requires high resolution structures of members of this particular group of enzymes (i.e., those described in Supplementary Fig. 2a) in complex with their natural substrate. To date, no published studies of these specific enzymes have yielded the necessary enzyme/substrate complexes. Our goal has been to bridge this significant gap in our mechanistic understanding.
In this work, we begin by adding a protein to the list of PTPs that lack the WPD motif (Supplementary Fig. 2a), i.e., an Arabidopsis thaliana ortholog of Siw14 that that is encoded by a gene at locus tag At1g0500020. Hereafter we refer to the protein by its alternate nomenclature: AtPFA-DSP1. We demonstrate that this PTP actively hydrolyzes PP-InsPs. This is a significant accomplishment in itself: little is known about the nature of phosphatases in plants that might act as signaling off-switches by hydrolyzing PP-InsPs21,22, even though these polyphosphates license molecular defenses against herbivorous larvae and necrotrophic pathogens23, and supervise homeostasis of the growth-limiting phosphate micronutrient24. Next, we describe the crystal structures of the catalytic core of AtPFA-DSP1 (Supplementary Table 1) in complex with PP-InsPs and also a fluorinated PP-InsP analog25 at resolutions of up to 1.7 Å. These structural complexes include a variety of pre-reactant-, reactant-, intermediate- and product-bound states. Analysis of these structures, in concert with descriptions of ligand specificity and kinetic properties, together provide a data-driven proposal for a PTP reaction cycle that does not utilize any amino-acid residue as a general acid. Instead, we posit that PP-InsP substrate itself provides a proton to the leaving phosphate group, via a water relay. Additionally, we conclude that the hydrolyzed phosphate remains trapped as an enzyme-product complex, until it can be released in a prisoner exchange with another PP-InsP molecule; the latter process is assisted by the WPD loop. Overall, our description of this reaction cycle dramatically expands understanding of the mechanistic significance of diversity in WPD loop chemistry, while not diverging from the overall context of conservation of structural elements. Our array of structural snapshots also represents a valuable resource for future practical and computational studies of non-canonical chemical barcodes within the WPD loop that tailor reactivity of individual PTP family members.
Results and discussion
AtPFA-DSP1 is an active and selective PP-InsP phosphatase
A previous study18, has demonstrated that AtPFA-DSP1 non-selectively hydrolyzes nucleotides and a variety of other phosphorylated substrates, although the authors acknowledged these molecules were unlikely to be physiologically relevant substrates. We confirmed slow rates of dephosphorylation for such molecules (at 10 µM; mean values (nmol min−1 mg−1) ± standard errors, followed by the number of independent experiments in parentheses; source data are provided as a Source Data file): ATP, 14 ± 2 (8); GTP, 13 ± 2 (9); phosphoribosyl pyrophosphate 15 ± 4 (5). Low activity towards phenyl phosphate 5.0 ± 1.5 (Fig. 1c) excludes protein phosphotyrosine as a natural substrate. Furthermore, we found up to two orders of magnitude higher rates of phosphatase activity against three naturally occurring PP-InsPs: 5-diphosphoinositol 1,2,3,4,6-pentakisphosphate (5-InsP7) (Fig. 1a), 1,5-bis-diphosphoinositol 2,3,4,6-tetrakisphosphate (1,5-InsP8) (Fig. 1b) and 6-diphosphoinositol 1,2,3,4,5-pentakisphosphate (6-InsP7) (Fig. 1c) Note that 6-InsP7 has only been definitively identified in Dictyostelids26, however, Arabidopsis and rice synthesize material identified as 6-InsP7 and/or (diphosphoinositol 1,2,3,5,6-pentakisphosphate (4-InsP7)27). We are not aware of a previous description of a standalone PP-InsP phosphatase expressed in any plant.
This enzyme also shows strong positional specificity. For example, the specificity constant (kcat/Km) for 5-diphosphoinositol 1,3,4,6-tetrakisphosphate (5-PP-InsP4), which has a 2-hydroxyl group, is approximately 6-fold lower than that for 5-InsP7 (see Fig. 1a, d), from which we conclude that the 2-phosphate of 5-InsP7 contributes to substrate recognition. Moreover, the enzyme has only weak activity against 4-InsP7 (Fig. 1e) and 1-diphosphoinositol 2,3,4,5,6-pentakisphosphate (1-InsP7) (Fig. 1f, g) and negligible activity towards either 2-diphosphoinositol 1,3,4,5,6-pentakisphosphate (2-InsP7), 3-diphosphoinositol 1,2,4,5,6-pentakisphosphate (3-InsP7) or inositol hexakisphosphate (InsP6) (Fig. 1f, h–j). Note that 2-InsP7 and 3-InsP7 have to date not been identified in any organism.
Overall structure of AtPFA-DSP1
A low resolution (3.5 Å) structure of an AtPFA-DSP1 protein construct has previously been described, but significantly, without any candidate natural substrate in the crystal complex18. Consequently, the latter structure is not instructive for understanding reaction mechanisms. Nevertheless, it provided a template for molecular replacement to help us solve the structure of our fully traceable AtPFA-DSP149–215 construct, to include several high-resolution crystal complexes (1.65–1.9 Å) (Supplementary Table 1, Supplementary Fig. 2b, c). There are two molecules in each asymmetric unit, although gel filtration analysis showed that the protein construct behaves as a monomer in solution (Supplementary Fig. 2d). The core structure of PFA-DSP149–215 is similar to the equivalent region of the ScSiw14 ortholog (RMSD = 0.621 Å, derived by superimposing 798 comparable atoms; Supplementary Fig. 2e, f). Both structures exhibit a canonical PTP fold, in which their catalytic sites are defined by the three substrate-binding loops. The most N-terminal of these structural elements in AtPFA-DSP1 is equivalent to the WPD loop of canonical PTPs, although it does not contain the eponymous tripeptide motif (Supplementary Fig. 2a, f). Note that this loop makes a significant contribution to active site topology, as does the equivalent loop in canonical PTPs (Supplementary Fig. 2a, f). The C-terminus of the WPD loop in AtPFA-DSP1 retains a highly conserved Pro that in other PTPs is believed to act as a hinge to help dictate the range of motion of this flexible structural element9. Two additional substrate binding loops are described here as the P-loop and α5–α6 loop; these are equivalent to the P-loop and Q-loop of other Cys-based PTPs3,4. The amino-acid sequences of these catalytically important loop structures are extremely well conserved in other plant orthologs (Supplementary Fig. 2g).
These particular crystals of AtPFA-DSP149–215 contain inorganic phosphate (Pi) in the catalytic center, proximal to the N-terminus of helix α4 (Fig. 2a). The oxygen atom that is positioned at the apex of the phosphate ion’s tetrahedral geometry points away from α4. This oxygen makes a hydrogen bond with the unprotonated Nδ1 of His155 (we will return to this point below). We denote this orientation of Pi as pose A, i.e., Pi(A). The three other oxygen atoms of this Pi are intensively coordinated by amide groups of P-loop residues Lys151, Arg152 Lys154, His155, and Arg156. The side chains of Arg156 and Cys150/Ser150 also make polar contacts with Pi(A) (Fig. 2a, b). A previous structural analysis of the ScSiw14 orthologue found the active site occupied by a sulfate ion19 that is in an orientation equivalent to the Pi(A) pose (see below).

Panels (a, b), show the phosphate ion in stick and ball format (phosphorus is orange and oxygen in red) within a mixed stick- and ribbon-style rendition of the catalytic center of wild type and the C150S protein constructs, respectively (nitrogen is blue, sulfur is yellow). The omit Fo-Fc electron density maps, contoured at 5σ, are shown in green mesh; broken black lines show polar contacts. Corresponding ligand–protein interactions created by Ligplot+ are shown below each graphic. Source data are provided for (a, b) as PDB accession codes 7MOK and 7MOD, respectively.
Analysis of enzyme/substrate crystal complexes
A central goal of the current study was to prepare structural snapshots of enzyme/substrate crystal complexes for a PTP that does not utilize a canonical catalytic acid. Deriving such crystals for AtPFA-DSP1 was initially problematic; we were unable to displace enzyme-associated Pi by soaking crystals of wild type enzyme with 5-InsP7. This observation also speaks to how tightly the enzyme holds on to the Pi product (and see below). We therefore used a substrate-trapping strategy3 by mutating Cys150 to Ser, which reduced 5-InsP7 phosphatase activity 4000-fold (Table 1; Supplementary Fig. 3). Pi persisted in crystals of the freshly isolated AtPFA-DSP149–150,C150S,151–215 mutant (Fig. 2b; Supplementary Fig. 4a), but it could now be successfully substituted by 5-InsP7 (Supplementary Table 1; Supplementary Fig. 4b), presumably because Pi is held less tightly than is the case with wild type protein.
The 5-InsP7 substrate is held in an 11.6 Å wide and 15.2 Å deep pocket by polar interactions between the 1-, 4-, 5β-, and 6-phosphates and multiple residues in the P-loop: Lys151, Arg152, Lys154, His155 and Arg156 (Fig. 3a–c). These data also reveal that the His155 imidazole ring is flipped (180° χ2 angle changes) relative to its orientation in the Pi-bound crystal structures (Fig. 3b, c; Supplementary Fig. 4a, b). We analogize such behavior by depicting His155 as a Janus residue, such that the catalytic core is overlooked by either of the two faces of the imidazole ring, depending upon the nature of the ligand: in this case, 5-InsP7 (Fig. 3a–c) or Pi(A) (Fig. 2a, b). The protonated Nε2 in His155 forms a polar contact with the PP-InsP substrate, and the Nδ1 forms a hydrogen bond with the hydroxyl group of Asp191, in which the carbonyl oxygen is further stabilized by Arg188 (Supplementary Fig. 4b). Support for the proposed catalytic significance of Asp191 and His155 emerged from mutagenic studies, which reduced activity by >90% (Table 1).

a Surface representation colored to match structural elements correspond to gray for P-loop, cyan for α5-α6 loop, purple for WPD-loop, and yellow for the remainder. The 5-InsP7 is shown in stick format; carbon is white, phosphorus is orange and oxygen is red. Phosphate groups are numbered according to standard nomenclature. Panel (b) shows a similar orientation of 5-InsP7, with key interacting residues in stick format; nitrogen is blue, and oxygen. Panel (c) is a rendering of the ligand–protein interactions created by Ligplot+. Polar contacts within 3.2 Å are depicted with broken green lines. Hydrophobic interactions are shown in grey eyelash style. Equivalent data are shown in panels (d, e) for 6-InsP7 as the ligand; the latter’s carbons are colored dark gray. Panel (f) superimposes 6-InsP7 (dark gray; numbers denote positions of 2- and 6-phosphates) upon 5-InsP7 (light gray; boxed numbers denote 2- and 5-phosphates). Panels (g, h) compare the chemical structures of the α-phosphono-α,α-difluoroacetamide group (PCF2Am; blue) and the 5-diphosphate group (PP; red)respectively. Panels (i, j), show binding interactions for 5-PCF2Am-InsP5 (carbon is pink, and fluorine is cyan). Panel (k) superimposes 5-PCF2Am-InsP5 upon 5-InsP7 using the same color schemes as in panels (i, f) The omit Fo-Fc electron density maps, contoured at 5σ, are shown in green mesh. Source data files are provided as PDB accession codes 7MOE, 7MOF and 7MOG.
The 1- and 2-phosphates of 5-InsP7 also make multiple polar interactions with Lys186 in the α5-α6 loop (Fig. 3b, c). Gly118 and Lys120 from the WPD-loop make contributions to ligand binding with van der Waals interactions. Ser150, which replaces the catalytic cysteine, is located at the bottom of the ligand binding pocket and it helps to trap 5-InsP7 through its polar contact with one of the terminal oxygen atoms from the 5-β-phosphate moiety (Fig. 3a–c). We further noted that if a 1-β-phosphate were to be added to 5-InsP7, it would be solvent exposed, thereby rationalizing why 1,5-InsP8 is also actively hydrolyzed by this enzyme (Fig. 1b). Interestingly, catalytically productive binding of 1-InsP7, 2-InsP7 and 3-InsP7 is hindered by P-loop residues that would clash with either the axial 2-diphosphate or 2-monophosphate. This helps to explain why 1-InsP7, 2-InsP7 and 3-InsP7 are not efficient substrates for AtPFA-DSP1 (Fig. 1f).
Our observation that AtPFA-DSP1 exhibits similar catalytic activities towards both 5-InsP7 and 6-InsP7 (Fig. 1c) is rationalized by our structural data: due to the symmetry properties of the myo-inositol ring, the orientation of key, structural recognition features of 5-InsP7–particularly the diphosphate group and the flanking phosphate groups–are spatially mimicked by 6-InsP7 as a consequence of the latter being rotated in the active site relative to the position of 5-InsP7 (Fig. 3d–f). Thus, residues that interact with 5-InsP7 have an equivalent function with 6-InsP7. Interestingly, the WPD-loop makes a contribution to 6-InsP7 binding: Lys120 has polar contact with the 2-phosphate, while Gly118 has van der Waals interactions (Fig. 3d, e). Dictyostelium discoideum is known to synthesize 6-InsP726; our accommodation of the latter into the active site of AtPFA-DSP1 may be a realistic structural model to rationalize putative 6-InsP7 phosphatase activity by a candidate DdPFA-DSP1 gene (DDB_G0285909). Arabidopsis and rice synthesize material identified as 6-InsP7 and/or 4-InsP727.
There is considerable interest in the development of metabolically resistant PP-InsP bioisosteres that yield useful structural information for inhibitor development, and may also assist in deriving recalcitrant crystal complexes (see28). One recent development has been to replace the scissile 5-β-phosphate of 5-InsP7 with a phosphonodifluoromethyl (PCF2) group to yield 5-PCF2Am-InsP5 (Fig. 3g–i)25; for the purpose of synthetic strategy and analogue stability, the 5-α-phosphate group is replaced by an acetamide linkage. Interestingly, the addition of a PCF2 group to substrate analogs of certain protein tyrosine phosphatases can increase ligand affinity29,30. Furthermore, we25 have previously hypothesized that the binding affinity of 5-PCF2Am-InsP5 might be enhanced for ligand/protein interactions that do not involve Mg2+ (as is the case for the capture of PP-InsPs by AtPFA-DSP1). Indeed, we obtained a crystal complex of AtPFA-DSP149–150,Cys150Ser,151–215 with 5-PCF2Am-InsP5 in the active site (Fig. 3i–k). The orientation of 5-PCF2Am-InsP5 is very similar to that of 5-InsP7, except that the 5-PCF2Am group makes a gain-of-function interaction between one fluorine atom and the guanidinium group of Arg156. Our data validate the value of 5-PCF2Am-InsP5 for the study of specific modes of PP-InsP/protein interactions.
We also obtained instructive crystals of AtPFA-DSP1 in complex with 5-PP-InsP4 (Fig. 4) in which the orientation of the ligand differed between the two asymmetric units. In one, 5-PP-InsP4 adopts a configuration that is very similar to that of 5-InsP7, with an extended α-β phosphoanhydride that projects its β-phosphate into the catalytic pocket (‘β-IN’ pose, Fig. 4a; Supplementary Fig. 4c–f). In the other asymmetric unit, the β-phosphate of 5-PP-InsP4 is diverted away from the catalytic pocket (‘β-OUT’ pose) at a near right angle or a cis conformation to the α-phosphate; the phosphorus atom of this β-phosphate is 4 Å displaced from its equivalent in the β-IN pose (Fig. 4a–f; Supplementary Fig. 4d, g, h). These conclusions are strengthened by the low values for the β-factors associated with the two alternate positions of the β-phosphate (Supplementary Fig. 4f, g). Remarkably, PP-InsP4 β-OUT substrate and Pi product are captured together in the same crystal complex (Fig. 4d, f; Supplementary Materials Fig. 4d). Bearing in mind that we could not derive crystals of the apoenzyme (i.e., either Pi and/or substrate is always present), we hypothesize that these crystal complexes describe two reaction states of a process that we analogize as a prisoner exchange: entry of PP-InsP substrate is tightly coupled to Pi exit. Interestingly, the WPD-loop appears to play a particularly significant role in delivering substrate to the exchange point: in the β-OUT configuration, Lys120 in this loop has three interactions with the substrate’s 2-hydroxyl and 3-phosphate groups, and Asn119 interacts with the 5-β-phosphate (Fig. 4d, f). All of these interactions are absent from the β-IN configuration (Fig. 4c, e). Also of interest in that the Janus residue His155 imidazole ring is flipped 180° in the β-OUT pose relative to its β-IN orientation (Supplementary Fig. 4e, h).

Panels (a, b) are surface representations of the ligand binding pocket, with 5-PP-InsP4 drawn in stick format, in poses labeled β-IN (pale lavender carbons) and β-OUT (green carbons), respectively; phosphorus is orange and oxygens are red. Note the presence of Pi(B) (in stick and ball format) in panel (b). Panels (c, d) show the corresponding stick depictions of 5-PP-InsP4 and its interacting amino-acid residues (nitrogens are colored blue). The Fo-Fc electron density maps (green mesh) are contoured at 3σ in panel (c) and 5σ in panel (d). Panels (e, f) are renderings of the corresponding ligand–protein interactions created by Ligplot+. The source data file is provided as PDB accession code 7MOH.
Another key observation to emerge from these experiments with AtPFA-DSP149–150,Cys150Ser,151–215 is that in the crystal complex that contains 5-PP-InsP4 substrate and Pi, the latter’s oxygen atom at the apex of the tetrahedral structure points towards the α4 helix (Supplementary Fig. 4d, 5a). We name this orientation as Pi(B), to distinguish it from Pi(A), in which the apex of the tetrahedron points away from the α4 helix (Fig. 2). We are aware of only one other PTP structure that contains Pi in a pose equivalent to Pi(B): this protein is a mutant version of Wzb (the catalytic Cys is mutated to Ala; Supplementary Fig. 5b), a low molecular weight PTP produced by the prokaryote pathogen Vibrio vulnificus31. On the other hand, two crystal complexes, PTPMT1 (PDB: 3RGO32) and MTMR6 (PDB: 2YF0) contain a Pi(B)-like conformation of a sulfate ion, in which the oxygen atom at the apex of the tetrahedron points towards the α helix and forms two hydrogen bonds with S109 or T343, respectively (Supplementary Fig 5c, d). In contrast, a sulfate ion in a crystal complex with ScSiw1419 is in the pose equivalent to that for Pi(A) (Supplementary Fig 5e).
We also observed the Pi(B) pose in crystals of wild type enzyme prepared in the absence of β-mercaptoethanol (Fig. 5a, b). In this Pi(B) pose, the side chain of Cys150 may adopt alternate conformations, one of which may permit an intramolecular disulfide bond with Cys92 (Fig. 5a, c). Another distinguishing feature of Pi(B) is that it makes an interaction with the backbone of His155 (Figs. 4f; 5a, b), whereas Pi(A) makes polar contact with the His155 side chain (Fig. 2). Furthermore, we noted that Pi(B) can make polar contacts with both the side chain and amide backbone of Thr157 (Fig. 5a, b) whereas Pi(A) does not interact with this residue (Fig. 2). The sidechain of Thr157 could also contribute to catalysis if it were to assist in lowering the pKa of Cys150 (see below). Evidence that a Thr157 is catalytically important was derived from analysis of a Thr157Ala mutant, which reduced enzyme activity >97% (Table 1).

a Pi(B), in stick and ball format (phosphorus is orange, oxygen is red), in a crystal structure complex with AtPFA-DSP149–215 (PDB accession code 7MOL) obtained in the absence of mercaptoethanol (see text). Broken lines depict polar interactions of Pi(B) (≤3.2 Å) with nearby residues (nitrogens are blue). The Fo-Fc electron density map (green mesh) is contoured at 5σ. b corresponding rendering of the ligand–protein interactions created by Ligplot+. c Pi(B) from panel (a) is superimposed upon Pi(A) (taken from Fig. 2a); the P-loops of the corresponding proteins are colored gray for Pi(B) and light brown/dark brown for Pi(A). Note the two conformations of Cys150, one of which may form a disulfide bond (see panel a). d, e the movement of the phosphorus atom is indicated by the orange trace, and each of the oxygen atoms of Pi(A) were arbitrarily colored either purple, light pink, red or green; this color scheme illustrates the predicted movements of each atom during molecular dynamics simulations, with reference to the plane of the backbone residues of the P-loop (blue rectangle; residues 151–156). Horizontal bars illustrate the switching between Pi poses A (black) and B (white). The coloration in this simulation has been transferred to Pi(B) in panels (c) and (e). Three additional replicates of the data in panel (d) are provided in Supplementary Fig. S8, and the initial 1000 ns of each of these aspects of all four simulations are animated in Supplementary Movies 1, 2, 3 and 4. Source data are provided as a Source Data file.
Pi rotation within the catalytic pocket
Our capture of two alternate static poses of Pi within the catalytic site (Fig. 2; Fig. 5a–c) prompted us to consider the possibility of there being dynamic interchange between them. To interrogate this idea, four independent 2µs molecular dynamics simulations of AtPFA-DSP149–215 were initiated with di-anionic Pi(A) in the catalytic pocket (i.e., structure 7MOK; Supplementary Table 1). Throughout each of these simulations, the root mean squared deviations of the heavy backbone atoms settled around 2 Å (Supplementary Fig 6a), indicative of a relatively stable overall protein configuration. From the root mean square fluctuations for Cα of the individual amino acids (Supplementary Fig 6b) it is concluded that the P-loop is a particularly stable entity. Moreover, dynamic cross correlation maps indicate no significant correlation between the movements of the P-loop and the considerably more dynamic WPD loop (Supplementary Fig 7). Thus, we used the relatively stable plane of P-loop backbone residues 151 to 156 as a reference point for monitoring the dynamics of the Pi ligand. Remarkably, these simulations show reorientations of Pi between two conformations that closely resembled those of Pi(A) and Pi(B) (Fig. 5d, e; Supplementary Fig. 8; Supplementary Movies 1–4), each of which has ionic interactions with P-loop residues (Supplementary Fig. 9). An analysis of the positional distribution of the phosphorus atom is consistent with a binary distribution of Pi between configurations A and B in the ratio 1:4 (Supplementary Fig. 10). Moreover, the one oxygen atom in Pi that is the most distant from the P-loop plane (as in Pi(A)) is only observed during 20% of the simulation time (Supplementary Fig. 10). The observed preference for the Pi(B) conformation found in our simulations is consistent with its stronger interaction free energy value (−81.6 ± 2.2 kcal/mol) compared to pose A (−68.1 ± 1.4 kcal/mol). Finally, in pose B, the phosphorus atom is on average 1.1Å closer to the P-loop plane compared to its position in pose A, which compares well with the difference of 1.5Å that was determined experimentally (Fig. 5c).
Contending with the conundrum of PTP-catalyzed substrate hydrolysis in the absence of an Asp/Glu catalytic acid
In canonical PTPs the WPD motif includes a highly conserved general acid—an Asp (or occasionally Glu)—that is typically considered to be an indispensable proton-donor to the leaving group (Supplementary Fig. 1). This motif is absent from AtPFA-DSP1, other plant orthologs and ScSiw14 (Supplementary Fig. 2a, f, g). We have not identified another candidate Asp/Glu from outside the WPD loop. Furthermore, the proximity of Arg156 to the highly negatively charged PP-InsP substrate should oppose any dramatic reduction in the value of its guanidinium pKa that would be required for it to act as a general acid33. Also, there is no precedent in the PTP field for an Arg to perform a catalytic acid function33.
It is therefore significant that our multiple PP-InsP/enzyme structures include a spatially conserved water molecule, Wat1 (Fig. 6a; Supplementary Fig. 11a, b), that forms a polar contact with the β-phosphate, a hydrogen bond with the diphosphate’s bridging oxygen atom, and a polar contact with an adjacent monophosphate group. Interestingly, Wat1 is not observed in a crystal complex with phenyl phosphate (Supplementary Fig. 11c), which is not an efficient substrate (Fig. 1c), even though the latter’s phosphate group is almost superimposable upon the β-phosphate of 5-InsP7 (Supplementary Fig. 11d). Thus, we posit that PP-InsPs recruit Wat1 to assist their own hydrolysis: for 5-InsP7 as substrate, we propose that a proton from the C-4 phosphate is relayed by Wat1 to the bridging oxygen atom of the diphosphate on the neighboring C-5, thereby stabilizing the phosphate leaving group (Fig. 7). This enzymatic requirement for a water molecule to shuttle a proton from a monophosphate to a diphosphate could make a significant contribution to catalytic regiospecificity while reducing the substrate’s intramolecular steric and electrostatic interference. This is a striking example of enzymatic adaptation to the unique physicochemical properties of the PP-InsP family.

a the relative positions of a catalytic water (Wat1; pink sphere) and 5-InsP7 (stick format) in a crystal complex with AtPFA-DSP149–150,C150S,151–215 (PDB accession code 7MOD); polar interactions are highlighted with broken black lines, with bond distances marked in Å. The omit Fo-Fc electron density map is contoured at 5σ and shown in green mesh. b a reaction intermediate that we designate to be a metaphosphate anion (ball and stick format; orange for phosphorus, red for oxygens). The crystal complex depicted in this panel was obtained by soaking 0.1 mM 5-InsP7 into AtPFA-DSP149–215 at pH 8.0 for 2 h (7MOM; Supplementary Table 1). Broken black lines designate polar interactions with residues (stick format; blue for nitrogen, yellow for sulfur). The omit Fo-Fc electron density map is contoured at 4σ and shown in green mesh. A putative reactive water molecule (Wat2) is shown as a purple sphere. Distances are described in Å. His155 is shown in Nε2-protonated τ tautomer state. c relative positions of Wat2 and the putative metaphosphate to illustrate distances between elements (Å); those within polar interaction distance are depicted as broken gray lines. The omit Fo-Fc electron density map is contoured at 4σ and shown in green mesh. Panel (d) is a rendering of the corresponding ligand–protein interactions created by Ligplot+. Panel (e) shows polar interactions (<3.2 Å; broken black lines) that Cys150 has with Thr157 and His149.

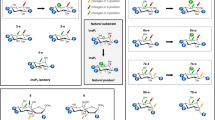
Key polar contacts are highlighted with black dashed lines. Coloring of oxygen atoms is only for illustrative purposes. State 1 depicts presumed canonical nucleophilic attack by Cys150 on the β-phosphate of enzyme-bound 5-InsP7. We presume a thiophosphate intermediate is formed, although we did not capture one in our crystals. Also shown is a water molecule (Wat1) that we postulate to shuttle a proton from the substrate’s 4-phosphate to the diphosphate’s bridging oxygen, with the release of InsP6 product. State 2 depicts the proposed activation by His155 of a second reactive water (Wat2) to facilitate its capture by an enzyme-stabilized metaphosphate intermediate. The resulting enzyme-bound Pi, rotating between its ‘A’ and ‘B’ orientations, are depicted in States 3a and 3b. Despite this limited mobility, Pi remains trapped within the active site until a prisoner exchange with fresh substrate, so that a new catalytic cycle can begin. Some cautionary notes: all protonation states are illustrative and not definitive (nevertheless, there is consensus that the phosphate group that is targeted for hydrolysis by PTPs is di-anionic9), and it has previously been argued a metaphosphate intermediate cannot exist38 (see main text for further discussion).
Furthermore, we superimposed the AtPFA-DSP1-bound 5-InsP7 upon the tyrosine from an Asp-Ala-Asp-Glu-Tyr-Leu-vanadate substrate analog/PTP1B crystal complex34; this Tyr overlays the 5- and 6-phosphates of 5-InsP7 (Supplementary Fig. 11e). Thus, some basic structural elements of AtPFA-DSP1 and PTP1B are well conserved geometrically, despite the huge physicochemical differences in the nature of their respective substrates. Nevertheless, these data lead to an important conclusion that it would be electrostatically and sterically incompatible for AtPFA-DSP1 to have a general acid that is spatially equivalent to Asp181 in PTP1B, as it would clash with the negatively charged PP-InsP substrate (Supplementary Fig. 11e). An additional viewpoint (building on the proposed proton relay discussed above) is that the 4-phosphate of 5-InsP7 functionally replaces the catalytic Asp residue.
Identification of a metaphosphate-like reaction intermediate in the AtPFA-DSP1 reaction cycle
In experiments in which crystals of wild type AtPFA-DSP149–215 were soaked with reduced concentrations of 5-InsP7 at pH 8.0, we observed a near-planar, triangular-shaped electron density consistent with a metaphosphate; all three of its presumed P–O bonds are an appropriate 1.5 Å in length (Fig. 6b–d). There was no evidence of a pentacovalent phosphorane that would have signified an associative reaction pathway35. The putative metaphosphate intermediate in wild type enzyme is stabilized by intensive polar contacts with a cluster of residues in the P-loop, Lys151, Arg152, Lys154, His155, and Arg156, including the latter’s positively charged guanidinium group (Fig. 6b–d; Fig. 7).
Computational studies with PTPs support the idea that a metaphosphate-like species occurs during the formation and subsequent hydrolysis of the canonical PTP Cys-thiophosphate intermediate9,36. If formation of the latter is also a characteristic of AtPFA-DSP1, the pKa of Cys would need to be significantly depressed37. Other PTPs accomplish this through a network of hydrogen bonds that the catalytic Cys has with residues in the P-loop and the contiguous α4 helix37. Thr157 and His149 could potentially contribute to such a phenomenon in AtPFA-DSP1 (Fig. 6e), although we have not observed a thiophosphate intermediate.
We acknowledge authoritative reviews of the phosphoryl transfer and phosphatase literature which discount the possibility that a metaphosphate can accumulate as a reaction intermediate1,38. Nevertheless, there are descriptions of a metaphosphate as an intermediate in the reaction cycle of fructose-1,6-bisphosphatase39,40 and an evolutionarily related vanadium chloroperoxidase41 which, to our knowledge, have never been disproved experimentally. On the other hand, a previous description of a pentacoordinate phosphorane intermediate in crystals of β-phosphoglucomutase42 is now recognized to be a case of mistaken identity: the species in question turned out to be MgF3−, which was contributed by the crystallization buffer43. Both Mg2+ and F− are present in our crystallization buffers. Therefore, we modeled 2mFo-Fc difference maps contoured at 1.5 σ; this analysis revealed that the longer Mg-F bond lengths of 1.8 Å extend beyond the center of the electron density and approach its boundary (Supplementary Fig 12a); further modeling with Fo-Fc difference maps shows that negative density features are especially prominent for two of the three fluoride atoms (Supplementary Fig 12b).
We also detected a metaphosphate-like molecule in a crystal complex obtained upon soaking 5-InsP7 into AtPFA-DSP149–150,Cys150Ser,151–215 at pH 10 for one day (Supplementary Figs. 12c, d, 13). Bearing in mind uncertainty over side chain pKa values in a protein microenvironment44, we posit that a finite degree of reactivity is licensed by an extended time of incubation of the protein at very alkaline pH, along with the proximity of Ser150 to the β-phosphorus atom of 5-InsP7 (Fig. 6a; Supplementary Fig. 13). The modeled Fo-Fc electron density difference for a metaphosphate is below 1 σ (Supplementary Fig. 12c), indicating that there is excellent agreement between the experimental data and the model. In contrast, the modeled Fo-Fc for MgF3- again showed significant disagreement of electron density features (Supplementary Fig. 12d).
We have considered how a metaphosphate intermediate might be incorporated into a proposed reaction pathway. A metaphosphate is an excellent target for capturing a polarized water molecule, to generate the Pi end-product45. We identified a potential candidate, Wat2, in both wild type and Cys150Ser mutant enzymes, and in both cases Wat2 could be activated by the His155 Janus residue (Fig. 6b–d; Fig. 7; Supplementary Fig. 13). The Nδ1 in His155 is in the Nε2-protonated τ tautomer state, stabilized by a carbonyl oxygen from the backbone of Lys186 (Fig. 6b; Supplementary Fig. 13). In addition, a π-cation interaction can be formed between Arg188 and His155 (Fig. 6b, Supplementary Fig. 13) which is expected to further assist the latter’s deprotonation46. Nevertheless, in the wild type enzyme, the 3.7 Å distance and 144° angle from Wat2 to the metaphosphate are not an optimal environment for this reaction, which in addition to the latter’s intensive contacts with P-loop residues (see above), are factors that may assist our crystallographic capture of this proposed intermediate state. In such an event, we propose His155 substitutes for the general base activity of the missing catalytic Asp/Glu), that normally supports this phase of the PTP reaction cycle (Fig. 7; Supplementary Fig. 1). Consistent with this idea, mutation of His155 reduced catalytic activity by 95–98% (Table 1).
In conclusion, our multiple structural snapshots include substrate/enzyme crystal complexes for a Cys-based PTP-phosphatase that lacks a functional canonical catalytic acid. We have linked together these individual structural data-sets (Supplementary Table 1) to construct a reaction cycle that extends the chemical continuum of the PTP family reaction cycle, while not departing from an overall conservation of fundamental structural elements (Fig. 7). Consequently, we can now rationalize how AtPFA-DSP1 is optimized for regiospecific and rapid hydrolysis of the β-phosphates of PP-InsP substrates.
In the plant kingdom there is a wide distribution of orthologs of AtPFA-DSP1 (Supplementary Fig. 2g); over-expression of PFA-DSPs in transgenic Arabidopsis and rice down-regulates immune responses to pathogens47. Plant immunity is enhanced by 1,5-InsP823, and the latter is a preferred substrate for these PFA-DSPs (Fig. 1). Consequently, we propose a reduction in cellular 1,5-InsP8 levels upon phosphatase overexpression can account for the negative impact of these enzymes upon plant immune responses. Similarly, we propose that impaired tolerance to drought upon overexpression of OsPFA-DSP in transgenic rice48 arises out of an accompanying decrease in PP-InsP levels. Pursuit of AtPFA-DSP1 functionality will require further experiments performed in vivo, although we note that the PP-InsP phosphatase activity of the yeast ortholog (ScSiw14) has previously been verified in a deletion mutant strain of yeast20.
Our structural data also enrich the PTP research field by providing a valuable resource for future practical and theoretical studies to interrogate the reaction cycles of PFA-DSP1s (e.g. Supplementary Fig. 2g), and those of other PTP family members that also operate efficiently without a catalytic acid, such as PTEN14,15, human CDC25 phosphatase49, MCE116 and PIR1/PIR17 (Supplementary Fig. 2a). Finally, our data represent a significant extension to the known chemical versatility of the WPD loop, thereby greatly extending our understanding of its important contributions to tailoring reactivity profiles for individual PTP family members.
Methods
Protein expression and purification
For expression of AtPFA-DSP1, we purchased a codon-optimized cDNA (Genscript Inc.) with the following sequence: ATGAAACTGGTTGAAAAAACCACCACCACCGAACAGGATAATGGTGAAGATTTTTGTCGTACCATTATTGAAGTTAGCGAAGTTAATCGTAATGTTTTTCAGGCACCGGGTGGTGAAGCAGATCCGTTTCGTGTTGTTAGCGGTGAAGAACTGCATCTGATTCCGCCGCTGAATTTTTCTATGGTGGATAATGGCATTTTTCGCTCTGGCTTTCCGGATTCTGCTAATTTTTCTTTTCTGCAGACCCTGGGCCTGCGCTCAATTATTTATCTGTGCCCGGAACCGTATCCGGAATCAAATCTGCAGTTTCTGAAAAGTAATGGTATTCGTCTGTTTCAGTTTGGTATTGAAGGTAATAAAGAACCGTTTGTTAATATTCCGGATCATAAAATTCGTATGGCACTGAAAGTGCTGCTGGATGAAAAAAATCATCCGGTGCTGATTCATTGTAAACGTGGCAAACATCGTACCGGCTGTCTGGTGGGCTGCCTGCGCAAACTGCAGAAATGGTGCCTGACCTCAATTTTTGATGAATATCAGCGCTTTGCGGCTGCGAAAGCCCGCGTGTCAGATCAGCGTTTTATGGAAATTTTTGATGTGAGCAGCTTTAGCCATATTCCGATGAGTTTTAGTTGTTCTATTCGC
The Gateway expression system (Invitrogen) was used to subclone into the pDest-566 vector the cDNAs that encode one of several versions of AtPFA-DSP1: either full-length enzyme, or a series of full-length enzymes with single site mutations (using a site-directed mutagenesis kit (Stratagene), or residues 49-215, or residues 49-215 in which Cys150 was mutated (see below). This vector also encodes a 6xHis tag, maltose-binding protein tag, and tobacco etch virus protease cleavage site at the N terminus. All mutants were verified by sequencing. Primers are listed as below (mutations in upper case). All proteins were expressed and purified similarly.
C150S 5ʹ- gaaaaaaatcatccggtgctgattcatTCTaaacgtggcaaacatcgtaccg
C150S 3ʹ- cgatgtttgccacgtttAGAatgaatcagcaccggatgatttttttcatc
H155D 5ʹ-ctgattcattgtaaacgtggcaaaGATcgtaccggctgtctggtggg
H155D 3ʹ-caccagacagccggtacgATCtttgccacgtttacaatgaatcagcac
H155E 5ʹ-ctgattcattgtaaacgtggcaaaGAAcgtaccggctgtctggtggg
H155E 3ʹ-caccagacagccggtacgTTCtttgccacgtttacaatgaatcagcac
H155L 5ʹ- ctgattcattgtaaacgtggcaaaCTGcgtaccggctgtctggtggg
H155L 3ʹ- caccagacagccggtacgCAGtttgccacgtttacaatgaatcagcac
T157A5ʹ- cattgtaaacgtggcaaacatcgtGCCggctgtctggtgggctg
T157A 3ʹ- ccaccagacagccGGCacgatgtttgccacgtttacaatgaatc
D191A 5ʹ-gcccgcgtgtcaGCtcagcgttttatggaaatttttgatgtgagc
D191A 3ʹ-caaaaatttccataaaacgctgaGCtgacacgcgggctttcg
The recombinant plasmids were transformed into DE3 competent E. coli cells (Stratagene) that were pretransformed with chaperone plasmid pGro7 (Takara Clontech). An overnight culture of the transformed E. coli cells was inoculated into nutrient-rich 2 × YT medium (16 g/liter Tryptone, 10 g/liter yeast extract, 5.0 g/liter NaCl) supplemented with 0.07% (w/v) l-arabinose at pH 7.5 and grown at 37 °C to an A595 of 0.7. Isopropyl β-d-thiogalactopyranoside (0.1 mM) was then added, and cultures were continued at 15 °C for 20 h.
The cells were harvested by centrifugation at 5000 x g for 10 min and disrupted using a constant cell disruption system (Constant Systems Ltd.) under 20 k.p.s.i. Recombinant wild type (WT) and mutant proteins were purified by several chromatographic procedures performed at 4 °C. First, the protein was applied to a nickel-nitrilotriacetic acid-agarose column (Qiagen), which was then washed with buffer containing 300 mM NaCl, 20 mM Tris-HCl (pH 7.2), 20 mM imidazole. Protein was eluted by increasing the imidazole concentration to 400 mM. Next, the eluate was applied to a HiTrapTM Heparin HP column (Cytiva) and eluted with 10 column volumes of a 50–2000 mM NaCl gradient in 20 mM Tris-HCl (pH 7.2). After cleavage using the tobacco etch virus protease, the protein was further purified using another HiTrap Heparin HP column followed by a SuperdexTM 200 gel filtration column (Cytiva) that was eluted with 150 mM NaCl, 20 mM Tris-HCl (pH 7.2). Purified proteins were concentrated to 0.4–10 mg ml−1 and stored in aliquot at −80 °C. It is likely Pi in the protein’s active site (see Figures) is captured during the bacterial cultures, since Pi was not added to either the purification buffers or the crystallization buffers (see below).
Crystallization
The crystallization of core catalytic domain of AtPFA-DSP1 (residues 49–215) was optimized by hanging drop vapor diffusion against a well buffer of 0.4 M NaCl, 50 mM β-mercaptoethanol at 25 °C (3 μl of 5.5 mg/ml protein plus 1 μl of well buffer in the crystallization drop). The formed crystals were soaked in 30% PEG400, 13 mM MgCl2, 33 mM NaF, 50 mM β-mercaptoethanol, 66 mM HEPES, pH 7.2, and 0.05–10 mM ligands. For some experiments, the soaking buffer was changed to either 66 mM Tris-HCl (pH 8.0) or 50 mM N-cyclohexyl-2-aminoethanesulfonic acid. Soaking was performed at 25 °C for up to six days. More details for each of the resulting crystal complexes are listed in Supplementary Table 1.
Data collection, structure determination, and refinement
Diffraction data were collected using Advanced Photon Source beam line 22-ID and 22-BM. All data were processed with the program HKL200050. The complex structures of AtPFA-DSP1 were determined by molecular replacement from the previously reported AtPFA-DSP1 structure that has a significantly lower atomic resolution and did not contain any potential substrates (PDB: 1XRI).The initial structure was further rebuilt with Coot51 and refined with REFMAC52 from the CCP4 package. The molecular graphics representations were prepared with the program PyMOL (Schrödinger, LLC). Atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 7MOD, 7MOE, 7MOF, 7MOG, 7MOH, 7MOI, 7MOJ, 7MOK, 7MOL and 7MOM.
Enzyme assays
The catalytic activity of full-length AtPFA-DSP1 was originally screened using 10 μM concentrations of each of the PP-InsPs and the other indicated organic phosphates. All of the InsP7 isomers, and the 1,5-InsP8, were chemically synthesized and characterized as described previously53,54. New syntheses of 5-InsP7 and 1,5-InsP8 were performed as described previously53,55, and purity was verified as >95% by capillary electrophoresis mass spectrometry56 (Supplementary Fig. 14a,b). Previous work25,54,57 has described the synthesis and characterization of the other InsP7 isomers that have been used in this study, as well as the 5-PP-InsP4, and the 5-PCF2Am-InsP5. All other organic phosphates were purchased from Sigma–Aldrich. Technical replicates of the same protein sample were each performed in independent assays. Reactions (100 μl; 30 °C; 30 min) contained 20 mM HEPES (pH 7.2), 100 mM KCl, 0.8 mM MgCl2, and 20 μM EDTA. Reactions were quenched with 100 μl of phosphate detection reagent (36:1 v/v of 2.6% sodium molybdate in 2.5 M HCl: 0.126% malachite green chloride). Pi release was quantified from the absorbance at 620 nm58. In some assays, 10 mM β-mercaptoethanol was added to the reaction buffer; there was less than a 5% change in enzyme activity. Subsequently, reaction kinetics were determined for the most efficiently hydrolyzed substrates (1,5-InsP8, 4-InsP7, 5-InsP7, 6-InsP7 and 5-PP-InsP4), at various concentrations as described in the figures. Data were analyzed using GraphPad Prism.
Mutant versions of AtPFA-DSP1 were assayed as described above except for the C150S version; its low activity could not be accurately determined with the colorimetric method. Instead, 10 μM 5-InsP7 substrate was spiked with [3H]-radiolabeled 5-InsP7 (prepared as previously described59), reactions were quenched with perchloric acid59, neutralized59, and analyzed using a 250 × 4.6 mm Synchropak Q100 HPLC column. The elution gradient was generated by mixing Buffer A (1 mm Na2EDTA) with Buffer B (Buffer A plus 2.5 M NH4H2PO4, pH 4.0) as follows: 0–1 min, 30% B; 1–21 min, 30–60% B, 21–24 min, 60–70% B. The eluate was mixed in-line with 2.5 volumes of MonoFlow 4 scintillation fluid (National Diagnostics), and radioactivity was assayed with a BetaRam 6 C detector (LabLogic) using Laura6 data collection software.
Molecular dynamics simulations
The starting configurations for the molecular dynamics trajectories were based on PDB ID 7MOK; missing atoms and protons were introduced by using the leap module of Amber.1860, 29 Na+ and 29 Cl− ions were added to provide the 100 mM effective ionic concentration, plus an additional three Cl− ions for charge neutralization. The system was solvated in a box of TIP3P water with the box boundary extending to 20 Å from the nearest peptide atom (resulting in 50,481 atoms in the simulation box). All Lys, Arg, Glu and Asp residues are considered to be in their charged states. His149 and His155 were considered δ-protonated due to their proposed hydrogen-bonding with ligand; remaining His residues were deemed ε-protonated. Prior to equilibration, the solvated system was sequentially subjected to (1) 500 ps belly dynamics with fixed peptide, (2) minimization (5,000 steps), (3) constant temperature (200 K) and constant pressure (1 atm) dynamics (~1 ns) at fixed protein to assure a reasonable starting density around 1 g/cc, (4) minimization (5,000 steps), (5) step-wise heating MD at constant volume (to bring the temperature up to 300 K in 3 ns), and (6) constant volume simulation for 10 ns with a constraint force constant of 10 kcal/mol applied only on backbone heavy atoms. After releasing all constraining forces within the next 20 ns of the equilibration period, sampling was increased by performing four independent, constant temperature (Langevin thermostat) constant volume molecular dynamics simulations for 2 μs each. All trajectories were calculated using the PMEMD module of Amber.18 with 1 fs time step. Long range coulombic interactions were handled using the PME method with a 10 Å cut-off for the direct interactions. The amino-acid parameters were selected from the FF14SB forcefield of Amber.18, the phosphate forcefield was selected from the gaff2 parameters in Amber.18, and the charges (Supplementary Table 2) were generated from single point B3LYP/6-31 G* calculations of an optimized geometry using Gaussian-0961. The partial atomic charges and the gaff2 atom types used for Pi in the study are provided in Supplementary Table 3. At the salt concentration of 100 mM, the MMGBSA module with the standard parameters was used to estimate binding energies from 100 samples selected from molecular dynamics simulations for each pose of Pi.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon reasonable request. Structural data generated during the course of this study are available at the PDB under accession codes 7MOD, 7MOE, 7MOF, 7MOG, 7MOH, 7MOI, 7MOJ, 7MOK, 7MOL, 7MOM. Previously reported structural data used in the course of this study are available at the PDB under the following accession codes: 1XR1, 2YF0, 3RGO, 3I7Z, 6E3B, 6BYF, 7DHF. Source data are provided with this paper.
References
Hengge, A. C. Kinetic isotope effects in the characterization of catalysis by protein tyrosine phosphatases. Biochim Biophys. Acta 1854, 1768–1775 (2015).
Alonso, A. & Pulido, R. The extended human PTPome: a growing tyrosine phosphatase family. FEBS J. 283, 1404–1429 (2015).
Tautz, L., Critton, D. A. & Grotegut, S. Protein tyrosine phosphatases: structure, function, and implication in human disease. Methods Mol. Biol. 1053, 179–221 (2013).
Tonks, N. K. Protein tyrosine phosphatases-from housekeeping enzymes to master regulators of signal transduction. FEBS J. 280, 346–378 (2013).
Bheri, M., Mahiwal, S., Sanyal, S. K. & Pandey, G. K. Plant protein phosphatases: what do we know about their mechanism of action? FEBS J. 288, 756–785 (2021).
Wang, H., Gu, C., Rolfes, R. J., Jessen, H. J. & Shears, S. B. Structural and biochemical characterization of Siw14: a protein-tyrosine phosphatase fold that metabolizes inositol pyrophosphates. J. Biol. Chem. 293, 6905–6914 (2018).
Sankhala, R. S., Lokareddy, R. K. & Cingolani, G. Structure of human PIR1, an atypical dual-specificity phosphatase. Biochemistry 53, 862–871 (2014).
Martins, A. & Shuman, S. Mechanism of phosphoanhydride cleavage by baculovirus phosphatase. J. Biol. Chem. 275, 35070–35076 (2000).
Crean, R. M., Biler, M., van der Kamp, M. W., Hengge, A. C. & Kamerlin, S. C. L. Loop dynamics and enzyme catalysis in protein tyrosine phosphatases. J. Am. Chem. Soc. 143, 3830–3845 (2021).
Groves, M. R., Yao, Z. J., Roller, P. P., Burke, T. R. Jr. & Barford, D. Structural basis for inhibition of the protein tyrosine phosphatase 1B by phosphotyrosine peptide mimetics. Biochemistry 37, 17773–17783 (1998).
Kuznetsov, V. I. & Hengge, A. C. New functional aspects of the atypical protein tyrosine phosphatase VHZ. Biochemistry 52, 8012–8025 (2013).
Yun, H. Y. et al. Structural study reveals the temperature-dependent conformational flexibility of Tk-PTP, a protein tyrosine phosphatase from Thermococcus kodakaraensis KOD1. PLoS One 13, e0197635 (2018).
Rudolph, J. Cdc25 phosphatases: structure, specificity, and mechanism. Biochemistry 46, 3595–3604 (2007).
Xiao, Y. et al. PTEN catalysis of phospholipid dephosphorylation reaction follows a two-step mechanism in which the conserved aspartate-92 does not function as the general acid-mechanistic analysis of a familial Cowden disease-associated PTEN mutation. Cell Signal 19, 1434–1445 (2007).
Rodriguez-Escudero, I. et al. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum. Mol. Genet 20, 4132–4142 (2011).
Changela, A., Ho, C. K., Martins, A., Shuman, S. & Mondragon, A. Structure and mechanism of the RNA triphosphatase component of mammalian mRNA capping enzyme. EMBO J. 20, 2575–2586 (2001).
Kincaid, R. P., Lam, V. L., Chirayil, R. P., Randall, G. & Sullivan, C. S. RNA triphosphatase DUSP11 enables exonuclease XRN-mediated restriction of hepatitis C virus. Proc. Natl Acad. Sci. USA 115, 8197–8202 (2018).
Aceti, D. J. et al. Structural and functional characterization of a novel phosphatase from the Arabidopsis thaliana gene locus At1g05000. Proteins 73, 241–253 (2008).
Florio, T. J., Lokareddy, R. K., Gillilan, R. E. & Cingolani, G. Molecular architecture of the inositol phosphatase Siw14. Biochemistry 58, 534–545 (2019).
Steidle, E. A. et al. A novel inositol pyrophosphate phosphatase in Saccharomyces cerevisiae: Siw14 selectively cleaves the beta-phosphate from 5-diphosphoinositol pentakisphosphate (5PP-IP5). J. Biol. Chem. 291, 6772–6783 (2016).
Lorenzo-Orts, L., Couto, D. & Hothorn, M. Identity and functions of inorganic and inositol polyphosphates in plants. N. Phytol. 225, 637–652 (2020).
Gunawardana, D., Likic, V. A. & Gayler, K. R. A comprehensive bioinformatics analysis of the nudix superfamily in Arabidopsis thaliana. Comp. Funct. Genomics 2009, 820381 (2009).
Laha, D. et al. VIH2 regulates the synthesis of inositol pyrophosphate InsP8 and Jasmonate-dependent defenses in Arabidopsis. Plant Cell 27, 1082–1097 (2015).
Zhu, J. et al. Two bifunctional inositol pyrophosphate kinases/phosphatases control plant phosphate homeostasis. Elife 8, e43582 (2019).
Riley, A. M., Wang, H., Shears, S. B. & Potter, B. V. L. Synthesis of an α-phosphono-α,α-difluoroacetamide analogue of the diphosphoinositol pentakisphosphate 5-InsP7. Medchemcomm 10, 1165–1172 (2019).
Laussmann, T., Reddy, K. M., Reddy, K. K., Falck, J. R. & Vogel, G. Diphopho-myo-inositol phosphates from Dictyostelium identified as D-6-diphospho-myo-inositol pentakisphosphate and D-5,6-bisdiphospho-myo-inositol tetrakisphosphate. Biochem. J. 322, 31–33 (1997).
Riemer, E. et al. ITPK1 is an InsP6/ADP phosphotransferase that controls phosphate signaling in Arabidopsis. Mo. Plant 14, 1864–1880 (2021).
Shears, S. B. & Wang, H. Metabolism and functions of inositol pyrophosphates: insights gained from the application of synthetic analogues. Molecules 25, https://doi.org/10.3390/molecules25194515 (2020).
Combs, A. P. Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J. Med Chem. 53, 2333–2344 (2010).
Burke, T. R. Jr. & Lee, K. Phosphotyrosyl mimetics in the development of signal transduction inhibitors. Acc. Chem. Res 36, 426–433 (2003).
Wang, X. & Ma, Q. Wzb of Vibrio vulnificus represents a new group of low molecular weight protein tyrosine phosphatases with a unique insertion in the W-loop. J Biol Chem 296, https://doi.org/10.1016/j.jbc.2021.100280 (2021).
Xiao, J. et al. Structural and functional analysis of PTPMT1, a phosphatase required for cardiolipin synthesis. Proc. Natl Acad. Sci. USA 108, 11860–11865 (2011).
Pinkston, J. et al. Significant loop motions in the SsoPTP protein tyrosine phosphatase allow for dual general acid functionality. Biochemistry 60, 2888–2901 (2021).
Brandao, T. A., Hengge, A. C. & Johnson, S. J. Insights into the reaction of protein-tyrosine phosphatase 1B: crystal structures for transition state analogs of both catalytic steps. J. Biol. Chem. 285, 15874–15883 (2010).
Kumara Swamy, K. C. & Satish Kumar, N. New features in pentacoordinate phosphorus chemistry. Acc. Chem. Res 39, 324–333 (2006).
Asthagiri, D., Liu, T., Noodleman, L., Van Etten, R. L. & Bashford, D. On the role of the conserved aspartate in the hydrolysis of the phosphocysteine intermediate of the low molecular weight tyrosine phosphatase. J. Am. Chem. Soc. 126, 12677–12684 (2004).
Tanner, J. J., Parsons, Z. D., Cummings, A. H., Zhou, H. & Gates, K. S. Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid. Redox Signal 15, 77–97 (2011).
Lassila, J. K., Zalatan, J. G. & Herschlag, D. Biological phosphoryl-transfer reactions: understanding mechanism and catalysis. Annu. Rev. Biochem 80, 669–702 (2011).
Choe, J. Y., Iancu, C. V., Fromm, H. J. & Honzatko, R. B. Metaphosphate in the active site of fructose-1,6-bisphosphatase. J. Biol. Chem. 278, 16015–16020 (2003).
Shi, R. et al. Crystal structures of human muscle fructose-1,6-bisphosphatase: novel quaternary states, enhanced AMP affinity, and allosteric signal transmission pathway. PLOS ONE 8, e71242 (2013).
de Macedo-Ribeiro, S., Renirie, R., Wever, R. & Messerschmidt, A. Crystal structure of a trapped phosphate intermediate in vanadium apochloroperoxidase catalyzing a dephosphorylation reaction. Biochemistry 47, 929–934 (2008).
Lahiri, S. D., Zhang, G., Dunaway-Mariano, D. & Allen, K. N. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science 299, 2067–2071 (2003).
Baxter, N. J. et al. A Trojan horse transition state analogue generated by MgF3- formation in an enzyme active site. Proc. Natl Acad. Sci. USA 103, 14732–14737 (2006).
Hofer, F., Kraml, J., Kahler, U., Kamenik, A. S. & Liedl, K. R. Catalytic site pKa values of aspartic, cysteine, and serine proteases: constant pH MD simulations. J. Chem. Inf. Modeling 60, 3030–3042 (2020).
Kiani, F. A. & Fischer, S. Comparing the catalytic strategy of ATP hydrolysis in biomolecular motors. Phys. Chem. Chem. Phys. 18, 20219–20233 (2016).
Liao, S. M., Du, Q. S., Meng, J. Z., Pang, Z. W. & Huang, R. B. The multiple roles of histidine in protein interactions. Chem. Cent. J. 7, 44 (2013).
He, H. et al. Two homologous putative protein tyrosine phosphatases, OsPFA-DSP2 and AtPFA-DSP4, negatively regulate the pathogen response in transgenic plants. PLOS ONE 7, e34995 (2012).
Liu, B. et al. OsPFA-DSP1, a rice protein tyrosine phosphatase, negatively regulates drought stress responses in transgenic tobacco and rice plants. Plant Cell Rep. 31, 1021–1032 (2012).
Kristjansdottir, K. & Rudolph, J. Cdc25 phosphatases and cancer. Chem. Biol. 11, 1043–1051 (2004).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr 60, 2126–2132 (2004).
Winn, M. D., Murshudov, G. N. & Papiz, M. Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 (2003).
Capolicchio, S., Wang, H., Thakor, D. T., Shears, S. B. & Jessen, H. J. Synthesis of densely phosphorylated bis-1,5-diphospho-myo-inositol tetrakisphosphate and its enantiomer by bidirectional P-anhydride formation. Angew. Chem. Int Ed. Engl. 53, 9508–9511 (2014).
Capolicchio, S., Thakor, D. T., Linden, A. & Jessen, H. J. Synthesis of unsymmetric diphospho-inositol polyphosphates. Angew. Chem. Int Ed. Engl. 52, 6912–6916 (2013).
Pavlovic, I. et al. Cellular delivery and photochemical release of a caged inositol-pyrophosphate induces PH-domain translocation in cellulo. Nat. Commun. 7, 10622 (2016).
Qiu, D. et al. Analysis of inositol phosphate metabolism by capillary electrophoresis electrospray ionization mass spectrometry. Nat. Commun. 11, 6035 (2020).
Wang, H. et al. Synthetic inositol phosphate analogs reveal that PPIP5K2 has a surface-mounted substrate capture site that is a target for drug discovery. Chem. Biol. 21, 689–699 (2014).
Hoenig, M., Lee, R. J. & Ferguson, D. C. A microtiter plate assay for inorganic phosphate. J. Biochem Biophys. Methods 19, 249–251 (1989).
Weaver, J. D., Wang, H. & Shears, S. B. The kinetic properties of a human PPIP5K reveal that its kinase activities are protected against the consequences of a deteriorating cellular bioenergetic environment. Biosci. Rep. 33, 228–241 (2013).
Case, D. A. et al. Amber 2018. University of California, San Francisco (2018).
Frisch, M. J. et al. (Wallingford, CT; 2016).
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences, and the German Research Foundation (DFG) under Germany’s Excellence Strategy (CIBSS – EXC-2189 – Project ID 390939984) and (DFG Grant JE 572/4-1) and the Wellcome Trust; BVLP is a Wellcome Trust Senior Investigator (grant 101010). We are grateful to the NIEHS Collaborative crystallography group, and the Advanced Photon Source (APS) SE Regional Collaborative Access Team (SERCAT) 22-ID and 22-BM beam lines, for assistance with crystallographic data collection. This work was funded in part by The Wellcome Trust. For the purpose of open access the authors have applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission.
Funding
Open Access funding provided by the National Institutes of Health (NIH).
Author information
Authors and Affiliations
Contributions
H.W., N.J., G.Z., A.M.R., B.V.L.P. and H.J.J. prepared critical reagents and performed experiments. L.P. performed computational studies. B.V.L.P., H.J.J. and S.B.S. supervised the studies. All authors contributed to data analysis and preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Gino Cingolani, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Perera, L., Jork, N. et al. A structural exposé of noncanonical molecular reactivity within the protein tyrosine phosphatase WPD loop. Nat Commun 13, 2231 (2022). https://doi.org/10.1038/s41467-022-29673-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-29673-y
This article is cited by
-
Substrate promiscuity of inositol 1,4,5-trisphosphate kinase driven by structurally-modified ligands and active site plasticity
Nature Communications (2024)
-
Loop dynamics and the evolution of enzyme activity
Nature Reviews Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
